
Abstract
Single cell trajectory analysis is a computational approach that orders cells along a pseudotime axis. This temporal modeling approach allows the characterization of transitional processes such as lineage development, response to insult, and tissue regeneration. The concept can also be applied to resolve spatial organization of cells within the originating tissue. Known as temporal and spatial transcriptomics, respectively, these methods belong to the most powerful analytical techniques for quantitative gene expression data currently available. Here, we discuss three different approaches: principal component analysis, the ‘Monocle’ algorithm, and self-organizing maps. We use a previously published qRT-PCR dataset of single neuroblast cells isolated from the developing mouse inner ear to highlight the basic features of the three methods and their individual limitations, as well as the distinct advantages that make them useful for research on the inner ear. The complex developmental morphogenesis of the inner ear and its specific challenges such as the paucity of cells as well as important open questions such as sensory hair cell regeneration render this organ a prime target for single cell trajectory analysis strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Organ development, disease progression, and regeneration are examples of biological processes that affect the state and identity of cells over time. In a simple case, this can describe the transition from an unspecified progenitor cell to a more specialized lineage-committed differentiated cell, for example a sensory hair cell of the inner ear. Such a change can occur unidirectionally or in bifurcating form as inner ear precursor cells give rise to either neurons or to supporting cells and hair cells, the two major types of inner ear sensory epithelia (Fig. 1a). As development progresses, expression of progenitor cell-associated genes will mostly decrease whereas markers that confer cell-type identity, as well as cytomorphological and physiological specializations, will become successively upregulated. These changes in gene expression are universally translated in dissimilar transcriptomes (that is the mixture of all RNA molecules present in a cell at a given time) and can be measured quantitatively using gene arrays, quantitative (q)RT-PCR, or whole transcriptome shotgun sequencing platforms (RNA-Seq). A typical approach to studying the dynamic behavior of development-associated gene expression has been time-series experiments in which dissected tissues are sampled from defined consecutive time points and collectively subjected to quantitative expression analyses. Usually, this strategy results in the identification of stage-specific markers and global genetic co-regulatory modules that may play a role in orchestrating tissue development (Spellman et al. 1998; reviewed in Bar-Joseph et al. 2012). Nevertheless, biological systems are inherently dynamic and heterogeneous in nature and consequently the aforementioned conventional investigations will fall short of accurately mirroring cellular progression over time. The major difficulty arises from the fact that the bulk of cell groups rather than individual cells are assessed, which disregards cellular heterogeneity as a universal trait. Inevitably, this leads to an incomplete picture of the developmental process, as, for example gene-to-gene correlations in small subsets of cells are challenging to identify in bulk samples. Additionally, averaging artifacts can mask biologically relevant processes and can negatively interfere with correct data interpretation as shown in a number of studies (Bengtsson et al. 2005; Levsky and Singer 2003; Toriello et al. 2008). Even in cases of presumed homogenous cell populations (e.g., cell lines or purified cell types), asynchronicity effects will lead to a mixtures of cells where each member of the group may execute the same developmental program, yet at different paces. The study of single cells, on the other hand, presents a promising alternative to sufficiently address some if not all listed issues. Single-cell analysis allows for the discrimination of distinct subpopulations of cells that are transitory in character and delineate specific cell states within otherwise homogeneously appearing populations. To better understand a cell’s progression from one state to another, it is essential to characterize the many and often obscure intermediate conditions that connect both states. Resolving this process down to a cell-to-cell scale and being able to record quantitative data puts the researcher in a favorable position to start tackling the conundrum of cellular differentiation.

Trajectory analysis: conceptual overview. a Trajectories describe a directional route, which can serve as a model to describe cellular differences. Shown is an unbranched as well as a branched (bifurcated) type of trajectory. Single cells are organized along the vector and because they gradually differ in their transcriptomes, they represent temporally defined processes or spatial organization. b Schematic overview of a simplified trajectory analysis workflow to resolve temporal dynamics of a biological process. Individual cells are profiled at three distinct consecutive time points. Subsequently, cells are ordered along different trajectories according to the experimental time points. Trajectories may be merged to give rise to a joined vector that comprehensively describes the biological process
Successful applications of single cell analysis require (1) the ability to isolate individual cells from the tissue source, and (2) a platform to process them rapidly and in parallel. With the advent of fluorescence-activated cell sorting (FACS) and the availability of different transgenic reporter mice, it is now routine to selectively enrich for specific cellular subsets of organs like the inner ear (Doetzlhofer et al. 2006; Herget et al. 2013; Jan et al. 2011; Sinkkonen et al. 2011). Conventional flow cytometers are capable of depositing individual cells into single wells of multiwell plates for subsequent analyses. Secondly, the introduction of microfluidic circuit devices (Melin and Quake 2007; Whitesides 2006) has paved the way for successful single cell applications in a number of fields including cell culture and nucleic acid quantitation. These ‘lab-on-a-chip’ instruments enable the simultaneous and reliable measurement of hundreds of different parameters in hundreds of individual cells (Blow 2009). Commercial availability as well as numerous established methodologies allow researchers to obtain quantitative gene expression data in a matter of hours. Lastly, continuous advancements in next-generation sequencing technologies (such as Illumina and Ion Torrent platforms) have contributed further to the field, which has led to a steady increase of cell-multiplexing capability and decrease of overall expenses (Wang et al. 2009). For research on the inner ear, this is good news all along because particularly molecular studies have been hampered for decades by the scarcity of tissue material.
What is single cell trajectory analysis and why is it important?
Single cell trajectory analysis strives to utilize biological heterogeneity among related cells that undergo changes over time. A trajectory describes a directional path along which individual cells can be arranged, such that their order represents for instance a temporally defined process. This can be achieved by applying mathematical analysis techniques aimed at identifying patterns in high-dimensional data—the data format distinctive of quantitative expression studies, such as the output of multiplex single cell qRT-PCR or RNA-Seq experiments. The reason why this strategy, that is generally referred to as ‘temporal transcriptomics’, works can be explained by the fact that no two individual cells are exactly the same even within most homogeneous cell populations. Sources for such an omnipresent degree of cellular heterogeneity have been identified en masse in the past and seem to have crucial impacts on cells of developing organs/organisms in general (Arias and Hayward 2006; Hayashi et al. 2008; Losick and Desplan 2008; Raj and van Oudenaarden 2008). For instance, extrinsic stimuli may trigger non-uniform responses of cells in a clonal or isogenic cell population. Niche compartments in vivo are illustrative examples in which cells may have different access to environmental determinants. Another example is cultured cells such as human myoblast cells that undergo induced differentiation and may respond differently based on cell-to-cell contact deviations or other reasons (Trapnell et al. 2014). As a result, cells profiled collectively at one static time point after the trigger always differ from another depending on the kind and rate of response. These often minuscule differences are reflected in successive changes of global gene expression that can be used to reconstruct temporal patterns (i.e. trajectories; Fig. 1b).
Compressing high-dimension data into a single dimension by formulating an in silico progression model results in a vector. Along it, individual cells are organized such that each of them resides at a particular stage of the process and therefore represents a singular pseudotime point. This means that, in a traditional time-series experiment, each respective time point would represent a separate time-series study by itself (for example, time points 1, 2, and 3 in Fig. 1b). If cellular differentiation is the underlying biological process, and if the genes that are assayed construe the various steps of the process, then there is a high likelihood that the resulting cell trajectory derived from a single time point will describe cell differentiation. Connecting trajectories of multiple time points can additionally enhance the biological integrity and coherence of the model. Variably chosen time intervals (e.g., hours, days, weeks) will lead to variable degrees of trajectory overlap and as a result can describe the differentiation process over multiple sampling time points across varying timescales (Fig. 1b). The power of this approach is that it reveals the order of molecular events as cells transit over time such as from a progenitor state into a differentiating and subsequently into a differentiated state. Quantitative information on select groups of genes (if multiplex qRT-PCR is being used) or on all detectable genes expressed in individual cells (for RNA-Seq datasets) is available for each single cell along the pseudotime-axis, and allows the researcher to extract knowledge with unprecedented efficiency and resolution. In turn, this contributes to a better understanding of how cells change from one state to another during the time period investigated and enables the deciphering of the mechanisms involved during these changes.
A possible limitation that could influence the sequence of individual cells along a trajectory relates to the characteristic process of transcription, which is stochastic to a certain extent and can happen in bursts (Raj and van Oudenaarden 2008). Specifically, the initiation of gene expression follows stochastic principles leading to random differences in transcript levels in cells that just start expressing a certain gene (van Roon et al. 1989). Additional random fluctuations in availability of proteins and factors involved in mRNA synthesis at any given time result in phenotypical differences between otherwise identical cells (McAdams and Arkin 1997). Once mRNA synthesis has reached a steady state, it is conceivable that the concentration of a specific transcript in an individual cell becomes mostly defined by the “burst” or pulse duration and its frequency. The low and high limits of transcript concentrations consequently are different in each individual cell and differ for each individual gene (Fig. 2a–c). The question of how much of the variation of gene expression levels between individual cells can be attributed to biological-associated heterogeneity rather than just ‘noise’ requires the utilization of a multidimensional approach that considers gene expression data from many closely related cells as well as many genes. In addition, the analysis methods described in this review do not reduce quantitative gene expression information to a binary code, but consider distinct expression level ranges (Fig. 2a–c), a principle that substantially increases the available complexity of relevant information. Ordering of cells along a trajectory consequently does not rely on binary information (i.e., on versus off) of a few genes, but takes into consideration information extracted from the quantitative transcript measures of many genes. As a consequence, the more genes of the assay (in case of multiplex single cell qRT-PCR) or the more detectable genes with RNA-Seq are correlated with a trajectory, the smaller the potential error introduced by stochastic gene expression. With the same plausibility, stochastic events will also even out with increasing sample sizes. We conclude that, although stochastic gene expression could affect the outcome of trajectory analyses at low sample sizes, and when only a few genes are taken into consideration, its effects become less significant and probably negligible as sample size and gene number increase.

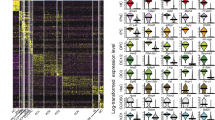
Violin plots of select transcript expression histograms in otic vesicle/neuroblast cells. a Expression distributions for “housekeeping genes” Actb and Gapdh in 382 individual cells isolated from the mouse otocyst and delaminating neuroblasts. Note that all cells express the two genes and that expression levels range between a low and high level on the log2 scale of expression. b Expression distributions of Notch2 and Pax2 mRNA show a unimodal distribution of cells that express the genes at different levels. A fraction of cells do not express these genes, illustrated by the peaks centered at zero. c Examples for multimodal expression such as Tbx2. MRNA is detectable in a majority of the 382 cells but with distinct peaks at different expression levels that likely reflect biological heterogeneity and not simply noise. Likewise, Eya2 transcripts center around two distinct expression level maxima when detected, indicating the presence of two distinct subpopulations of cells (bimodal distribution)
Temporal transcriptomics in inner ear research
In the developing inner ear, we recently utilized trajectory analysis to describe the lineage progression of neuroblasts (Durruthy-Durruthy et al. 2014a). These neural precursor cells are born in the ventro-anterior region of the otocyst, a transiently existing vesicle that forms in mice during the 10th day post-fertilization. They delaminate from the otocyst and migrate ventro-medially. As development proceeds, neuroblasts accumulate, proliferate, and eventually differentiate into the neurons that ultimately form the ganglia innervating the organ of Corti and the vestibular organs of the inner ear (Rubel and Fritzsch 2002). To analyze not only the neuroblast lineage but also the cell population that constitutes the otocyst, we used a Pax2-Cre transgenic mouse line to permanently label cells of the otic lineage in a fluorescent reporter mouse line. A total of 384 cells were sorted by FACS and subsequently examined by multiplex qRT-PCR for expression of 96 preselected otic genes. A number of clustering approaches and dimension-reduction techniques, such as principal component analysis (PCA), revealed that about 30 % of all cells were of neuronal lineage identity (Durruthy-Durruthy et al. 2014a). This group was further partitioned into two related subgroups, one with prevalent expression of genes found in early neuroblasts such as Neurog1 (Ma et al. 1998), whereas the second group was defined by markers that become upregulated, as neuron precursors migrate and differentiate, for example Isl1 (Li et al. 2004). PCA seeks to identify the directions defined by vectors in multi-dimensional variable space along which the variance of the data is greatest. Known as principal components, these vectors are ordered ascendingly according to their overall contribution to variability. We noted that by far the majority of variability of neuroblast-associated cells was preserved in the very first component, which suggested that only one single dimension might adequately visualize differences between cells. Upon projection of neuronal cells onto the first component, we interpreted the resulting trajectory as a measure of time (Fig. 3a, b), as described in Durruthy-Durruthy et al. (2014a). Cells ordered along the pseudotime-line displayed the transition from an early neuroblast state, likely representing the newly emerging or early delaminating phase towards a differentiated state where cells began displaying neural identity. Although most genes in this particular study were not included to exclusively describe the neuroblast lineage per se, we succeeded in identifying a number of interesting co-regulatory motifs as well as regulatory components indicated by expression of signaling pathway-associated markers. Ultimately, a more targeted collection of genes will be necessary to thoroughly resolve specific events of neuroblast differentiation starting from the moment of cell fate initiation, delamination, migration, accumulation, transient proliferation, and bifurcation into cochlear and vestibular fates, experiments that are currently under way.

Comparison of three different unsupervised, exploratory approaches to study progression of otic neurogenesis over time along a trajectory. a Schematic overview of the mathematical principle that governs each of the following approaches: principal component analysis (PCA, left), independent component analysis (ICA, center), and self-organizing maps (right). For each case, hypothetical data points are shown and their spread across a two-dimensional (2D) coordinate system. Left: In PCA, red arrows indicate the two principal components (PC) of the data and their orthogonal relationship. PC1 follows the direction of the largest variability of the data. Center: Red arrows refer to the two independent components of the data. The drawing suggests that they present the original data more accurately than PCA as IC1 and IC2 are not required to stand perpendicular to each other. Right: The final 2D-lattice (red) of 9 neurons is shown as it spans across the 2D-data space after the hypothetical training process of the artificial neural network. Topological features of the data are preserved in the neuron-organization of the self-organizing maps. b Visualization of expression data for three marker genes for each of the three approaches. Top left: Adopted from Durruthy-Durruthy et al. (2014a). Individual points represent cells and are color-coded according to gene expression levels. Cells are ordered along the first principal component vector. Right: Individual cells are shown in red and organized across a pseudo-time axis after the ‘Monocle’ algorithm. Distribution of cells is fitted (line) and shows additional information of the dynamics of otic neurogenesis. Bottom left: Self-organizing maps color-coded with mean expression levels for each of the three markers. The map consists of 15 neurons of which each contains a variable number of individual cells
The question that arises from this first example of single cell trajectory analysis of otic derivatives is whether one-dimensional PCA is robust enough to be applicable to other biological systems inside and outside of the inner ear. In the following section, we introduce alternative methods for establishing single cell trajectories and apply them to our existing dataset from Durruthy-Durruthy et al. (2014a, b). We will discuss the major differences between them as well as advantages that each procedure offers for potential applications to cell trajectories in the developing, regenerating, and mature inner ear.
Limitations of PCA-based trajectory analyses
A disadvantage of one-dimensional PCA lies in its inability to resolve processes following trends that are not of a unidirectional nature. Lineage bifurcations, which are common events in developing organs, as well as disturbances that arise from secondary and higher order biological processes, cannot be ascribed by multi-dimensional data reduction to a single vector/trajectory. This is because one-dimensional PCA requires the majority of variability inherent to the dataset to be represented by a single principal component. Therefore, in more complex scenarios, it is important to acknowledge the interlaced structure of assorted biological sequences of events that might take place in parallel and can fully or partially overlap. Moreover, for some processes, we will not have a priori knowledge about distinguishing genes making it challenging to define differential trajectories. These conceptual difficulties require alternative mathematical algorithms.
A specific example of aforementioned complexity addresses the study of sensory hair cell regeneration in non-mammalian vertebrate utricles, an ongoing project in our laboratory. Here, we presume that ototoxic insult resulting in hair cell loss leads to a series of events orchestrated by dying hair cells and surviving supporting cells that initiate a highly efficient regenerative program. The chicken inner ear utilizes two general mechanisms of hair cell regeneration (Adler and Raphael 1996; Cafaro et al. 2007; Corwin and Cotanche 1988; Roberson et al. 1992, 2004; Ryals and Rubel 1988): (1) the direct conversion of supporting cells into hair cells, a process also known as direct transdifferentiation, and (2) the asymmetric division of supporting cells into new hair cells and supporting cells. We hypothesize that both processes will be carried out by different classes of supporting cells, but the time course and genetic programs regulating initiation, progression, and termination of the different modes of hair cell regeneration will transpire in distinctively different lineage trajectories. We speculate direct transdifferentiation will follow a unidirectional path, whereas asymmetric division of supporting cells will be featured in more complex aspects of the data with multiple biological processes implicated (cell cycle re-entry, cell division, bifurcating lineage, and cell differentiation). Unraveling these heterogeneous supporting cell groups and assembling different lineage trajectories in this specific case will require new inventive analytical approaches that go beyond simple cluster analysis followed by PCA. In this review, we will not be able to solve the intricacies of hair cell regeneration, although it has been part of an important and highly relevant debate of our field. However, we are able to highlight the potential of single cell analysis and how it can profit our domain, as researchers will undoubtedly be confronted with high-dimensional datasets in the upcoming years.
‘Monocle’ and one-dimensional self-organizing maps as alternatives
Other procedures to order cells or populations of cells on a temporal scale exist and bypass the limitations of PCA-based algorithms (el Amir et al. 2013; Bendall et al. 2011, 2014).
‘Monocle’ is a recently introduced method that places whole-transcriptome profiles of single cells along an artificial temporal curve in an orderly fashion (Trapnell et al. 2014). Using individual primary human skeletal muscle myoblasts, Trapnell and colleagues impressively showcased the power of their unsupervised approach when applied to single cells by improving temporal resolution during a dynamic biological process such as differentiation. Assuming that cellular differentiation can be described as a continuous procedure with several intermediate states, ‘Monocle’ aims to model this process by identifying a starting and end point(s) of cell trajectories in high-dimensional space. As the number of dimensions directly correlates with the number of genes by which an individual cell is characterized, ‘Monocle’ implements—similarly to one-dimensional PCA—a dimension-reduction step prior to cell ordering. Yet, instead of recognizing the principal components of the data structure, their approach attempts to establish the independent components of the data using independent component analysis (ICA) (Hyvarinen 1997; Hyvarinen and Oja 2000). Although ICA is generally regarded as a generalization of PCA as it calculates vectors in multi-dimensional space as linear combinations, both operations are fundamentally different from an analytical perspective (Draper et al. 2003). Whereas PCA assumes the data to be normally distributed and organized linearly, ICA expects non-Gaussian data dispersion. Likewise, the overall goal of PCA is to identify the direction in high-dimensional space along which data variance is maximized so that the lower-dimension data-projection error is minimized (Jolliffe 2002). In contrast, ICA intends to minimize statistical dependence among vectors (thus, they are as independent from each other as possible) and coincidently maximize the non-normal distribution of vector elements. As a result, components derived from PCA have an associated importance value (called eigenvalue), are arranged accordingly, and have an orthogonal relationship, contrary to components computed from ICA, which exhibit equal significance and are constructed non-orthogonally (Fig. 3a, left, center). These mathematical differences can have far-reaching consequences in successfully recognizing patterns in differentially structured biological data that relate to processes like differentiation, regeneration, or disease progression.
An additional criterion distinguishing ‘Monocle’ from 1D-PCA is the modality of cell ordering. Whereas in PCA (Fig. 3b, top left; Durruthy-Durruthy et al. 2014a), the sequence of cells along the trajectory is simply the result of data projection, ‘Monocle’ makes use of so-called minimum spanning trees to connect cells along the longest possible path involving as many cells as possible (Trapnell et al. 2014). This strategy is particularly useful in dissecting biological processes that cannot be explained as unbranched processes and may occur in parallel. Notably, during differentiation, a progenitor cell may give rise to more than one lineage, leading to two or more lineage trajectories. This level of complexity, generally termed bifurcation, can be described using ‘Monocle’. In addition, Monocle allows for subsequent cluster analysis comparing two or more groups of cells as a function of pseudotime and identifying differentially expressed genes.
To test how the algorithm performs on datasets other than those Trapnell and colleagues described in their study, we utilized single cell qRT-PCR data from our neuroblast-associated cell group that was assembled by a multitude of clustering assessments. We focused on the three hallmark genes that possibly best describe the progress of early inner ear neurogenesis. The protocol involves the initial selection of two input parameters, namely (1) the genes whose expression data are used to ‘order’ all cells, and (2) the number of paths on which one expects the cells to associate. ‘Ordering genes’ as the authors refer to them are supposed to comprise a group of markers that best explain the dynamic behavior of the biological process, hence their expression profiles across single cells vary considerably. We designated Neurog1, Neurod1, and Isl1 for sequence determination, and visualized their expression levels on three equivalent trajectories. Due to the lack of distinctive markers, we chose only one trajectory path without bifurcating attributes. In a similar manner to published data (Durruthy-Durruthy et al. 2014a), neuroblast-specifier Neurog1 becomes gradually downregulated along the axis as one follows individual cells from left to right (Fig. 3b, right). Markers that are expressed at later stages of otic neurogenesis markedly label cells on the right side of the model, mirroring the further advanced stage of maturation. Interestingly, Neurod1 and Isl1 show a synchronous order of events, defined by an increase of expression, followed by a decrease, which is then again succeeded by marker upregulation towards the end of the axis. Altogether, the trend of all three markers, although not immediately detectable for Neurod1 and Isl1 in our 1D-PCA model, coincides with our published findings. Nonetheless, in this particular sample, Monocle facilitates a readier assessment of differences in gene expression along the trajectory.
As a reminder, regardless of the approach employed, the series of cells along the trajectory does not necessarily indicate how similar two cells are, nor does it allow determining a particular stage of the process. It does, however, permit the conclusion that a cell that precedes another cell is less advanced in the process of investigation, and vice versa.
Notwithstanding the fact that 1D-PCA and Monocle differ in various mathematical ways, they have one commonality that may limit their overall utility in recognizing additional biologically relevant patterns: both assume the variables to conform a linear relationship, which is not always the case and prevents the identification of motifs that are of non-linear character. A simple example is the correlative but non-linear behavior of expression levels of two genes. A continuous increase of expression of a specific gene could be accompanied by a discontinuous (such as exponential or binary) change of expression of the second gene.
As non-linear but correlated relationships have implications in many biological systems, we propose as a third practical alternative the use of self-organizing maps (SOMs), a subtype of artificial neural networks (Kohonen et al. 2001). Neural network terminology refers to a general operating principle of the sensory nervous system where input signals are projected onto particular territories in the brain. For instance, neighboring photoreceptors in the retina when stimulated elicit a response in the visual cortex of the cerebellum with a distinct topographical arrangement. Surprisingly, SOMs have not been as widely used as PCA in analysis of multivariate datasets derived from quantitative RNA measurements of single or bulk cells. Tamayo and colleagues were one of the first to apply SOMs to describe hematopoietic differentiation and yeast cell-cycle regulation in several well-studied cell-line models (Tamayo et al. 1999). Instead of classifying samples, the authors were able to group genes in functionally related cohorts and conclusively demonstrated how SOMs can greatly support data interpretation. The key distinction to the afore-discussed procedures is that dimension reduction in SOMs operates in a topology-preserving mode. Thus, neighborhood relationships and distance associations between single cells in high-dimensional space are conserved when presented in a lower-dimensional format. Considering data points (cells) positioned in a multi-variable coordinate system with objects positioned further away from each other the more different they are, may effectively help in capturing dissimilarities. Usually, the low-dimension space comprises a 2D-lattice that is composed of interconnecting nodes (called neurons) which itself encompasses similar, classified objects, such as cells. The degree of ‘similarity’ thus refers to how close two cells are in multivariate space where their location is exclusively determined by their expression profiles (Fig. 3a, right).
A central feature of artificial neural networks is their capability to learn, unlike non-machine-learning algorithms such as ICA or PCA. In SOMs, this learning phase happens adaptively such that observations (i.e., cells) of an input layer are successively presented to a pre-defined number of adjoined neurons in an output layer. Generally, the number of neurons is smaller than the number of input cells. If expression data are provided, this reiterative learning process ensures that nearby cells are eventually classified in the same or adjacently located nodes/neurons. After training and mapping is completed, the resulting maps can serve as a platform for various clustering approaches to identify distinct territories that distinguish groups of cells.
To illustrate the potential of this strategy, we tested the qRT-PCR data from the identified neuroblast population. We chose a one-dimensional 15 × 1 neuron-lattice as output layout and exemplary visualized expression data of the previously introduced neuroblast markers (Fig. 3b, bottom left). In comparison to 1D-PCA and Monocle, the output of this SOM analysis shows an analogous expression distribution of all three genes, characterized by contrasting gradients of markers Neurog1 and Isl1, and largely unchanged expression of Neurod1 across all nodes. Even though one loses single cell resolution as nodes represent classifier objects that include cells in close high-dimension proximity, we conjecture that self-organizing maps, especially when examining hundreds or thousands of cells, will offer an invaluable tool to parse multivariate expression data from single cells of developing organs. Furthermore, SOMs are suitable for subsequent cluster analysis that in turn may promote data interpretation.
Conclusion and outlook
The wonderfully complex morphology and physiology of the inner ear offers a multitude of applications for single cell trajectory analysis. Trajectories do not necessarily have to be of a temporal nature but could also be applied to systems with spatial coordinates, a concept that is referred to as ‘spatial transcriptomics’. Rather than early versus late, opposing ends of the output curve could represent dorsal versus ventral, for instance. Recent projects that utilize high-dimensional quantitative transcriptome data to reconstruct the mouse otocyst in the form of a hollow sphere (Durruthy-Durruthy et al. 2014a, b) illustrate how single cell spatial transcriptomics can have the potential to greatly impact research in inner ear biology. A prime example for another useful application of spatial transcriptomics is the molecular analysis of the tonotopic organization of the cochlear duct in mammals. Gradual changes in hair cell morphology as well as physiologically tonotopic gradients along the basal-to-apical cochlear axis manifest themselves in changes of hair cell mechanoelectrical transduction channel properties, calcium buffers, and synapses (Holton and Hudspeth 1983; Liberman et al. 2011; Mutai et al. 2005; Ricci et al. 2000, 2003). Utilizing single cell trajectory analysis of qRT-PCR or RNA-Seq data, these spatially encrypted variations can be studied at high resolution. We envision that such experiments can contribute to a better understanding of how molecular gradients bestow hair cells with highly specialized and gradually changing features, allowing them to maintain physiological properties that are in perfect alignment with their position along the cochlear frequency map. Of course, this exploration will not halt at inner and outer hair cells, it can be extended to analyze different supporting cell subtypes as well as to study processes that govern tonotopy establishment during development.
The ability to profile thousands of genes in individual cells has remarkably improved our understanding of organ development (Brunskill et al. 2014; Durruthy-Durruthy et al. 2014a), tissue homeostasis (Buczacki et al. 2013), and regeneration (Pina et al. 2012) in various biological systems. Whereas nowadays expression profiles of hundreds of single cells are routinely assayed in one experiment, soon this number will climb to the thousands, if not much higher. Methodologies that are proficient at dealing with this mounting data are needed, and their successful implementation will inevitably determine the pace and direction of the development of single cell research in the future.
The concept discussed here of computationally constructed single cell trajectories may be universally applied and is (hopefully) particularly promising for the inner ear research community. Understanding lineage formation and progression, such as of the prosensory domain cells in the vestibular system, the cochlea, as well as the aforementioned neuroblasts, are prerequisites in developing cell-based therapeutic approaches. Likewise, we envision that this methodology will allow researchers to identify the distinct steps that initiate, execute, maintain, and terminate the process of hair cell regeneration in the non-mammalian inner ear. This knowledge, in turn, when paired with future studies conducted in mammals, which in the adult show dysfunctional regenerative abilities, could indeed speed up the process of finding novel cures for hearing loss. There are many additional possibilities, and we would like to conclude with advising overexcited researchers like ourselves to exploit different existing and emerging novel data analysis methods, which without doubt will increasingly improve our understanding of biology and favorably impact the progress of medicine in future years.
References
Adler HJ, Raphael Y (1996) New hair cells arise from supporting cell conversion in the acoustically damaged chick inner ear. Neurosci Lett 205:17–20
Arias AM, Hayward P (2006) Filtering transcriptional noise during development: concepts and mechanisms. Nat Rev Genet 7:34–44
Bar-Joseph Z, Gitter A, Simon I (2012) Studying and modelling dynamic biological processes using time-series gene expression data. Nat Rev Genet 13:552–564
Bendall SC, Simonds EF, Qiu P, el Amir AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP (2011) Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332:687–696
Bendall SC, Davis KL, el Amir AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe’er D (2014) Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157:714–725
Bengtsson M, Stahlberg A, Rorsman P, Kubista M (2005) Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res 15:1388–1392
Blow N (2009) Microfluidics: the great divide. Nat Methods 6:683–685
Brunskill EW, Park JS, Chung E, Chen F, Magella B, Potter SS (2014) Single cell dissection of early kidney development: multilineage priming. Development 141:3093–3101
Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ (2013) Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495:65–69
Cafaro J, Lee GS, Stone JS (2007) Atoh1 expression defines activated progenitors and differentiating hair cells during avian hair cell regeneration. Dev Dyn 236:156–170
Corwin JT, Cotanche DA (1988) Regeneration of sensory hair cells after acoustic trauma. Science 240:1772–1774
Doetzlhofer A, White P, Lee YS, Groves A, Segil N (2006) Prospective identification and purification of hair cell and supporting cell progenitors from the embryonic cochlea. Brain Res 1091:282–288
Draper BA, Kyungim B, Stewart Bartlett M, Beveridge JR (2003) Regognizing faces with PCA and ICA. Comp Vision Image Underst 91:115–137
Durruthy-Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, Laske RD, Altman R, Heller S (2014a) Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell 157:964–978
Durruthy-Durruthy R, Gottlieb A, Heller S (2014b) Three-dimensional computational reconstruction of tissues with hollow spherical morphologies using single cell gene expression data. Nat Protocols. doi:10.1038/nprot.2015.022
el Amir AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D (2013) viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31:545–552
Hayashi K, Lopes SM, Tang F, Surani MA (2008) Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell 3:391–401
Herget M, Scheibinger M, Guo Z, Jan TA, Adams CM, Cheng AG, Heller S (2013) A simple method for purification of vestibular hair cells and non-sensory cells, and application for proteomic analysis. PLoS ONE 8:e66026
Holton T, Hudspeth AJ (1983) A micromechanical contribution to cochlear tuning and tonotopic organization. Science 222:508–510
Hyvarinen A (1997) Independent component analysis by minimization of mutual information, vol Report A46. Helsinki University of Technology, Helsinki
Hyvarinen A, Oja E (2000) Independent component analysis: algorithms and applications. Neural Netw 13:411–430
Jan TA, Chai R, Sayyid ZN, Cheng AG (2011) Isolating LacZ-expressing cells from mouse inner ear tissues using flow cytometry. J Vis Exp e3432
Jolliffe IT (2002) Principal Component Analysis. Springer, New York
Kohonen T, Kaski S, Somervuo P, Lagus K, Oja M, Paatero V (2001) Self-organizing maps. Springer Series in Information Sciences, vol 30. Springer, Berlin, pp 114–122
Levsky JM, Singer RH (2003) Gene expression and the myth of the average cell. Trends Cell Biol 13:4–6
Li H, Liu H, Sage C, Huang M, Chen ZY, Heller S (2004) Islet-1 expression in the developing chicken inner ear. J Comp Neurol 477:1–10
Liberman LD, Wang H, Liberman MC (2011) Opposing gradients of ribbon size and AMPA receptor expression underlie sensitivity differences among cochlear-nerve/hair-cell synapses. J Neurosci 31:801–808
Losick R, Desplan C (2008) Stochasticity and cell fate. Science 320:65–68
Ma Q, Chen Z, del Barco BI, de la Pompa JL, Anderson DJ (1998) neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 20:469–482
McAdams HH, Arkin A (1997) Stochastic mechanisms in gene expression. Proc Natl Acad Sci U S A 94:814–819
Melin J, Quake SR (2007) Microfluidic large-scale integration: the evolution of design rules for biological automation. Annu Rev Biophys Biomol Struct 36:213–231
Mutai H, Mann S, Heller S (2005) Identification of chicken transmembrane channel-like (TMC) genes: expression analysis in the cochlea. Neuroscience 132:1115–1122
Pina C, Fugazza C, Tipping AJ, Brown J, Soneji S, Teles J, Peterson C, Enver T (2012) Inferring rules of lineage commitment in haematopoiesis. Nat Cell Biol 14:287–294
Raj A, van Oudenaarden A (2008) Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135:216–226
Ricci AJ, Gray-Keller M, Fettiplace R (2000) Tonotopic variations of calcium signalling in turtle auditory hair cells. J Physiol 524(Pt 2):423–436
Ricci AJ, Crawford AC, Fettiplace R (2003) Tonotopic variation in the conductance of the hair cell mechanotransducer channel. Neuron 40:983–990
Roberson DF, Weisleder P, Bohrer PS, Rubel EW (1992) Ongoing production of sensory cells in the vestibular epithelium of the chick. Hear Res 57:166–174
Roberson DW, Alosi JA, Cotanche DA (2004) Direct transdifferentiation gives rise to the earliest new hair cells in regenerating avian auditory epithelium. J Neurosci Res 78:461–471
Rubel EW, Fritzsch B (2002) Auditory system development: primary auditory neurons and their targets. Annu Rev Neurosci 25:51–101
Ryals BM, Rubel EW (1988) Hair cell regeneration after acoustic trauma in adult Coturnix quail. Science 240:1774–1776
Sinkkonen ST, Chai R, Jan TA, Hartman BH, Laske RD, Gahlen F, Sinkkonen W, Cheng AG, Oshima K, Heller S (2011) Intrinsic regenerative potential of murine cochlear supporting cells. Sci Rep 1:26
Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B (1998) Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 9:3273–3297
Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR (1999) Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci U S A 96:2907–2912
Toriello NM, Douglas ES, Thaitrong N, Hsiao SC, Francis MB, Bertozzi CR, Mathies RA (2008) Integrated microfluidic bioprocessor for single-cell gene expression analysis. Proc Natl Acad Sci U S A 105:20173–20178
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32:381–386
van Roon MA, Aten JA, van Oven CH, Charles R, Lamers WH (1989) The initiation of hepatocyte-specific gene expression within embryonic hepatocytes is a stochastic event. Dev Biol 136:508–516
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Whitesides GM (2006) The origins and the future of microfluidics. Nature 442:368–373
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Durruthy-Durruthy, R., Heller, S. Applications for single cell trajectory analysis in inner ear development and regeneration. Cell Tissue Res 361, 49–57 (2015). https://doi.org/10.1007/s00441-014-2079-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-014-2079-2