
Abstract
The endothelial layer of blood vessels controls the passage of cells and solutes from the blood into the surrounding tissue. Crucial for this regulation is the integrity of endothelial cell–cell junctions. Various molecular mechanisms control junctional integrity of the endothelial layer including GTPases, modulation of the actomyosin cytoskeleton and phosphorylation and dephosphorylation of junctional proteins. Several kinases and phosphatases have been identified that are good candidates for the regulation of the endothelial barrier function. For some of them, in vivo evidence has recently been presented that highlights their importance in either the regulation of vascular permeability or leukocyte extravasation. This review will summarize current knowledge about the regulation of endothelial junctions by kinases and phosphatases. In particular, the role of the endothelial specific phosphatase VE-PTP in the context of endothelial cell contact stability will be highlighted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The endothelial barrier
The integrity of blood vessels is crucial for tissue homeostasis and appropriate functioning of the immune system. Consequently, vascular permeability and the extravasation of leukocytes are tightly regulated. A central player in controlling vascular integrity is the endothelium of the blood vessel wall, which serves as a semipermeable barrier between the blood and the surrounding tissue. Solutes and leukocytes can pass the endothelial barrier either by a trans-cellular route directly through the body of an endothelial cell or through a para-cellular pathway by mechanisms that reversibly open and close endothelial junctions. The constitutive passage of solutes through resting endothelium is mainly mediated by a transcellular pathway, whereas inflammation-induced enhancement of vascular permeability mainly relies on the opening of endothelial junctions (Majno and Palade 1961; Mehta and Malik 2006; Schulte et al. 2011). Leukocytes can also directly pass through endothelial cells or through junctions, as has been demonstrated in vitro as well as in vivo (Carman and Springer 2004; Feng et al. 1998; Ley et al. 2007; Schoefl 1972; Vestweber 2007). However, recent in vivo studies analyzing large numbers of extravasating leukocytes revealed that the paracellular route is the major pathway in several tissues (Küppers et al. 2013; Schulte et al. 2011; Woodfin et al. 2011). Thus, the control of endothelial junctions is of central importance for inflammation-induced vascular permeability and leukocyte recruitment.
Several transmembrane adhesion receptors at endothelial junctions are involved in leukocyte diapedesis (transmigration) but only some of them play a role for the regulation of junctional integrity (Muller 2011; Vestweber 2007). PECAM-1 was the first identified diapedesis-mediating adhesion receptor that binds in a homophilic way to PECAM-1 on leukocytes (Muller et al. 1993). Other receptors that are also found at endothelial contacts and on leukocytes and support the diapedesis process are CD99 and CD99L2 (Bixel et al. 2004, 2007; Schenkel et al. 2002). At least the latter does not seem to support leukocyte diapedesis via homophilic molecular interactions, although a heterophilic ligand has not yet been identified (Seelige et al. 2013). None of these three proteins are involved in the regulation of endothelial junctions, as antibodies against them or gene deficiency do not seem to modulate vascular permeability.
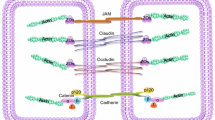
The prototype of an endothelial transmembrane protein that is located at junctions and plays a role for leukocyte diapedesis and vascular permeability is VE-cadherin (Dejana and Vestweber 2013; Vestweber et al. 2009). VE-cadherin is of dominant importance for the stability of endothelial junctions since antibodies against it can enhance vascular permeability and leukocyte extravasation (Corada et al. 1999; Gotsch et al. 1997) and gene deficiency disrupts vascular integrity (Carmeliet et al. 1999; Gory-Faure et al. 1999). VE-cadherin is a major component of endothelial composite junctions that consist in intermingled adherens and tight junctions in many vascular beds (Dejana et al. 2008). VE-cadherin is linked to the catenins inside the cells that connect it with the actin cytoskeleton, an essential requirement for its ability to form intact endothelial junctions (Kemler 1993).
The junctional adhesion molecules (JAMs) form a subgroup of the Ig-supergene family, which are closely located to endothelial and epithelial tight junctions and can mediate homophilic and heterophilic interactions (Bradfield et al. 2007; Monteiro and Parkos 2012; Weber et al. 2007). JAM-A and JAM-C participate in leukocyte extravasation (Martin-Padura et al. 1998; Woodfin et al. 2011), whereas JAM-A is known to affect junctional stability in epithelial cells (Laukoetter et al. 2007) and JAM-C was suggested to affect VE-cadherin function (Orlova et al. 2006). In contrast to the JAMs, ESAM is a more distantly related tight junction-associated member of this family that is selectively expressed only in endothelial cells and that also supports leukocyte extravasation and the induction of vascular permeability (Wegmann et al. 2006). The typical tight junction components such as occludin and some of the claudins are of course also expressed in endothelial cells but participation in the regulation of leukocyte extravasation has not yet been described and permeability regulating activities are focused on small molecular weight components (Furuse et al. 1993, 1998; Morita et al. 1999).
Tyrosine phosphorylation and cell contact stability
Several studies have suggested that the increase of tyrosine phosphorylation of junction associated proteins correlates with a decrease in junctional integrity. Inhibitors of tyrosine phosphatases, with very broad specificity for almost all phosphatases, deregulate junction integrity in epithelial (Staddon et al. 1995) and endothelial cells (Young et al. 2003).
In agreement with this, it was demonstrated that vascular endothelial growth factor (VEGF) leads to strong tyrosine phosphorylation of the adherens junction molecules VE-cadherin, β-catenin and plakoglobin, which correlated with an increase in endothelial permeability (Esser et al. 1998). Moreover, the same study also described that PECAM-1 was tyrosine phosphorylated upon VEGF stimulation. Importantly, the cadherin association of the catenins was not altered and the cadherin–catenin complex retained its junctional localization, indicating that catenin and cytoskeletal association might not be affected by tyrosine phosphorylation. In line with this study, the inhibition of the kinase Src prevented the VEGF-induced increase in permeability, showing that a kinase is involved in the VEGF-induced opening of endothelial junctions (Weis et al. 2004).
Thrombin, another permeability-increasing agent, promotes tyrosine phosphorylation of VE-cadherin-associated β-catenin, plakoglobin and p120-catenin (Ukropec et al. 2000). Additional studies revealed that other permeability-increasing agents such as histamine (Andriopoulou et al. 1999), tumor necrosis factor-α (TNF-α) (Angelini et al. 2006) and platelet-activating factor (PAF) (Hudry-Clergeon et al. 2005) also induce tyrosine phosphorylation of VE-cadherin, β-catenin, γ-catenin and p120-catenin. Taken together, these studies suggest a correlation between the induction of permeability triggered by a variety of different stimuli and the induction of tyrosine phosphorylation of components of the VE-cadherin–catenin complex.
These studies prompted a more detailed analysis of certain tyrosine residues within the cytoplasmic tail of VE-cadherin. It has been shown that certain tyrosine/phenylalanine (Y/F) point mutants (Y658F and Y731F) of VE-cadherin, when overexpressed in cultured endothelial cells, inhibited transmigration of myeloid cells (Allingham et al. 2007). In another report, it was found that overexpression of each of three VE-cadherin mutants (Y731F, Y645F and Y733F) but not the Y658F VE-cadherin mutant, reduced transmigration of lymphocytes through transfected endothelial cell monolayers (Turowski et al. 2008). Analyzing the relevance of Y658 and Y731 of VE-cadherin for VEGF-induced permeability, it was shown that an Y658/731F VE-cadherin double mutant inhibited the VEGF effect (Monaghan-Benson and Burridge 2009). Parts of the VEGF-induced signaling mechanism leading to the phosphorylation of the VE-cadherin–catenin complex were revealed as activation of Rac-1 and subsequent production of reactive oxygen species (ROS). As a possible consequence of this, β-catenin dissociated from VE-cadherin (Monaghan-Benson and Burridge 2009). The importance of the cadherin–catenin complex for the adhesive function of cadherins makes the dissociation of this complex indeed an attractive target for mechanisms that counteract cadherin-mediated cell adhesion. Besides the study above, several other studies have reported that catenin phosphorylation correlated with the dissociation of β-catenin (Lilien and Balsamo 2005; Piedra et al. 2001, 2003) and plakoglobin (Miravet et al. 2003) from various cadherins. VEGF-induced tyrosine phosphorylation of β-catenin was also found to dissociate this catenin from VE-cadherin (Chen et al. 2012). Interestingly, however, tyrosine phosphorylation of α-catenin was reported to correlate with enhanced binding to β-catenin (Burks and Agazie 2006). In addition, other groups have found that stimuli that trigger enhanced vascular permeability and tyrosine phosphorylation of the VE-cadherin–catenin complex do not necessarily lead to the dissociation of β-catenin from VE-cadherin (Adam et al. 2010; Andriopoulou et al. 1999; Konstantoulaki et al. 2003; Nottebaum et al. 2008; Timmerman et al. 2012). Despite these differences, most of these studies have in common that they argue for a role of tyrosine phosphorylation of the VE-cadherin–catenin complex in the regulation of endothelial junctions.
Kinases that influence endothelial barrier integrity
Several kinases have been identified that affect endothelial barrier integrity and phosphorylate proteins at endothelial junctions. The first of them to be described were members of the Src kinase family. VEGF-stimulated induction of vascular leaks was blocked in mice deficient for either Src or the Src family member Yes, whereas Fyn-deficiency had no effect (Eliceiri et al. 1999). Furthermore, blocking of endothelial Src also resulted in the inhibition of neutrophil transmigration through endothelial monolayers (Allingham et al. 2007). Binding of the catenin p120 to VE-cadherin was able to reduce Src-mediated VE-cadherin phosphorylation and this again resulted in decreased transendothelial migration of leukocytes (Alcaide et al. 2008). Various tyrosine residues of VE-cadherin have been described as being phosphorylated, directly or indirectly, by the activity of Src. Whereas Y658 and Y731 were described as targets downstream of Src in studies using corresponding commercially available site-specific anti phospho-tyrosine antibodies (Monaghan-Benson and Burridge 2009), a study based on peptide mapping suggested that Y685 was the exclusive tyrosine residue being phosphorylated upon VEGF mediated stimulation of Src (Wallez et al. 2007). VEGF was also described to stimulate a Src-dependent signaling cascade that leads to the activation of p21-activated kinase (PAK) followed by the phosphorylation of serine 665 on VE-cadherin, which creates a binding site for the association of β-arrestin. This in turn initiates clathrin-dependent endocytosis of VE-cadherin leading to the weakening of endothelial junctions (Gavard and Gutkind 2006). More recently, it was demonstrated that shear-induced junctional Src-activation leads to the phosphorylation of Y658 and Y685 of VE-cadherin in veins but not in arteries. Src-inhibition in this study blocked VE-cadherin phosphorylation and bradykinin-induced permeability (Orsenigo et al. 2012). Taken together, these studies suggest various mechanisms whereby Src stimulates phosphorylation of VE-cadherin, which in turn leads to weakening of endothelial junctions. However, it has also been reported that Src-mediated tyrosine phosphorylation of VE-cadherin alone is not sufficient to induce contact opening, as shown by overexpression of dominant-negative c-terminal Src kinase (Csk) (Adam et al. 2010). Csk inhibits Src by phosphorylation at Y527 (Okada and Nakagawa 1989). Interestingly, Src-mediated phosphorylation of Y685 of VE-cadherin creates a specific binding site for Csk (Baumeister et al. 2005). This interaction was implicated in cell density-dependent inhibition of cell growth. Whether binding of this negative regulator of Src might also initiate a negative feed-back loop that might serve to restrict junction opening is not yet known.
Apart from Src-family kinases, the redox-sensitive proline-rich tyrosine kinase 2 (Pyk2) is also involved in modulating endothelial integrity. Inhibition of Pyk2 prevents β-catenin phosphorylation and the Rac1-mediated loss of endothelial cell contact stability, a signaling pathway that is initiated upon loss of VE-cadherin function (van Buul et al. 2005). In addition, Pyk2 mediates the ICAM-1-triggered phosphorylation of VE-cadherin and downregulation of Pyk2 activity results in decreased leukocyte transmigration (Allingham et al. 2007). Also, the Pyk2-related focal adhesion kinase (FAK) has been shown to phosphorylate β-catenin and this β-catenin phosphorylation upon FAK-recruitment to VE-cadherin is necessary for VEGF-induced permeability (Chen et al. 2012). On the other hand, FAK was also reported to support the strengthening of endothelial junctions upon stimulation with sphingosine-1-phosphate (Belvitch and Dudek 2012).
Not only cytosolic kinases but also receptor tyrosine kinases (RTKs) are able to influence the stability of endothelial junctions. One is the VEGF receptor-2 (VEGFR-2) and another the angiopoietin receptor Tie-2. As already stated, stimulation of VEGFR-2 by VEGF results in VE-cadherin phosphorylation and loosening of endothelial contacts (Esser et al. 1998) and furthermore increases angiogenesis (Detmar et al. 1998). Ang1/Tie-2 signaling in turn increases the barrier function of endothelial junctions (Gamble et al. 2000; Mammoto et al. 2007). In addition to physiological Ang1/Tie-2 signaling, a short synthetic peptide that activates Tie-2 also increases the barrier function of cell contacts (David et al. 2011; Kumpers et al. 2011). Ang1-mediated Tie-2 signaling is capable of counteracting the VEGF-induced permeability increase in blood vessels (Thurston et al. 1999). Overexpression of Ang1 in vivo strongly stabilizes the endothelial barrier function, rendering the endothelium insensitive to increases in VEGF-induced permeability. Thus, the interplay between VEGFR-2-signaling and Tie-2-signaling is crucial for controlling endothelial barrier function.
Collectively, these reports illustrate that diverse kinases and therefore numerous regulatory pathways are involved in the control of endothelial barrier integrity. To ensure precise regulation of endothelial junction opening, the kinase-mediated phosphorylation has to be balanced by phosphatase-mediated dephosphorylation.
PTPs counteract kinases that influence endothelial barrier function
Protein-tyrosine-phosphatases (PTPs) can be grouped into classical PTPs, dual-specific PTPs and low-molecular-weight PTPs (Alonso et al.; 2004, Kappert et al. 2005). The classical PTPs, hereafter referred to as PTPs, can further be divided into membrane-spanning receptor PTPs (RPTPs) and cytosolic non-receptor PTPs (NRPTPs) or cytosolic PTPs. The latter consist in the catalytic domain and additional sequences that regulate their activity or localization. RPTPs display high variability in their extracellular region and possess one or two intracellular phosphatase domains. Extracellular ligands of RPTPs are to date largely unknown. However, RPTPκ and also RPTPμ can interact in trans in a homophilic way (Brady-Kalnay et al. 1993; Gebbink et al. 1993; Sap et al. 1994) and DEP-1 (CD148) interacts with components of matrigel (Sorby et al. 2001). Furthermore, heparan sulfate proteoglycans were shown to bind to RPTP-σ (Aricescu et al., 2002; Johnson et al. 2006).
Higher cell density and decreased phosphorylation of junctional proteins is accompanied by increased phosphatase activity in the membrane fraction (Gaits et al. 1995) and by increased phosphatase expression and higher cell contact localization of several phosphatases, including DEP-1, RPTPμ, and VE-PTP (Campan et al. 1996; Gaits et al. 1995; Nottebaum et al. 2008; Östman et al. 1994). Furthermore, a large scale of all PTPs by sodium orthovanadate or phenylarsine oxide results in higher phosphorylation of the cadherin–catenin complex and simultaneously increases transendothelial permeability and leukocyte transmigration (Young et al. 2003). Thus, cytosolic PTPs and RPTPs are important for the regulation of junctional integrity in endothelial cells. Several phosphatases are known to interact with junctional proteins and thus influence endothelial barrier function, among which are PTP1B, SHP-1, SHP-2, RPTPμ, DEP-1, and VE-PTP.
The first PTP to be discovered was the cytosolic PTP1B in the late 1980s (Charbonneau et al. 1989; Tonks et al. 1988a, b). Later, it was found that PTP1B binds to N-cadherin and dephosphorylates β-catenin, thus maintaining N-cadherin-mediated adhesion (Balsamo et al. 1996, 1998). PTP1B is in addition known to interfere with VEGF-mediated VEGFR-2-signaling, since it counter-regulates VEGF-induced phosphorylation of VEGFR-2. In line with this, PTP1B is involved in stabilizing VE-cadherin-mediated cell–cell adhesions by reducing VE-cadherin tyrosine phosphorylation (Nakamura et al. 2008).
SHP-2 is a cytosolic PTP that was reported to associate via β-catenin with the VE-cadherin–catenin complex. Thrombin stimulation increases SHP-2 phosphorylation and dissociates SHP-2 from the VE-cadherin–β-catenin complex, resulting in increased phosphorylation of the cadherin–catenin complex and decreased endothelial barrier integrity (Ukropec et al. 2000). SHP-2 function is necessary for maintaining endothelial cell contact integrity (Grinnell et al. 2009) and is moreover involved in the recovery of endothelial junctions after thrombin stimulation (Timmerman et al. 2012). SHP-2 furthermore interacts with the phosphorylated PECAM-1 cytoplasmic domain (Jackson et al. 1997; Masuda et al. 1997) and regulates its phosphorylation status (Cao et al. 1998), which in turn influences the association of SHP-2 to PECAM-1 (Cao et al. 1998; Newman and Newman 2003).
RPTPμ was probably the first PTP shown to interact with various cadherins, among them E-, N- and R-cadherin, as was analyzed in different cell types (Brady-Kalnay et al. 1995, 1998). RPTPμ also binds directly to VE-cadherin at endothelial cell contacts, which leads to its dephosphorylation and is accompanied by increased endothelial barrier function (Sui et al. 2005).
DEP-1, a member of the R3 subfamily of RPTPs, has also been reported to influence endothelial junction integrity. DEP-1 expression and activity are strongly enhanced upon increasing cell density (Östman et al. 1994), where DEP-1 plays a role in contact inhibition of growth by interfering with VEGFR-2 triggered cell proliferation (Lampugnani et al. 2003). On the other hand, it has been shown that DEP-1 dephosphorylates the inhibitory Y529 of Src and thereby supports VEGF-induced and Src-mediated stimulation of endothelial permeability (Spring et al. 2012). In addition, DEP-1 interacts with occludin and DEP-1 overexpression enhances epithelial barrier function, during tight junction assembly (Sallee and Burridge 2009). These studies indicate the complex roles of DEP-1 in regulating endothelial junction integrity. To further elucidate DEP-1 functions, gene-deficient mice were generated by different approaches. However, depending on the approach, results differed. Takahashi et al. (2003) observed that an in-frame replacement of DEP-1 cytoplasmic sequences with enhanced green fluorescent protein leads to embryonic lethality at embryonic day E11.5 due to disorganized vascular structures and growth retardation. Mutant yolk sacs and embryos exhibited strong defects in vascular remodeling. In contrast, DEP-1-deficient mice, generated by targeted disruption of the DEP-1 gene directly after the signal peptide sequence, were viable, healthy and fertile and showed no signs of embryonic defects. No obvious alterations in anatomy, life span, or spontaneous tumor appearance were detected (Trapasso et al. 2006). In line with this, DEP-1-loss-of-function mice generated by constitutive deletion of the DEP-1 transmembrane exon also did not display embryonic lethality (Zhu et al. 2008). While, in the first study, a mutant form of DEP-1 lacking the phosphatase domain is still present in the plasma membrane, no cell surface-located DEP-1 remains in the last two studies. Dominant negative effects in the former study are possible but have not yet been analyzed in detail.
The impact of VE-PTP on endothelial junction stability
Like DEP-1, VE-PTP belongs to the R3 subtype of RPTPs. VE-PTP is so far the only known phosphatase that is exclusively expressed in endothelial cells (Baumer et al. 2006; Fachinger et al. 1999). It consists in 17 FNIII-like extracellular domains, a transmembrane region and a single cytoplasmic phosphatase domain. VE-PTP is the murine homologue of human RPTPβ (Fachinger et al. 1999) and is crucial for embryonic development, as VE-PTP-deficient mice die around embryonic day E9.5/E10.0 due to severe vascular malformations (Baumer et al. 2006; Dominguez et al. 2007). Since the first vascular plexus develops normally in the absence of VE-PTP, this phosphatase is dispensable for the initiation of vasculogenesis but is essential for the following maturation and remodeling of vessel structures during angiogenesis (Baumer et al. 2006; Carra et al. 2012; Dominguez et al. 2007).
Our group and others have analyzed the role of VE-PTP in regulating the endothelial barrier function in more detail during the last years. An important interaction partner of VE-PTP is the adhesion molecule VE-cadherin (Nawroth et al. 2002; Nottebaum et al. 2008). VE-cadherin specifically co-precipitates with VE-PTP and, for this interaction, the membrane proximal extracellular domains of both proteins are sufficient (Nawroth et al. 2002). When elucidating the influence of VE-PTP on VE-cadherin function in more detail, it was shown that VE-PTP expression reverses VEGFR-2-induced tyrosine phosphorylation of VE-cadherin and increases the VE-cadherin-mediated barrier function (Nawroth et al. 2002). Furthermore, we found that VE-PTP is redistributed to endothelial contacts with increasing cell density, which is accompanied by an increased interaction of VE-PTP with VE-cadherin (Nottebaum et al. 2008). Downregulation of VE-PTP expression reduces VE-cadherin adhesiveness, leading to increased transendothelial permeability and leukocyte transendothelial migration. In addition, VE-PTP downregulation increases tyrosine phosphorylation of plakoglobin, which was identified as a direct substrate of VE-PTP. Plakoglobin is crucial for the contact-stabilizing function of VE-PTP (Nottebaum et al. 2008).
Importantly, VEGF-induced endothelial permeability correlates with the dissociation of VE-PTP from VE-cadherin. This dissociation was also detected upon leukocyte binding to TNF-α-inflamed endothelium and was accompanied by increased tyrosine phosphorylation of the VE-cadherin–catenin complex (Nottebaum et al. 2008). Recently, we were able to show that this VE-PTP-VE-cadherin dissociation is in fact necessary for efficient opening of endothelial contacts in vivo (Broermann et al. 2011). We demonstrated that specific stabilization of the VE-PTP-VE-cadherin interaction results in a lack of permeability induction after VEGF- or LPS-stimulation and reduces leukocyte extravasation in IL-1β- or LPS-stimulated tissues (Broermann et al. 2011). Stabilization of the VE-PTP-VE-cadherin association was achieved by fusing two additional protein domains (FKBP and FRB*) to the C-terminus of either VE-cadherin or VE-PTP, respectively. These domains contained different binding sites for a small molecular weight chemical compound (rapalog) that was able to strongly stabilize the interaction between VE-PTP and VE-cadherin. To analyze the effect of these modifications in vivo, we inserted the cDNAs for VE-cadherin-FKBP and VE-PTP-FRB* into the VE-cadherin gene locus of mice, thereby replacing endogenous VE-cadherin and ensuring endothelial specific expression. We found that administering the rapalog compound to these knock-in mice strongly inhibited the induction of vascular permeability by VEGF and LPS as well as the cytotkine-stimulated recruitment of neutrophils in vivo, whereas no such affect was seen with this compound in wild-type mice (Broermann et al. 2011). This established that the dissociation of VE-PTP from VE-cadherin is required for the opening of endothelial junctions in vivo.
Recently, the endothelial leukocyte-binding receptor and the downstream signaling pathway that trigger the dissociation of VE-PTP from VE-cadherin were identified. We found that binding of lymphocytes to VCAM-1 is necessary to induce this dissociation (Vockel and Vestweber 2013). In addition, we found that the signaling steps involved and required for this process comprised the activation of Rac1, the production of reactive oxygen species (ROS) via NADPH-oxidase and the activation of the kinase Pyk. Importantly, the same signaling cascade was also required for the VEGF-induced dissociation of VE-PTP from VE-cadherin (Vockel and Vestweber 2013). This is in good agreement with previous studies that showed that VEGF-induced phosphorylation of the VE-cadherin–catenin complex also required Rac activation and ROS production (Monaghan-Benson and Burridge 2009). The actual dissociation of VE-PTP from VE-cadherin is probably mediated by the binding of a phosphorylated substrate to VE-PTP, which leads to an allosteric change that dissociates the extracellular domains of these two proteins. The idea for this was based on the finding that a phosphatase-dead trapping mutant of VE-PTP could not bind to VE-cadherin, although robust binding of VE-PTP and VE-cadherin is mediated via their extracellular domains. This initiated the hypothesis that binding of other substrates to the phosphatase domain of VE-PTP would inhibit the association with VE-cadherin via allosteric effects. Testing this hypothesis, it was indeed possible to show that a model substrate of VE-PTP comprising a phosphorylated peptide of Tie-2 when introduced into endothelial cells via a fused Tat peptide could trigger the dissociation of VE-PTP from VE-cadherin, whereas the non-phosphorylated substrate had no such effect. Collectively, these results establish a Rac1, NADPH-oxidase, Pyk-dependent signaling pathway that contributes to the VEGF- and lymphocyte-induced destabilization of endothelial junctions via dissociation of VE-PTP from VE-cadherin (Fig. 1).

Proposed signaling mechanism for the lymphocyte-induced dissociation of VE-PTP from VE-cadherin. Lymphocyte-binding to VCAM-1 or stimulation by VEGF triggers the production of reactive oxygen species (ROS) via Rac1-mediated activation of NADPH oxidase (NOX). This leads to activation of the redox-sensitive kinase Pyk2 that triggers directly or indirectly the phosphorylation of a VE-PTP substrate that in turn binds to VE-PTP. This binding may cause structural or conformational changes across the membrane that lead to the detachment of the extracellular domain of VE-PTP from VE-cadherin. This facilitates phosphorylation of components or associated factors of the VE-cadherin–catenin complex that participates in the destabilization of endothelial cell contacts. This figure was originally published in (Vockel and Vestweber 2013), ©The American Society of Hematology
VE-PTP has also been reported to associate with the VEGFR-2. Although this interaction does not support direct co-immunoprecipitation, it could be documented based on proximity ligation assays (Mellberg et al. 2009). In line with this, silencing of VE-PTP was accompanied by enhanced VEGFR-2 phosphorylation and downstream signaling. VE-PTP was also implicated in Tie-2-mediated balancing of VEGFR-2 effects and was suggested to be relevant for endothelial cell polarity and vessel lumen formation (Hayashi et al. 2013).
The tyrosine kinase receptor Tie-2 was actually the first VE-PTP-binding partner that was identified (Fachinger et al. 1999). According to this study, VE-PTP substrate trapping mutants co-precipitated with Tie-2 but not with VEGFR-2, and VE-PTP specifically dephosphorylated Tie-2. When elucidating the role of the VE-PTP–Tie-2 interaction in more detail, we found that antibodies against VE-PTP dissociate it from Tie-2 and trigger VE-PTP endocytosis whereas VE-cadherin-bound VE-PTP was not affected (Winderlich et al. 2009). This release of VE-PTP from Tie-2 led to the activation of this kinase receptor. This in turn activated Erk and Akt signaling and promoted cell proliferation, leading to circumferential growth and widening of vessel structures in newborn mice (Winderlich et al. 2009). These results recapitulated embryonic defects that had been described in VE-PTP gene-deficient mice (Baumer et al. 2006; Dominguez et al. 2007). In conclusion, this study showed that VE-PTP balances the activity of Tie-2, which helps in determining vessel diameter and remodeling of the vasculature (Winderlich et al. 2009).
It is well documented that Angiopoietin-1 (Ang-1) strongly supports stabilization of endothelial junctions and counteracts VEGF-induced vascular permeability in vivo (Thurston et al. 1999). This raises the question whether VE-PTP counteracts endothelial cell contact integrity by deactivating Tie-2. A first hint for this was suggested in a recent report (Goel et al. 2013). To understand the physiological relevance of VE-PTP for the regulation of vascular permeability it will be important to analyze this in more detail in the future. Interestingly, Ang-1 stimulation induces the redistribution of Tie-2 to junctions where it probably connects Tie-2 of neighboring cells in trans (Saharinen et al. 2008). Together with Tie-2, the associated VE-PTP molecules are also redistributed to junctions. It will be interesting to analyze whether and how this re-distribution is relevant for the increase in junctional integrity.
Concluding remarks
As illustrated in this review, a complex set of membrane receptors and adhesion molecules as well as signal transducing kinases and phosphatases are involved in the regulation of endothelial junctions. These molecular mechanisms regulate the subcellular distribution, clustering cell surface expression of adhesion molecules such as VE-cadherin and their cytoskeletal linkage. In addition, these signaling mechanisms and others, such as GTPases and additional scaffolding proteins, regulate cytoskeletal activities that modulate the formation of cortical actin fibers and radial actin stress fibers. It is the interplay of these mechanisms that finally determines endothelial junction integrity. A deeper understanding of these mechanisms will allow the development of new strategies to interfere with a loss of junctional integrity of the vascular endothelium and thereby stop vascular leaks and harmful leukocyte invasion in inflammation.
References
Adam AP, Sharenko AL, Pumiglia K, Vincent PA (2010) SRC-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem 285:7045–7055
Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW (2008) p120-catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood 112:2770–2779
Allingham MJ, van Buul JD, Burridge K (2007) ICAM-1-mediated, Src- and Pyk2-dependentvascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol 179:4053–4064
Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T (2004) Protein tyrosine phosphatases in the human genome. Cell 117:699–711
Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E (1999) Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol 19:2286–2297
Angelini DJ, Hyun S-W, Grigoryev DN, Garg P, Gong P, Singh IS, Jeffery AP, Hasday D, Goldblum SE (2006) TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol Lung Cell Mol Physiol 291:L1232–L1245
Aricescu AR, McKinnell IW, Halfter W, Stoker AW (2002) Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol Cell Biol 22:1881–1892
Balsamo J, Leung T, Ernst H, Zanin MK, Hoffman S, Lilien J (1996) Regulated binding of PTP1B-like phosphatase to N-cadherin: control of cadherin-mediated adhesion by dephosphorylation of beta-catenin. J Cell Biol 134:801–813
Balsamo J, Arregui C, Leung T, Lilien J (1998) The nonreceptor protein tyrosine phosphatase PTP1B binds to the cytoplasmic domain of N-cadherin and regulates the cadherin-actin linkage. J Cell Biol 143:523–532
Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D (2005) Association of Csk to VE-cadherin and inhibition of cell proliferation. EMBO J 24:1686–1695
Baumer S, Keller L, Holtmann A, Funke R, August B, Gamp A, Wolburg H, Wolburg-Buchholz K, Deutsch U, Vestweber D (2006) Vascular endothelial cell specific phospho-tyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood 107:4754–4762
Belvitch P, Dudek SM (2012) Role of FAK in S1P-regulated endothelial permeability. Microvasc Res 83:22–30
Bixel G, Kloep S, Butz S, Petri B, Engelhardt B, Vestweber D (2004) Mouse CD99 participates in T cell recruitment into inflamed skin. Blood 104:3205–3213
Bixel MG, Petri B, Khandoga AG, Khandoga A, Wolburg-Buchholz K, Wolburg H, Marz S, Krombach F, Vestweber D (2007) A CD99-related antigen on endothelial cells mediates neutrophil but not lymphocyte extravasation in vivo. Blood 109:5327–5336
Bradfield PF, Nourshargh S, Aurrand-Lions M, Imhof BA (2007) JAM family and related proteins in leukocyte migration. Arterioscler Thromb Vasc Biol 27:2104–2112
Brady-Kalnay SM, Flint AJ, Tonks NK (1993) Homophilic binding of PTP mu, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation. J Cell Biol 122:961–972
Brady-Kalnay SM, Rimm DL, Tonks NK (1995) Receptor protein tyrosine phosphatase PTPmu associates with cadherins and catenins in vivo. J Cell Biol 130:977–986
Brady-Kalnay SM, Mourton T, Nixon JP, Pietz GE, Kinch M, Chen H, Brackenbury R, Rimm DL, Del Vecchio RL, Tonks NK (1998) Dynamic interaction of PTPmu with multiple cadherins in vivo. J Cell Biol 141:287–296
Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, Vestweber D (2011) Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med 208:2393–2401
Burks J, Agazie YM (2006) Modulation of alpha-catenin Tyr phosphorylation by SHP2 positively effects cell transformation induced by the constitutively active FGFR3. Oncogene 114
Campan M, Yoshizumi M, Seidah NG, Lee ME, Bianchi C, Haber E (1996) Increased proteolytic processing of protein tyrosine phosphatase mu in confluent vascular endothelial cells: the role of PC5, a member of the subtilisin family. Biochemistry 35:3797–3802
Cao MY, Huber M, Beauchemin N, Famiglietti J, Albelda SM, Veillette A (1998) Regulation of mouse PECAM-1 tyrosine phosphorylation by the Src and Csk families of protein-tyrosine kinases. J Biol Chem 273:15765–15772
Carman CV, Springer TA (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 167:377–388
Carmeliet P, Lampugnani M-G, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert J-M, Collen D, Dejana E (1999) Targeted Deficiency or Cytosolic Truncation of the VE-cadherin Gene in Mice Impairs VEGF-Mediated Endothelial Survival and Angiogenesis. Cell 98:147–157
Carra S, Foglia E, Cermenati S, Bresciani E, Giampietro C, Lora Lamia C, Dejana E, Beltrame M, Cotelli F (2012) Ve-ptp modulates vascular integrity by promoting adherens junction maturation. PLoS One 7:e51245
Charbonneau H, Tonks NK, Kumar S, Diltz CD, Harrylock M, Cool DE, Krebs EG, Fischer EH, Walsh KA (1989) Human placenta protein-tyrosine-phosphatase: amino acid sequence and relationship to a family of receptor-like proteins. Proc Natl Acad Sci USA 86:5252–5256
Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, Guan JL, Acevedo LM, Weis SM, Cheresh DA, Schlaepfer DD (2012) VEGF-induced vascular permeability is mediated by FAK. Dev Cell 22:146–157
Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, McDonald DM, Ward PA, Dejana E (1999) Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci USA 96:9815–9820
David S, Park JK, Meurs M, Zijlstra JG, Koenecke C, Schrimpf C, Shushakova N, Gueler F, Haller H, Kumpers P (2011) Acute administration of recombinant Angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine 55:251–259
Dejana E, Vestweber D (2013) The Role of VE-cadherin in Vascular Morphogenesis and Permeability Control. In: Roy Fv (ed) The Molecular Biology of cadherins, Elsevier, Amsterdam, (in press)
Dejana E, Orsenigo F, Lampugnani MG (2008) The role of adherens junctions and VE-cadheri in the control of vascular permeability. J Cell Sci 121:2115–2122
Detmar M, Brown LF, Schon MP, Elicker BM, Velasco P, Richard L, Fukumura D, Monsky W, Claffey KP, Jain RK (1998) Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J Invest Dermatol 111:1–6
Dominguez MG, Hughes VC, Pan L, Simmons M, Daly C, Anderson K, Noguera-Troise I, Murphy AJ, Valenzuela DM, Davis S, Thurston G, Yancopoulos GD, Gale NW (2007) Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci USA 104:3243–3248
Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA (1999) Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell 4:915–924
Esser S, Lampugnani MG, Corada M, Dejana E, Risau W (1998) Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 111(Pt 13):1853–1865
Fachinger G, Deutsch U, Risau W (1999) Functional interaction of vascular endothelial-protein tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene 18:5948–5953
Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM (1998) Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J Exp Med 187:903–915
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123:1777–1788
Furuse M, Sasaki H, Fujimoto K, Tsukita S (1998) A single gene product, claudin-1 or −2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 143:391–401
Gaits F, Li RY, Ragab A, Ragab Thomas JM, Chap H (1995) Increase in receptor-like protein tyrosine phosphatase activity and expression level on density-dependent growth arrest of endothelial cells. Biochem J 311:97–103
Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas L, Rudge J, Yancopoulos G, Vadas MA (2000) Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res 87:603–607
Gavard J, Gutkind JS (2006) VEGF controls endothelial-cell permeability by promoting the β arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8:1223–1234
Gebbink MF, Zondag GC, Wubbolts RW, Beijersbergen RL, van Etten I, Moolenaar WH (1993) Cell-cell adhesion mediated by a receptor-like protein tyrosine phosphatase. J Biol Chem 268:16101–16104
Goel S, Gupta N, Walcott BP, Snuderl M, Kesler CT, Kirkpatrick ND, Heishi T, Huang Y, Martin JD, Ager E, Samuel R, Wang S, Yazbek J, Vakoc BJ, Peterson RT, Padera TP, Duda DG, Fukumura D, Jain RK (2013) Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J Natl Cancer Inst 105(1188):1201
Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, Huber P (1999) Role of vascular endothelial-cadherin in vascular morphogenesis. Development 126:2093–2102
Gotsch U, Borges E, Bosse R, Böggemeyer E, Simon M, Mossmann H, Vestweber D (1997) VE cadherin antibody accelerates neutrophil recruiment in vivo. J Cell Sci 110:583–588
Grinnell KL, Casserly B, Harrington EO (2009) Role of protein tyrosine phosphatase SHP2 in barrier function of pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol 298:L361–L370
Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, Hellberg C, Mellberg S, Koch S, Dimberg A, Koh GY, Dejana E, Belting HG, Affolter M, Thurston G, Holmgren L, Vestweber D, Claesson-Welsh L (2013) VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun 4:1672
Hudry-Clergeon H, Stengel D, Ninio E, Vilgrain I (2005) Platelet-activating factor increases VE cadherin tyrosine phosphorylation in mouse endothelial cells and its association with the PtdIns3′-kinase. FASEB J 19:512–520
Jackson DE, Ward CM, Wang R, Newman PJ (1997) The protein-tyrosine phosphatase SHP-2 binds platelet/endothelial cell adhesion molecule-1 (PECAM-1) and forms a distinct signaling complex during platelet aggregation. Evidence for a mechanistic link between PECAM-1- and integrin-mediated cellular signaling. J Biol Chem 272:6986–6993
Johnson KG, Tenney AP, Ghose A, Duckworth AM, Higashi ME, Parfitt K, Marcu O, Heslip TR, Marsh JL, Schwarz TL, Flanagan JG, Van Vactor D (2006) The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron 49:517–531
Kappert K, Peters KG, Bohmer FD, Ostman A (2005) Tyrosine phosphatases in vessel wall signaling. Cardiovasc Res 65:587–598
Kemler R (1993) From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet 9:317–321
Konstantoulaki M, Kouklis P, Malik AB (2003) Protein kinase C modifications of VE-cadherin, p120, and beta-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am J Physiol Lung Cell Mol Physiol 285:L434–L442
Kumpers P, Gueler F, David S, Slyke PV, Dumont DJ, Park JK, Bockmeyer CL, Parikh SM, Pavenstadt H, Haller H, Shushakova N (2011) The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care 15:R261
Küppers V, Vestweber D, Schulte D (2013) Locking endothelial junctions blocks leukocyte extravasation, but not in all tissues. Tissue Barriers 1:eLocation ID: e23805
Lampugnani MG, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, Dejana E (2003) Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP 1/CD148. J Cell Biol 161:793–804
Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA (2007) JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 204:3067–3076
Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689
Lilien J, Balsamo J (2005) The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of b-catenin. Curr Opin Cell Biol 17:459–465
Majno G, Palade GE (1961) Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol 11:571–605
Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, Ingber DE, Sukhatme VP (2007) Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem 282:23910–23918
Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127
Masuda M, Osawa M, Shigematsu H, Harada N, Fujiwara K (1997) Platelet endothelial cell adhesion molecule-1 is a major SH-PTP2 binding protein in vascular endothelial cells. FEBS Lett 408:331–336
Mehta D, Malik AB (2006) Signaling Mechanisms regulating endothelial permeability. Physiol Rev 86:279–367
Mellberg S, Dimberg A, Bahram F, Hayashi M, Rennel E, Ameur A, Westholm JO, Larsson E, Lindahl P, Cross MJ, Claesson-Welsh L (2009) Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J 23:1490–1502
Miravet S, Piedra J, Castano J, Raurell I, Franci C, Dunach M, Garcia de Herros A (2003) Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Mol Cell Biol 23:7391–7402
Monaghan-Benson E, Burridge K (2009) The regulation of vascular endothelial growth factor induced microvascular permeability requires Rac and reactive oxygen species. J Biol Chem 284:25602–25611
Monteiro AC, Parkos CA (2012) Intracellular mediators of JAM-A-dependent epithelial barrier function. Ann NY Acad Sci 1257:115–124
Morita K, Furuse M, Fujimoto K, Tsukita S (1999) Claudin multigene family encoding four transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci USA 96:511–516
Muller WA (2011) Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 6:323–344
Muller WA, Weigl SA, Deng X, Phillips DM (1993) PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med 178:449–460
Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, Razvi M, Kini V, Mahadev K, Goldstein BJ, McKinney R, Fukai T, Ushio-Fukai M (2008) Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res 102:1182–1191
Nawroth R, Poell G, Ranft A, Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U, Vestweber D (2002) VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J 21:4885–4895
Newman PJ, Newman DK (2003) Signal transduction pathways mediated by PECAM-1: new roles for an old molecule in platelet and vascular cell biology. Arterioscler Thromb Vasc Biol 23:953–964
Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, Filippova K, Lyck R, Engelhardt B, Kamenyeva O, Bixel MG, Butz S, Vestweber D (2008) VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med 205:2929–2945
Okada M, Nakagawa H (1989) A protein tyrosine kinase involved in regulation of pp 60c-src function. J Biol Chem 264:20886–20893
Orlova VV, Economopoulou M, Lupu F, Santoso S, Chavakis T (2006) Junctional adhesion molecule-C regulates vascular endothelial permeability by modulating VE-cadherin-mediated cell-cell contacts. J Exp Med 203:2703–2714
Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Young Koh G, Franco D, Kurtcuoglu V, Poulikakos D, Baluk P, McDonald D, Grazia Lampugnani M, Dejana E (2012) Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 3:1208
Östman A, Yang Q, Tonks NK (1994) Expression of DEP-1, a receptor-like protein-tyrosine phosphatase, is enhanced with increasing cell density. Proc Natl Acad Sci USA 91:9680–9684
Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG (2001) Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem 276:20436–20443
Piedra J, Miravet S, Castano J, Palmer HG, Heisterkamp N, Garcia de Herreros A, Dunach M (2003) p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin Interaction. Mol Cell Biol 23:2287–2297
Saharinen P, Eklund L, Miettinen J, Wirkkala R, Anisimov A, Winderlich M, Nottebaum A, Vestweber D, Deutsch U, Koh GY, Olsen BR, Alitalo K (2008) Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol 10:527–537
Sallee JL, Burridge K (2009) Density-enhanced phosphatase 1 regulates phosphorylation of tightjunction proteins and enhances barrier function of epithelial cells. J Biol Chem 284:14997–15006
Sap J, Jiang YP, Friedlander D, Grumet M, Schlessinger J (1994) Receptor tyrosine phosphatase R-PTP-kappa mediates homophilic binding. Mol Cell Biol 14:1–9
Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA (2002) CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol 3:143–150
Schoefl GI (1972) The migration of lymphocytes across the vascular endothelium in lymphoid tissue. A reexamination. J Exp Med 136:568–588
Schulte D, Küppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, Vestweber D (2011) Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J 30:4157–4170
Seelige R, Natsch C, März S, Jing D, Frye M, Butz S, Vestweber D (2013) Endothelial-specific gene ablation of CD99L2 impairs leukocyte extravasation in vivo. J Immunol 190:892–896
Sorby M, Sandstrom J, Ostman A (2001) An extracellular ligand increases the specific activity of the receptor-like protein tyrosine phosphatase DEP-1. Oncogene 20:5219–5224
Spring K, Chabot C, Langlois S, Lapointe L, Trinh NT, Caron C, Hebda JK, Gavard J, Elchebly M, Royal I (2012) Tyrosine phosphorylation of DEP-1/CD148 as a mechanism controlling Src kinase activation, endothelial cell permeability, invasion, and capillary formation. Blood 120:2745–2756
Staddon JM, Herrenknecht K, Smales C, Rubin LL (1995) Evidence that tyrosine phosphorylation may increase tight junction permeability. J Cell Sci 108(Pt 2):609–619
Sui XF, Kiser TD, Hyun SW, Angelini DJ, Del Vecchio RL, Young BA, Hasday JD, Romer LH, Passaniti A, Tonks NK, Goldblum SE (2005) Receptor protein tyrosine phosphatase micro regulates the paracellular pathway in human lung microvascular endothelia. Am J Pathol 166:1247–1258
Takahashi T, Takahashi K, St John PL, Fleming PA, Tomemori T, Watanabe T, Abrahamson DR, Drake CJ, Shirasawa T, Daniel TO (2003) A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol 23:1817–1831
Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM (1999) Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 286:2511–2514
Timmerman I, Hoogenboezem M, Bennett AM, Geerts D, Hordijk PL, van Buul JD (2012) The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of beta-catenin phosphorylation. Mol Biol Cell 23:4212–4225
Tonks NK, Diltz CD, Fischer EH (1988a) Characterization of the major protein-tyrosine- phosphatases of human placenta. J Biol Chem 263:6731–6737
Tonks NK, Diltz CD, Fischer EH (1988b) Purification of the major protein-tyrosine- phosphatases of human placenta. J Biol Chem 263:6722–6730
Trapasso F, Drusco A, Costinean S, Alder H, Aqeilan RI, Iuliano R, Gaudio E, Raso C, Zanesi N, Croce CM, Fusco A (2006) Genetic ablation of Ptprj, a mouse cancer susceptibility gene, results in normal growth and development and does not predispose to spontaneous tumorigenesis. DNA Cell Biol 25:376–382
Turowski P, Martinelli R, Crawford R, Wateridge D, Papagiorgiou A-P, Lampugnani MG, Gamp AC, Vestweber D, Adamson P, Dejana E, Greenwood J (2008) Phosphorylation of Vascular Endothelial Cadherin Controls Lymphocyte Emigration. J Cell Sci 121:29–37
Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ (2000) SHP2 association with VE- cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem 275:5983–5986
van Buul JD, Anthony EC, Fernandez-Borja M, Burridge K, Hordijk PL (2005) Proline-rich tyrosine kinase 2 (Pyk2) mediates vascular endothelial-cadherin-based cell-cell adhesion by regulating beta-catenin tyrosine phosphorylation. J Biol Chem 280:21129–21136
Vestweber D (2007) Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev 218:178–196
Vestweber D, Winderlich M, Cagna G, Nottebaum AF (2009) Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol 19:8–15
Vockel M, Vestweber D (2013) How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood 122:2512–2522
Wallez Y, Cand F, Cruzalegui F, Wernstedt C, Souchelnytskyi S, Vilgrain I, Huber P (2007) Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene 26:1067–1077
Weber C, Fraemohs L, Dejana E (2007) The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol 7:467–477
Wegmann F, Petri J, Khandoga AG, Moser C, Khandoga A, Volkery S, Li H, Nasdala I, Brandau O, Fässler R, Butz S, Krombach F, Vestweber D (2006) ESAM supports neutrophil extravasation, activation of Rho and VEGF-induced vascular permeability. J Exp Med 203:1671–1677
Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D, Cheresh D (2004) Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest 113:885–894
Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, Deutsch U, Nottebaum AF, Vestweber D (2009) VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol 185:657–671
Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S (2011) The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol 12:761–769
Young BA, Sui X, Kiser TD, Hyun SW, Wang P, Sakarya S, Angelini DJ, Schaphorst KL, Hasday JD, Cross AS, Romer LH, Passaniti A, Goldblum SE (2003) Protein tyrosine phosphatase activity regulates endothelial cell-cell interactions, the paracellular pathway, and capillary tube stability. Am J Physiol Lung Cell Mol Physiol 285:L63–L75
Zhu JW, Brdicka T, Katsumoto TR, Lin J, Weiss A (2008) Structurally distinct phosphatases CD45 and CD148 both regulate B cell and macrophage immunoreceptor signaling. Immunity 28:183–196
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Küppers, V., Vockel, M., Nottebaum, A.F. et al. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res 355, 577–586 (2014). https://doi.org/10.1007/s00441-014-1812-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-014-1812-1