
Abstract
Parkinson’s disease (PD) is characterized by the progressive loss of dopaminergic neurons in the substantia nigra leading to the major clinical and pharmacological abnormalities of PD. In order to establish causal or protective treatments for PD, it is necessary to identify the cascade of deleterious events that lead to the dysfunction and death of dopaminergic neurons. Based on genetic, neuropathological, and biochemical data in patients and experimental animal models, dysfunction of the ubiquitin-proteasome pathway, protein aggregation, mitochondrial dysfunction, oxidative stress, activation of the c-Jun N-terminal kinase pathway, and inflammation have all been identified as important pathways leading to excitotoxic and apoptotic death of dopaminergic neurons. Toxin-based and genetically engineered animal models allow (1) the study of the significance of these aspects and their interaction with each other and (2) the development of causal treatments to stop disease progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Neuropathological and neuroanatomical characteristics
The pathological hallmarks of Parkinson’s disease (PD) are the loss of nigrostriatal dopaminergic neurons and the presence of intraneuronal proteinaceous cytoplasmic inclusions, termed “Lewy Bodies” (LBs). During the lifetime of a patient, the diagnosis of PD is made on clinical grounds, but definitive diagnosis requires the identification of both LBs and neurodegeneration in the substantia nigra pars compacta (SNpc).
Degeneration of dopaminergic neurons in PD
The cell bodies of the nigrostriatal neurons are located in the SNpc and project primarily to the putamen. The loss of these neurons, which normally contain conspicuous amounts of neuromelanin (Marsden 1983), produces the classic gross neuropathological finding of SNpc de-pigmentation. The pattern of SNpc cell loss appears to parallel the expression level of the dopamine transporter (DAT) mRNA (Uhl et al. 1994) and is consistent with the finding that the depletion of dopamine (DA) is most pronounced in the dorsolateral putamen (Bernheimer et al. 1973), the main site of projection for these neurons. At the onset of symptoms, DA in the putamen is depleted by approximately 80%, and approximately 60% of the SNpc dopaminergic neurons have been lost. The mesolimbic dopaminergic neurons, the cell bodies of which reside adjacent to the SNpc in the ventral tegmental area (VTA), are much less affected in PD (Uhl et al. 1985). Consequently, there is significantly less depletion of DA in the caudate (Price et al. 1978), the main site of projection for these neurons.
The neuropil of the SNpc is composed of axon projections from the striatum and globus pallidus. It stains strongly for calbindin D28K, and most dopaminergic cell bodies reside within this calbindin-rich neuropil (Damier et al. 1999a). However, the most susceptible neurons in PD tend to be in calbindin-poor areas of the substantia nigra (Damier et al. 1999b). Thus, cell loss is concentrated in ventrolateral and caudal portions of the SNpc, whereas during normal aging, the dorsomedial aspect of the SNpc is affected (Fearnley and Lees 1991). Therefore, even though age is an important risk factor for PD, neurodegeneration in PD appears to be a specific process distinct from normal aging.
Interestingly, the degree of terminal loss in the striatum appears to be more pronounced than the magnitude of SNpc dopaminergic neuron loss (Bernheimer et al. 1973). This suggests that striatal nerve terminals are the primary target of the degenerative process and that neuronal death in PD may result from a “dying back” process. Experimental support for this concept includes the observations that, in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated monkeys, the destruction of striatal terminals precedes that of SNpc cell bodies (Herkenham et al. 1991), and that, in MPTP-treated mice, protection of the striatal terminals prevents the loss of SNpc dopaminergic neurons (Wu et al. 2003).
Lewy bodies
LBs are spherical eosinophilic cytoplasmic protein aggregates composed of numerous proteins, including α-synuclein, parkin, ubiquitin, synphilin, and neurofilaments. They have a diameter of more than 15 μm and an organized structure with a dense hyaline core surrounded by a clear halo. Electron microscopy has revealed a dense granulovesicular core surrounded by a ring of radiating fibrils of 8–10 nm (Duffy and Tennyson 1965; Pappolla 1986).
LBs are found in all affected brain regions (Forno 1996; Spillantini et al. 1998). However, they are not specific for PD and are also found in a disease called “dementia with Lewy bodies”, in Alzheimer’s disease (AD), and, as an incidental pathological finding, in healthy people of advanced age (Gibb and Lees 1988). The role of LBs in neuronal death is controversial (see below), as are the reasons for their increased frequency in AD and the relationship of incidental LB to the occurrence of PD.
It should be noted that intracellular protein aggregates not only give rise to the large LBs located in the soma of neurons. Protein aggregates can also be formed in dendrites, resulting in local swellings termed Lewy neurites.
Non-dopaminergic pathology in PD
Although dopaminergic neuron loss is characteristic for PD, the neurodegeneration extends well beyond dopaminergic neurons. Neurodegeneration and LB formation are found in noradrenergic (locus coeruleus), serotonergic (raphe), and cholinergic (nucleus basalis of Meynert, dorsal motor nucleus of vagus) systems, and in the cerebral cortex (especially the cingulate and entorhinal cortices), olfactory bulb, and autonomic nervous system. The degeneration of hippocampal structures and cholinergic cortical inputs most likely explains the high rate of dementia that accompanies PD, particularly in older patients. However, the lesions in cholinergic, serotonergic, and noradrenergic pathways are not as clearly characterized as those in the dopaminergic systems. Whereas involvement of these neurochemical systems is generally thought to occur in more severe or late-stage disease, the temporal relationship of damage to specific neurochemical systems is not well established. For example, some PD patients develop depression months or years prior to the onset of motor symptoms, which could be attributable to an early involvement of non-dopaminergic pathways.
Pathogenesis of PD
Whatever insult initially provokes neurodegeneration, studies of toxic PD models and the functions of genes implicated in inherited forms of PD suggest two major hypotheses regarding the pathogenesis of the disease: one hypothesis posits that misfolding and aggregation of proteins are instrumental in the death of SNpc dopaminergic neurons, whereas the other proposes that the culprit is mitochondrial dysfunction and consequent oxidative stress, including toxic oxidized dopamine species. These pathogenic factors are not mutually exclusive, and one of the key aims of current PD research is to elucidate the sequence in which they act and whether points of interaction between these pathways are relevant to the demise of SNpc dopaminergic neurons. Potential points of interaction are summarized in Fig. 1. A second uncertain issue is whether the multiple molecular cell death-related pathways activated during PD neurodegeneration ultimately engage in one common downstream mechanism, such as apoptosis, or whether they remain highly divergent. Clearly, this issue is of great consequence in determining possible therapeutic strategies for PD.

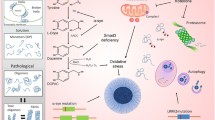
Hypothetical pathway of genetic (red) and metabolic (black) abnormalities leading to the death of dopaminergic neurons. Proteins, of which α-synuclein is only an example, undergo permanent degradation. The ubiquitin-proteasome pathway is the most important mediator of this turnover. Defects in this pathway, such as the increased generation of misfolded proteins or an impairment of ubiquitination or proteasomal function, may result in the accumulation and aggregation of misfolded proteins. Mutations in the α-synuclein gene lead to an increased tendency of spontaneous aggregation. The same applies for exogenic toxins (i.e., free radicals, 1-methyl-4-phenylpyridinium). Since parkin has E3 ubiquitin ligase activity, loss-of-function mutations may result in impaired ubiquitination and accumulation of (non-ubiquitinated) proteins that cannot be degraded. Mutations in ubiquitin C-terminal hydrolase L1 (UCH-L1) may alter the ubiquitin ligase activity and the de-ubiquitination function of polyubiquitin. Misfolded proteins may act cytotoxically directly or may be sequestered into Lewy bodies. Whether this sequestration provides neuroprotection or whether Lewy bodies are themselves toxic remains to be clarified. PTEN-induced protein kinase 1 (PINK-1) has recently been identified as causing autosomal recessive PD, is localized to mitochondria, and may contribute to the well-established mitochondrial dysfunction in PD
Protein aggregation and misfolding
The abnormal deposition of protein in brain tissue is a feature of several age-related neurodegenerative diseases, including PD, AD, and Huntington’s disease (Schulz and Dichgans 1999). Although the composition and location of protein aggregates differ between diseases, the existence of protein aggregates in most age-related neurodegenerative diseases suggests that protein deposition per se, or some related event, is toxic to neurons. Protein deposition may cause damage directly, may interfere with intracellular trafficking, or may sequester proteins that are important for cell survival.
Work by Braak and colleagues (2003) has shown that not every subtype of neurons has the capacity or the requirement to form protein aggregates. Moreover, the susceptible neuronal populations are affected in a uniform sequence in which the dorsal vagal nucleus, locus coeruleus, SNpc, mesocortex, and neocortex acquire protein aggregates in this stereotypic temporal order (Braak et al. 2003). However, these histological changes correlate poorly with both neuronal cell loss, which is most pronounced in the SNpc, and with functional deficits, which typically are asymmetrical and vary greatly between individuals.
Together with other data, these observations suggest that there is no direct link between the formation of protein inclusions and cell death (Saudou et al. 1998; Marx et al. 2003). Following current hypotheses, it is probably not the protein inclusions themselves but their fibrillary precursors that mediate toxicity (Bucciantini et al. 2002; Caughey and Lansbury 2003). In this case, LBs may even provide protection by the sequestration of ubiquitinated proteins, and toxicity may only occur in the absence of further sequestration capacity.
Evidence from familial PD
Aggregation of proteins results if the capacity of the cell to degrade proteins is impaired. The significance of the protein degradation pathway involving ubiquitination and the proteasome for the pathogenesis of PD has been highlighted by the identification of gene mutations in autosomally dominant or autosomally recessive inherited PD (Table 1). Mutations in any one of the first three PD genes identified, viz., α-synuclein, parkin, or ubiquitin C-terminal hydrolase L1 (UCH-L1) are thought to impair this pathway, as we and others have recently reviewed (Chung et al. 2001; McNaught et al. 2001; Krüger et al. 2002).
Mutations in α-synuclein favor the aggregation of α-synuclein and may promote LB formation. Loss-of-function mutations of parkin abolish its ubiquitin E3 ligase activity, which is required for the polyubiquitination necessary to target proteins to the proteasome for degradation. This failure may lead to the accumulation of misfolded proteins that are substrates of parkin. Whether or not α-synuclein is a parkin substrate is still under debate. Whereas the evidence that α-synuclein is directly ubiquitinated is sparse, a new 22-kDa glycosylated form of α-synuclein (αSp22) has been identified as a parkin substrate in normal human brain but not in other species (Shimura et al. 2001). In contrast to normal parkin, mutant parkin associated with autosomal recessive PD fails to bind αSp22.
An Ile93Met mutation in the UCH-L1 gene is thought to cause autosomal recessive PD in one family of German descent (Leroy et al. 1998). The UCH-L1 protein is found in LBs in PD, and its function relates it to the ubiquitin-proteasome pathway, because it is involved in ubiquitin re-utilization after processing of the target proteins by the proteasome complex (Hershko and Ciechanover 1992). Aside from its de-ubiquitinating function, UCH-L1 exerts a previously unrecognized ubiquitin ligase activity upon dimerization (Liu et al. 2002). Both the I93M mutation and a S18Y polymorphism alter UCH-L1 ligase activity in a manner consistent with the hypothesis that impaired activity of the ubiquitin-proteasome system is critical in PD pathogenesis; UCH-L1 ligase activity is decreased by the pathogenic I93M mutation and increased by the protective S18Y polymorphism (Liu et al. 2002).
Animal models based on familial PD
Post-mortem human brains often contain artifacts attributable to autopsy delay and typically show end stage disease rather than an evolving disease process. Therefore, animal models are needed to study the pathogenesis of PD. Murine models expressing mutated α-synuclein show cytoplasmic protein aggregation and the formation of protein inclusions that are similar to those observed in PD. They also exhibit behavioral abnormalities late in animal life (Kahle et al. 2000; Rathke-Hartlieb et al. 2001; Giasson et al. 2002; Kahle et al. 2002). However, protein aggregation occurs distant from the SNpc, and no degeneration of dopaminergic neurons is seen. Similarly, the number of SNpc dopaminergic neurons does not decline in parkin-deficient mice (Goldberg et al. 2003; Itier et al. 2003).
For studies aiming at the mechanisms of and possible protective treatment against dopaminergic cell death, these genetically engineered models have the limitation that their visible pathology (inclusions of aggregated protein) are most likely irrelevant for neurodegeneration, the actual toxins (oligomere, soluble, fibrillary protein aggregates) are not readily accessible for histological or quantitative assays, and neuronal cell loss as the primary outcome measure is not present.
MPTP model of PD
Because of the limitation cited above, the best animal model for neurodegeneration in PD is still the toxicity of MPTP. It produces clinical, biochemical, and neuropathological changes reminiscent of those occurring in idiopathic PD. Several cell death mechanisms have been implicated in MPTP toxicity, including an inhibition of complex I in the mitochondrial electron transport chain, the generation of reactive oxygen species (ROS), inflammation, the activation of excitatory amino acid receptors, apoptosis, and autophagia (Dawson 2000; Beal 2001). This animal model is probably the best, if not the only, way experimentally to determine whether the neuropathological and biochemical abnormalities found in PD brains actually cause the dysfunction and death of dopaminergic neurons. Since these mechanisms have been reviewed substantially in the past, we focus here on mitochondrial dysfunction, ROS, and apoptosis. Inflammation is reviewed in another part of this review series (Teismann and Schulz 2004).
Mitochondrial complex I is the principal source of free radicals in the cell (Lenaz 1998). The respiration chain consumes nearly 100% of molecular oxygen, and powerful oxidants are produced as byproducts. A large body of evidence has established mitochondrial involvement in the pathogenesis of PD. First, MPTP toxicity, which produces parkinsonism in humans and laboratory animals, is mediated by the inhibition of respiratory chain complex I (Heikkila et al. 1985). Second, complex I deficiency and oxidative damage have been demonstrated in the substantia nigra of PD patients (Bindoff et al. 1989; Schapira et al. 1990; Mann et al. 1992; Janetzky et al. 1994; Hattori et al. 1991). Cybrids containing mtDNA from PD platelets also show reduced complex I activity (Gu et al. 1998), strongly suggesting that inherited and/or somatic mtDNA mutations might be responsible for the biochemical phenotype in PD. As maternally inherited forms of PD or parkinsonism with complex I deficiency have been reported (Swerdlow et al. 1998; Simon et al. 1999), these mutations might represent the primary cause of the disease in rare cases. Recently, mutations in a protein kinase, PINK-1, have been identified as causing autosomally recessive inherited PD (Valente et al. 2004). PINK-1, which carries a mitochondrial import sequence, has been localized to mitochondria, and the mutations causing PD also cause sensitization to cellular stress (Valente et al. 2004). Although the exact function of this kinase und the biochemical consequences of the detected mutations are unknown, a clearly inherited form of PD has, for the first time, been directly linked to mitochondria. The product of the fourth PD gene discovered, DJ-1 (Bonifati et al. 2003), also appears to accumulate in mitochondria (Bonifati et al. 2003) and has been implicated as a cellular monitor of oxidative stress (Mitsumoto and Nakagawa 2001; Mitsumoto et al. 2001).
As mentioned above, the inhibition of complex I increases the production of superoxide (•O2), a ROS that may form toxic hydroxyl radicals or react with nitric oxide (NO•) to form the highly toxic peroxynitrite. These molecules may cause cellular damage by the oxidation of nucleic acids, proteins, and lipids. Several biological markers of oxidative damage are elevated in the SNpc of PD brains (Schulz et al. 2000). Moreover, the content of the antioxidant glutathione is reduced in the SNpc of PD patients (Sian et al. 1994) consistent with an increased ROS production in PD or a primary reduction of the protective mechanisms against ROS.
Interference with the generation of these ROS has been shown to be protective against MPTP toxicity in a variety of experiments. Superoxide dismutase (SOD) is the main detoxifying enzyme for superoxide. Transgenic mice constitutively overexpressing Cu/Zn SOD (Przedborski et al. 1992) are resistant to MPTP toxicity, whereas mice with a partial deficiency of manganese SOD (Andreassen et al. 2001) or glutathion peroxidase knock-out mice (Klivenyi et al. 2000; Zhang et al. 2000) show increased toxicity. Conversion of hydrogen peroxide to toxic hydroxyl radicals is prevented by the glutathione system. Consequently, depletion of glutathion potentiates MPP+ toxicity in vivo (Wüllner et al. 1996). Pharmacological inhibition or genetic ablation of neuronal nitric oxide synthase (nNOS) results in protection from MPTP toxicity (Schulz et al. 1995; Przedborski et al. 1996). In addition, a genetic deficiency of inducible nitric oxide synthase (iNOS), which is induced in glia cells following MPTP toxicity, is protective (Liberatore et al. 1999; Dehmer et al. 2000)
A variety of crucial biomolecules, including lipids, proteins, and DNA, can be damaged by ROS, thereby potentially leading to neurodegeneration. Indeed, increased lipid peroxidation and DNA damage (in the form of OH8dG) are found in PD brains (Dexter et al. 1994). One target of these reactive species may be the electron transport chain itself, leading to mitochondrial damage and further production of ROS. Phospholipase A2 can be activated by phospholipid peroxidation and is involved in the propagation of oxidative cell injury by free radicals. Mice deficient in phospholipase A2 are less susceptible to the detrimental effects of MPTP (Klivenyi et al. 1998).
Oxidative stress might contribute to neurotrophic factor κB (NF-κB) activation and nuclear translocation, which is reported to be elevated in PD. NF-κB may increase proinflammatory mediators, such as tumor necrosis factor α or transforming growth factor β, in glial cells, further enhancing the formation of free radicals. However, translocation of NF-κB to the nucleus occurs not only in glia cells, but also in dopaminergic neurons following MPTP toxicity (Dehmer et al. 2004). Treatment with agonists of the peroxisome proliferator-activated receptor γ blocks NF-κB activation by increasing the expression of the inhibitory protein κBα (IκBα).
Oxidative stress by dopamine metabolism
Neurodegeneration by oxidative stress may also explain the predominant degeneration of dopaminergic neurons, as the metabolism of dopamine can generate free radicals and other ROS. The enzymatic oxidation of dopamine and of its deaminated metabolites (3,4-dihydroxybenzoic acid and homovanillic acid), catalyzed by monoamine oxidase, leads to the formation of hydrogen peroxide (H2O2). H2O2 can be inactivated by catalase or by glutathione peroxidase. Because catalase is compartmentalized into peroxisomes, the detoxification of cytosolic and mitochondrial peroxides depends predominantly on glutathione peroxidase. If it is not inactivated, H2O2 can react with Fe2+ and form the highly reactive and cytotoxic hydroxyl radical (•OH) via the Fenton reaction. As mentioned above, a variety of crucial biomolecules, including lipids, proteins, and DNA, can be damaged by ROS, thereby potentially leading to neurodegeneration.
Degeneration of dopaminergic neurons and oxidative stress by dopamine metabolism are likely to be self-perpetuating, because synaptic dopamine depletion caused by a decrease in dopamine neurons leads to a compensatory increase in dopamine turnover, with increased formation of H2O2. This hypothesis is supported by experimental studies demonstrating that enhanced dopamine turnover is associated with increased formation of oxidized glutathione; this, in turn, can be prevented by inhibitors of dopamine metabolism (Spina and Cohen 1989).
Does the replacement of dopamine in patients with PD by administration of L-Dopa exacerbate the progression of the disease by increasing oxidative stress? Several studies have shown that dopamine is toxic to primary mesencephalic dopaminergic neurons or dopaminergic cell lines in culture. However, this may be an artifact of cell culture, caused by the extracellular generation of ROS. Indeed, the co-culture of mesencephalic dopaminergic neurons with astrocytes enhances survival and prevents L-Dopa-induced death (Drukarch et al. 1998; McNaught and Jenner 1999; Zietlow et al. 1999), and L-Dopa treatment of rats does not induce toxicity in partially 6-hydroxydopamine (6-OHDA)-denervated PD animal models (Murer et al. 1998). In normal rats or 6-OHDA-denervated animals, systemic administration of dopamine or stereotaxic injection of dopamine into the striatum does not induce the production of hydroxyl radicals as measured by the salicylate assay. However, in animals in which an inhibitor of oxidative phosphorylation is co-injected into the striatum, a massive increase of ROS occurs (Xia et al. 2001b). Because an inhibition of oxidative phosphorylation has repeatedly been reported in PD patients, the current data cannot rule out that L-Dopa treatment contributes to disease progression in PD.
Points of interaction between protein aggregation and oxidative stress
Oxidative damage to α-synclein can enhance its ability to misfold and aggregate (Giasson et al. 2000). Indeed, oxidative dimer formation has been shown to be the critical rate-limiting step for the aggregation and fibrillogenesis of mutant α-synclein (Krishnan et al. 2003). Toxic protofibrils are stabilized by the formation of dopamine-synuclein adducts (Conway et al. 2001), and after MPTP treatment, synuclein is a preferential target for oxidative modification (Przedborski et al. 2001). Thus, the generation of oxidative stress by MPTP, paraquat, and rotenone leads to synuclein aggregation (Kowall et al. 2000; Lee et al. 2002; Meredith et al. 2002). H2O2 treatment has been shown to reduce the ubiquitination of protein (Jahngen-Hodge et al. 1997), which should reduce physiological protein degradation.
On the other hand, the expression of mutant synuclein leads to increased protein carbonylation and increased toxicity of MPP+, the active metabolite of MPTP (Lee et al. 2001), whereas parkin inhibits protein carbonylation (Hyun et al. 2002). A decrease of proteasomal activity, which has been observed in the substantia nigra of PD brains (McNaught and Jenner 2001), increases neuronal vulnerability to normally subtoxic levels of free radicals and amplifies energy depletion following complex I inhibition (Höglinger et al. 2003). Accordingly, the cytotoxicity of proteasome inhibitors has been shown to be enhanced by dopamine (Fornai et al. 2003). Taken together, these experiments show that tight interactions exist between the two pathways to PD.
Mode of cell death: a common downstream pathway?
How do cells ultimately die in PD? Does a common downstream pathway mediate all PD-related cell loss, or is there significant heterogeneity in the pathways activated in different sick neurons in a single patient, or among different patients with PD? The answers to these questions are important for the rational development of therapeutic strategies against PD.
In human brain, apoptosis has been considered to be an important mediator of cell death and to contribute to the degeneration of dopaminergic SNpc neurons during the pathogenesis of PD. Two studies have reported that 5%–8% of neurons in the SNpc of PD patients show DNA-end labeling, an apoptosis marker; a third study has reported characteristic chromatin changes seen by electron microscopy in 6% of the melanin-containing neurons (Mochizuki et al. 1996; Anglade et al. 1997; Tompkins et al. 1997). However, the significance of morphological features suggestive of apoptosis has remained controversial, and other groups have failed to detect apoptotic changes in the SNpc (Kosel et al. 1997; Banati et al. 1998; Wüllner et al. 1999), possibly because apoptotic DNA fragments have a relatively short half-life. The detection of activated caspase-3 and caspase-8 and the appearance of substrate cleavage products (molecular markers of apoptotic cell death) support the hypothesis that apoptosis and processed caspases are important mediators of neuronal cell death in neurodegenerative diseases (Hartmann et al. 2000, 2001b).
MPP+ toxicity involves the activation of caspases in vitro (Dodel et al. 1998; von Coelln et al. 2001) and in vivo (Yang et al. 1998; Eberhardt et al. 2000; Hartmann et al. 2000) under most, but not all, experimental conditions (Lotharius et al. 1999; Hartmann et al. 2001b; Han et al. 2003). The most likely explanation for the discrepancy is the severity of the insult. The more acute and severe insults will result in caspase-independent cell death, whereas chronic insults will result in caspase-dependent apoptosis. Probably, the remaining concentration of ATP determines the mode of cell death (Hartmann et al. 2001b; Han et al. 2003), because ATP is necessary for the activation of caspase-9 in the mitochondrial activation pathway.
In mice, chronic administration of MPTP induces apoptotic cell death in dopaminergic SNpc neurons. Transgenic mice expressing a dominant-negative mutant of interleukin-1β (synonymous with caspase-1)-converting enzyme are relatively resistant to MPTP toxicity (Klevenyi et al. 1999). Furthermore, the overexpression of the anti-apoptic protein, Bcl-2, prevents the activation of caspases and provides protection against MPTP toxicity (Yang et al. 1998). In transgenic mice expressing p35 (a broad-spectrum viral caspase inhibitor), cell loss after MPTP treatment is reduced (Viswanath et al. 2000). In these mice, the activation of caspase-3, caspase-8, and caspase-9, the release of cytochrome c, and the cleavage of Bid (a pro-apoptotic Bcl-2 family member) after MPTP injections are reduced compared with wild-type mice (Viswanath et al. 2001).
Study of the effects of MPP+ in PC12 cells and primary mesencephalic culture has established a temporal sequence of activation after cytochrome c release from caspase-9 to caspase-3 and finally caspase-8. All changes are prevented by a caspase-9 inhibitor (LEHD-CHO). A caspase-8 inhibitor (IETD-CHO) decreases caspase-3 or caspase-9 activation only slightly. Bid is cleaved by caspase-8 and promotes cytochrome c release (Viswanath et al. 2001). The activation of caspase-8 occurs in a minority of neurons and glial cells in parkinsonian SNpc and in nigral neurons after MPTP treatment of mice (Hartmann et al. 2001b). These data are compatible with a model of cytochrome c-induced caspase-9 activation leading to a caspase-3 activation that mediates the effector phase of apoptosis and with an amplification loop involving caspase-8 (Viswanath et al. 2001).
In addition to the endogenous activation of caspases by cytochrome c, there exists an exogenous receptor-activated pathway. Fas is one of these pro-apoptotic receptors and may activate caspase-8 (Hengartner 2000). Whether Fas is induced following MPTP/MPP+ toxicity is a subject of controversy (Gomez et al. 2001; Hayley et al. 2004). In our hands, Fas ligand (FasL) does not kill primary dopaminergic neurons in culture (R. von Coelln and J.B. Schulz, unpublished), and MPTP toxicity is not attenuated in lpr and gld mice, which are Fas-defective or FasL-defective, respectively (S. Rathke-Hartlieb and J.B. Schulz, unpublished). A recent report of Fas-deficient mice that are more resistant to MPTP toxicity than wild-types (Hayley et al. 2004) may be explained by the Fas-deficient mice having a pre-existing deficit of dopamine and its metabolites. Even though the conversion of MPTP to MPP+ in these mice is normal, the uptake of MPP+ into synaptic vesicles is probably reduced.
Therefore, as in most paradigms of neuronal apoptosis, the mitochondrial activation pathway appears to be more important than the Fas-dependent one in PD. The mitochondrial activation pathway requires the release of cytochrome c from mitochondria in connection with the opening of the mitochondrial transition pore. Cytochrome c then forms a tertiary complex with Apaf-1 and caspase-9 in the cytosol, and in the presence of ATP, this leads to the activation of caspase-9. MPP+ has been reported to be able to induce the opening of the mitochondrial transition pore (Cassarino et al. 1999). Virus-mediated expression of a dominant-negative form of Apaf-1, consisting of the wild-type caspase recruitment domain (CARD), provides protection against dopaminergic cell loss and caspase activation in the mouse MPTP model (Mochizuki et al. 2001). Neurons expressing non-activated caspase-3 seem to be particularly prone to early degeneration as compared with controls; their number is low in parkinsonian SNpc, whereas a higher number of cells with activated caspase-3 (6.5% vs 3.9%) has been observed (Hartmann et al. 2000). Consistent with this hypothesis, as MPP+-induced cell loss proceeds in culture, the number of neurons expressing activated caspase-3 declines rapidly.
There is growing evidence that, following protein misfolding, cell death occurs by apoptosis. Proteasomal inhibition induces the formation of protein inclusions and apoptotic cell death in cultured embryonic neurons (Qiu et al. 2000; Rideout et al. 2002). Apoptotic cell death has also been shown in N27 and HEK 293 cells following the overexpression of mutant α-synuclein (Zhou and Freed 2004). In vivo, the striatal administration of a proteasome inhibitor causes the selective degeneration of dopaminergic neurons and axon terminals, the appearance of apoptotic bodies, and the formation of cytoplasmic inclusions (Fornai et al. 2003). Even though more work is needed, these data suggest that apoptotic cell death may be a common downstream pathway for different models of PD and for PD itself.
Therapeutic strategies
In research on PD animal models, neuroprotective strategies often overlap with experiments aimed at dissecting the molecular pathway leading from, for example, MPTP administration to cell death. The challenge faced by researchers is to identify strategies that interfere early enough in the proposed pathway to be potent causal interventions, but that occur late enough to act within the common downstream pathway between the model and PD itself. As cell death in PD most likely occurs through caspase-dependent apoptosis, the inhibition of this cascade has been intensively studied. This strategy has the advantage of being independent of the model used.
Caspase inhibition is achieved by tripeptide or tetrapeptide inhibitors or by viral proteins and their mammalian homologs with different substrate specificity (Deveraux and Reed 1999; Robertson et al. 2000). Peptide caspase inhibitors (zVAD-fmk or selectice caspase-3 inhibitors) protect primary mesencephalic cultures against MPP+ (Dodel et al. 1998; Eberhardt et al. 2000; Bilsland et al. 2002). However, the loss of [3H]dopamine uptake as a marker for dendritic function is not reversed (Eberhardt et al. 2000). Recently, more detailed analysis has provided evidence that, although caspase inhibitors are protective against MPP+ toxicity in primary dopaminergic neurons or dopaminergic cell lines in culture, this rescue may be temporary, may cause a switch from apoptosis to necrosis, or may not result in functional benefit (Choi et al. 1999; Eberhardt et al. 2000; Hartmann et al. 2001b). An inhibitor of caspase-1-like enzymes was not effective against MPP+ in primary mesencephalic or cerebellar granule cells (Du et al. 1997; Bilsland et al. 2002).
Inhibitors of apoptosis proteins (IAP) were first discovered as viral proteins and were shown to suppress the defensive apoptotic host response to viral infection; ectopic expression in mammalian cells blocks apoptosis (Deveraux and Reed 1999). These inhibitors share one or several baculoviral IAP repeat (BIR) domains. Many of them also have a RING domain (dispensible for anti-apoptotic effects), and some of them possess a CARD. They block caspase activity by directly binding to specific pro-caspases or active caspases (Deveraux et al. 1998). The baculoviral protein p35, for example, is a broad inhibitor of caspase function, whereas cowpox virus product (CrmA) inhibits primarily caspase-1 and caspase-8. In humans, at least six homologous proteins have been discovered: NAIP, cIAP1/HIAP-2, cIAP2/HIAP-1, Survivin, Bruce, and X-linked IAP (XIAP).
We have compared, in cellular models, the efficacy of different adenoviral constructs (AdV-XIAP, AdV-HIAP1, AdV-HIAP2, AdV-NAIP, AdV-p35, AdV-crmA) against apoptotic stimuli and have found XIAP expression to be the most effective (Simons et al. 1999; Kügler et al. 2000; Gerhardt et al. 2001). XIAP preferably blocks the activation of caspase-3, caspase-6, and caspase-7 by inhibiting the processing of procaspase-9. XIAP contains a RING finger domain and three BIR domains, of which BIR-3 is assumed to associate with caspase-9, and BIR-1/2 with caspase-3 and caspase-7 (Robertson et al. 2000); it is ubiquitously expressed in human tissues but is sequestered from caspases by the Smac/Diablo and Omi/HtrAZ proteins under normal circumstances. In some instances, an intact c-Jun NH2-terminal kinase 1 (JNK1) signaling pathway seems to be required for its anti-apoptotic function (Sanna et al. 1998, 2002). Interestingly, XIAP has recently been identified as a ubiquitin ligase, providing additional cross-talk to the ubiquitin-proteasome system.
Transfection of the nigrostriatal pathway with an Ad-XIAP leads to strong expression of XIAP protein in the striatum and in dopaminergic neurons of the SNpc (Eberhardt et al. 2000). Expression of XIAP provides protection against the MPTP-induced loss of tyrosine-hydroxylase-positive neurons but not against the reduction of striatal catecholamine concentrations, suggesting a dissociation between neuronal survival and the loss of neuritic function. Additional studies in primary mesencephalic cultures have provided evidence that caspase inhibition by zVAD-fmk rescues the tyrosine hydroxylase-positive somata, but not their neurites and synapses, from MPP+-induced toxicity and 6-OHDA-induced toxicity (Eberhardt et al. 2000; von Coelln et al. 2001). In contrast, the adenovirus-mediated expression of glial-cell-derived neurotrophic factor (GDNF) results in higher striatal catecholamine concentrations but does not protect against the MPTP-induced loss of dopaminergic neurons. The combination of adenoviral gene transfer of XIAP and GDNF has synergistic effects: the MPTP-induced loss of tyrosine hydroxylase-positive neurons is almost completely blocked, and the dopamine concentrations in the striatum are fully restored (Eberhardt et al. 2000).
In order to rescue functional neurons before irreversible damage has occurred, recent research has focused on the mechanisms by which pro-apoptotic factors may be released from mitochondria to activate the caspase cascade. Prostate apoptosis response-4 (Par-4) was originally identified as being upregulated in prostate tumor cells undergoing apoptosis but is now known to be essential in developmental and pathological neuronal death (Guo et al. 1997; Mattson 2000). Levels of Par-4 increase rapidly in response to various apoptotic stimuli through enhanced translation of Par-4 mRNA. A leucine zipper domain in the carboxy-terminus of Par-4 is essential for its pro-apoptotic function, and the interactions of Par-4 with other proteins, including protein kinase Cζ and Bcl-2, through this zipper may be central to the mechanisms by which Par-4 induces mitochondrial dysfunction. Levels of Par-4 are selectively increased before death in dopaminergic neurons of SNpc in PD brain and in mice and monkeys following MPTP treatment (Duan et al. 1999). In culture, the blocking of Par-4 induction by antisense treatment provides protection.
The pro-apoptotic protein Bax may have a central role in mediating mitochondria-dependent apoptosis in neurons (Deckwerth et al. 1996). Models of Bax activation indicate that its oligomerization results in a homo-multimeric pore (Saito et al. 2000), a VDAC-containing pore (Shimizu et al. 1999), or permeabilization of the mitochondrial outer membrane (Kluck et al. 1999) to release cytochrome c. Following MPTP treatment, Bax is upregulated in the SNpc (Hartmann et al. 2001a). This upregulation appears to be of functional relevance, since mutant mice lacking Bax are significantly more resistant to MPTP toxicity than are their wild-type littermates (Vila et al. 2001). Collectively, the results indicate that Bax plays a pivotal role in SNpc dopaminergic neuronal death in the MPTP mouse model, probably by acting in injured neurons before the onset of irreversible cell death events.
One way in which the new transcription of early-death-inducing genes, including Bax, Par-4, or Bim (Putcha et al. 2001; Whitfield et al. 2001), that lead to the translocation of cytochrome c from mitochondria may occur is via the activation of the mitogen-activated protein (MAP) kinase pathway. Saporito and colleagues have shown that, in the MPTP model, the JNK pathway is activated and that the pharmacological inhibition of this pathway with CEP1347 is neuroprotective (Saporito et al. 1999, 2000). We have recently investigated the role of the pro-apoptotic JNK signaling cascade in SH-SY5Y human neuroblastoma cells in vitro and in mice in vivo (Xia et al. 2001a). MPTP/MPP+ lead to the sequential phosphorylation and activation of JNK kinase MKK4, JNK, and c-Jun, the activation of caspases, and apoptosis. In mice, adenoviral gene transfer of the JNK-binding domain of JNK-interacting protein-1 (a scaffold protein and inhibitor of JNK) inhibits this cascade downstream of MKK4 phosphorylation and blocks JNK, c-Jun, and caspase activation, the death of dopaminergic neurons, and the loss of catecholamines in the striatum. Furthermore, the gene transfer results in behavioral benefit. Therefore, the inhibition of the JNK pathway offers a new treatment strategy for PD by blocking the death signaling pathway upstream of the execution of apoptosis in dopaminergic neurons and thus provides a therapeutic advantage over the direct inhibition of caspases.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) has been shown to play a role in apoptosis in some cellular models. Age-induced and cytosine-arabinoside-induced apoptosis in cerebellar granule cells and age-induced apoptosis in cerebral cortical cultures is associated with increased expression of GAPDH and is prevented by treatment with GAPDH antisense oligonucleotides (Ishitani and Chuang 1996; Ishitani et al. 1996a, 1996b). Cell death-associated nuclear translocation of GAPDH and antisense protection occurs in several neuronal and non-neuronal systems (Ishitani et al. 1997; Saunders et al. 1999; Shashidharan et al. 1999). Downregulation of GAPDH expression by antisense oligonucleotides protects mesencephalic dopaminergic neurons from MPP+ toxicity (Fukuhara et al. 2001). CGP3466 (dibenzo[b,f]oxepin-10-ylmethyl-methyl-prop-2-ynyl-amine) is structurally related to R-(−)-deprenyl and shares its ability to bind to GAPDH and rescue neurons in several in vitro and in vivo paradigms (Kragten et al. 1998; Carlile et al. 2000). It also protects against MPTP-induced and 6-OHDA-induced toxicity and behavioral deficits in vivo, without affecting monoamine oxidase B activity (Andringa and Cools 2000; Andringa et al. 2000; Waldmeier et al. 2000). Even though this looks promising, crucial experiments, including the establishment of GAPDH upregulation and nuclear translocation by MPTP and the effects of CGP3446 on these changes, still need to be carried out in dopaminergic neurons.
Conclusions
Although several mechanisms and downstream mediators of dopaminergic cell death have been elucidated during the last decade, the identification of genes causing PD will allow us to investigate the initial events that cause neuronal dysfunction and lead to cell death. Investigations of MPTP toxicity have shown that blocking apoptosis and inflammation by pharmacological or genetic means often prevents the death of dopaminergic neurons, but not their terminals. This shortcoming may be solved by combining a protective anti-apoptotic treatment with a neurorestorative (e.g., neurotrophic) treatment. One may argue that blocking the final demise of the cell is too late in clinical terms and will not restore the metabolic dysfunction of dopaminergic neurons. Once we understand the initial pathogenetic steps that are initiated by the identified mutant gene, we should be able to develop therapeutic approaches that aim to interfere at the beginning of this deleterious cascade and that may lead to full protection and metabolic function.
References
Andreassen OA, Ferrante RJ, Dedeoglu A, Albers DW, Klivenyi P, Carlson EJ, Epstein CJ, Beal MF (2001) Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp Neurol 167:189–195
Andringa G, Cools AR (2000) The neuroprotective effects of CGP 3466B in the best in vivo model of Parkinson’s disease, the bilaterally MPTP-treated rhesus monkey. J Neural Transm Suppl 60:215–225
Andringa G, Oosten RV van, Unger W, Hafmans TG, Veening J, Stoof JC, Cools AR (2000) Systemic administration of the propargylamine CGP 3466B prevents behavioural and morphological deficits in rats with 6-hydroxydopamine-induced lesions in the substantia nigra. Eur J Neurosci 12:3033–3043
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12:25–31
Banati RB, Daniel SE, Blunt SB (1998) Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord 13:221–227
Beal MF (2001) Experimental models of Parkinson’s disease. Nat Rev Neurosci 2:325–334
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 20:415–455
Bilsland J, Roy S, Xanthoudakis S, Nicholson DW, Han Y, Grimm E, Hefti F, Harper SJ (2002) Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci 22:2637–2649
Bindoff LA, Birch-Machin M, Cartlidge NEF, Parker WD Jr, Turnbull DM (1989) Mitochondrial function in Parkinson’s disease. Lancet I:49
Bonifati V, Rizzu P, Baren MJ van, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, Swieten JC van, Brice A, Meco G, Duijn CM van, Oostra BA, Heutink P (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259
Braak H, Del Tredici K, Rub U, Vos RA de, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416:507–511
Carlile GW, Chalmers-Redman RM, Tatton NA, Pong A, Borden KE, Tatton WG (2000) Reduced apoptosis after nerve growth factor and serum withdrawal: conversion of tetrameric glyceraldehyde-3-phosphate dehydrogenase to a dimer. Mol Pharmacol 57:2–12
Cassarino DS, Parks JK, Parker WD Jr, Bennett JP Jr (1999) The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim Biophys Acta 1453:49–62
Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298
Choi WS, Yoon SY, Oh TH, Choi EJ, O’Malley KL, Oh YJ (1999) Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J Neurosci Res 57:86–94
Chung KK, Dawson VL, Dawson TM (2001) The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci 24:S7–S14
Coelln R von, Kügler S, Bahr M, Weller M, Dichgans J, Schulz JB (2001) Rescue from death but not from functional impairment: caspase inhibition protects dopaminergic cells against 6-hydroxydopamine-induced apoptosis but not against the loss of their terminals. J Neurochem 77:263–273
Conway KA, Rochet JC, Bieganski RM, Lansbury PT Jr (2001) Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294:1346–1349
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999a) The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D (28K) immunohistochemistry. Brain 122:1421–1436
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999b) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122:1437–1448
Dawson TM (2000) New animal models for Parkinson’s disease. Cell 101:115–118
Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Snider WD, Korsmeyer SJ (1996) Bax is required for neuronal death after trophic factor deprivation and during development. Neuron 17:401–411
Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB (2000) Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem 74:2213–2216
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with IκBα induction and block of NFκB and iNOS activation. J Neurochem 88:494–501
Deveraux QL, Reed JC (1999) IAP family proteins—suppressors of apoptosis. Genes Dev 13:239–252
Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17:2215–2223
Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ, Schapira A, Jenner P, Marsen CD (1994) Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol 35:38–44
Dodel RC, Du Y, Bales KR, Ling ZD, Carvey PM, Paul SM (1998) Peptide inhibitors of caspase-3-like proteases attenuate 1-methyl-4-phenylpyridinum-induced toxicity of cultured fetal rat mesencephalic dopamine neurons. Neuroscience 86:701–707
Drukarch B, Schepens E, Stoof JC, Langeveld CH, Van Muiswinkel FL (1998) Astrocyte-enhanced neuronal survival is mediated by scavenging of extracellular reactive oxygen species. Free Radic Biol Med 25:217–220
Du Y, Dodel RC, Bales KR, Jemmerson R, Hamilton-Byrd E, Paul SM (1997) Involvement of caspase-3-like cysteine protease in 1-methyl-4-phenylpyridinium-mediated apoptosis of cultured cerebellar granule neurons. J Neurochem 69:1382–1388
Duan W, Zhang Z, Gash DM, Mattson MP (1999) Participation of prostate apoptosis response-4 in degeneration of dopaminergic neurons in models of Parkinson’s disease. Ann Neurol 46:587–597
Duffy PE, Tennyson VM (1965) Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and locus coeruleus in Parkinson’s disease. Exp Neurol 24:398–414
Eberhardt O, Coelln RV, Kügler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E, Haid S, Isenmann S, Gravel C, Srinivasan A, Bahr M, Weller M, Dichgans J, Schulz JB (2000) Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J Neurosci 20:9126–9134
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114:2283–2301
Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti CL, Ruffoli R, Soldani P, Ruggieri S, Alessandri MG, Paparelli A (2003) Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J Neurosci 23:8955–8966
Forno LS (1996) Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55:259–272
Fukuhara Y, Takeshima T, Kashiwaya Y, Shimoda K, Ishitani R, Nakashima K (2001) GAPDH knockdown rescues mesencephalic dopaminergic neurons from MPP+-induced apoptosis. Neuroreport 12:2049–2052
Gerhardt E, Kügler S, Leist M, Beier C, Berliocchi L, Volbracht C, Weller M, Bähr M, Nicotera P, Schulz JB (2001) Cascade of caspase-activation in potassium-deprived cerebellar granule neurons: targets for treatment with peptide and protein inhibitors of apoptosis. Mol Cell Neurosci 17:717–731
Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290:985–989
Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34:521–533
Gibb WRG, Lees AJ (1988) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 51:745–752
Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635
Gomez C, Reiriz J, Pique M, Gil J, Ferrer I, Ambrosio S (2001) Low concentrations of 1-methyl-4-phenylpyridinium ion induce caspase-mediated apoptosis in human SH-SY5Y neuroblastoma cells. J Neurosci Res 63:421–428
Gu M, Cooper JM, Taanman JW, Schapira AH (1998) Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann Neurol 44:177–186
Guo N, Krutzsch HC, Inman JK, Roberts DD (1997) Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res 57:1735–1742
Han BS, Hong HS, Choi WS, Markelonis GJ, Oh TH, Oh YJ (2003) Caspase-dependent and -independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J Neurosci 23:5069–5078
Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC (2000) Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc Natl Acad Sci USA 97:2875–2880
Hartmann A, Michel PP, Troadec JD, Mouatt-Prigent A, Faucheux BA, Ruberg M, Agid Y, Hirsch EC (2001a) Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J Neurochem 76:1785–1793
Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC (2001b) Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J Neurosci 21:2247–2255
Hattori N, Tanaka M, Ozawa T, Mizuno Y (1991) Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann Neurol 30:563–571
Hayley S, Crocker SJ, Smith PD, Shree T, Jackson-Lewis V, Przedborski S, Mount M, Slack R, Anisman H, Park DS (2004) Regulation of dopaminergic loss by Fas in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J Neurosci 24:2045–2053
Heikkila RE, Nicklas WJ, Duvoisin RC (1985) Dopaminergic toxicity after the stereotaxic administration of the 1-methyl-4-phenylpyridinium ion (MPP+) to rats. Neurosci Lett 59:135–140
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776
Herkenham M, Groen BG, Lynn AB, De Costa BR, Richfield EK (1991) Neuronal localization of cannabinoid receptors and second messengers in mutant mouse cerebellum. Brain Res 552:301–310
Hershko A, Ciechanover A (1992) The ubiquitin system for protein degradation. Annu Rev Biochem 61:761–807
Höglinger GU, Carrard G, Michel PP, Medja F, Lombes A, Ruberg M, Friguet B, Hirsch EC (2003) Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J Neurochem 86:1297–1307
Hyun DH, Lee M, Hattori N, Kubo S, Mizuno Y, Halliwell B, Jenner P (2002) Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J Biol Chem 277:28572–28577
Ishitani R, Chuang DM (1996) Glyceraldehyde-3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons. Proc Natl Acad Sci USA 93:9937–9941
Ishitani R, Kimura M, Sunaga K, Katsube N, Tanaka M, Chuang DM (1996a) An antisense oligodeoxynucleotide to glyceraldehyde-3-phosphate dehydrogenase blocks age-induced apoptosis of mature cerebrocortical neurons in culture. J Pharmacol Exp Ther 278:447–454
Ishitani R, Sunaga K, Hirano A, Saunders P, Katsube N, Chuang DM (1996b) Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. J Neurochem 66:928–935
Ishitani R, Sunaga K, Tanaka M, Aishita H, Chuang DM (1997) Overexpression of glyceraldehyde-3-phosphate dehydrogenase is involved in low K+-induced apoptosis but not necrosis of cultured cerebellar granule cells. Mol Pharmacol 51:542–550
Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, Sanchez M, Denefle P, Benavides J, Tremp G, Rooney TA, Brice A, Garcia de Yebenes J (2003) Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 12:2277–2291
Jahngen-Hodge J, Obin MS, Gong X, Shang F, Nowell TR Jr, Gong J, Abasi H, Blumberg J, Taylor A (1997) Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem 272:28218–28226
Janetzky B, Hauck S, Youdim M, Riederer P, Jellinger K, Pantucek F, Zochling R, Boissl KW, Reichmann H (1994) Unaltered aconitase activity, but decreased complex-I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci Lett 169:126–128
Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, Putten H van der, Probst A, Kremmer E, Kretzschmar HA, Haass C (2000) Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J Neurosci 20:6365–6373
Kahle PJ, Haass C, Kretzschmar HA, Neumann M (2002) Structure/function of alpha-synuclein in health and disease: rational development of animal models for Parkinson’s and related diseases. J Neurochem 82:449–457
Klevenyi P, Andreassen O, Ferrante RJ, Schleicher JR Jr, Friedlander RM, Beal MF (1999) Transgenic mice expressing a dominant negative mutant interleukin-1beta converting enzyme show resistance to MPTP neurotoxicity. Neuroreport 10:635–638
Klivenyi P, Beal MF, Ferrante RJ, Andreassen OA, Wermer M, Chin MR, Bonventre JV (1998) Mice deficient in group IV cytosolic phospholipase A2 are resistant to MPTP neurotoxicity. J Neurochem 71:2634–2637
Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF (2000) Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci 20:1–7
Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barber MJ, Green DR, Newmeyer DD (1999) The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol 147:809–822
Kosel S, Egensperger R, Voneitzen U, Mehraein P, Graeber MB (1997) On the question of apoptosis in the parkinsonian substantia nigra. Acta Neuropathol 93:105–108
Kowall NW, Hantraye P, Brouillet E, Beal MF, McKee AC, Ferrante RJ (2000) MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 11:211–213
Kragten E, Lalande I, Zimmermann K, Roggo S, Schindler P, Muller D, Oostrum J van, Waldmeier P, Furst P (1998) Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(−)-deprenyl. J Biol Chem 273:5821–5828
Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, Citron M, Carpenter JF (2003) Oxidative dimer formation is the critical rate-limiting step for Parkinson’s disease alpha-synuclein fibrillogenesis. Biochemistry 42:829–837
Krüger R, Eberhardt O, Riess O, Schulz JB (2002) Parkinson’s disease: one biochemical pathway to fit all genes? Trends Mol Med 8:236–240
Kügler S, Straten G, Kreppel F, Isenmann S, Liston P, Bahr M (2000) The X-linked inhibitor of apoptosis (XIAP) prevents cell death in axotomized CNS neurons in vivo. Cell Death Differ 7:815–824
Lee M, Hyun D, Halliwell B, Jenner P (2001) Effect of the overexpression of wild-type or mutant alpha-synuclein on cell susceptibility to insult. J Neurochem 76:998–1009
Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ (2002) Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem 277:5411–5417
Lenaz G (1998) Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta 1366:53–67
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998) The ubiquitin pathway in Parkinson’s disease. Nature 395:451–452
Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S (1999) Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med 5:1403–1409
Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT Jr (2002) The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 111:209–218
Lotharius J, Dugan LL, O’Malley KL (1999) Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci 19:1284–1293
Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD (1992) Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain 115:333–342
Marsden CD (1983) Neuromelanin and Parkinson’s disease. J Neural Transm Suppl 19:121–141
Marx FP, Holzmann C, Strauss KM, Li L, Eberhardt O, Gerhardt E, Cookson MR, Hernandez D, Farrer MJ, Kachergus J, Engelender S, Ross CA, Berger K, Schöls L, Schulz JB, Riess O, Krüger R (2003) Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson’s disease. Hum Mol Genet 12:1223–1231
Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1:120–129
McNaught KS, Jenner P (1999) Altered glial function causes neuronal death and increases neuronal susceptibility to 1-methyl-4-phenylpyridinium- and 6-hydroxydopamine-induced toxicity in astrocytic/ventral mesencephalic co-cultures. J Neurochem 73:2469–2476
McNaught KS, Jenner P (2001) Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci Lett 297:191–194
McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P (2001) Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat Rev Neurosci 2:589–594
Meredith GE, Totterdell S, Petroske E, Santa Cruz K, Callison RC Jr, Lau YS (2002) Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson’s disease. Brain Res 956:156–165
Mitsumoto A, Nakagawa Y (2001) DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic Res 35:885–893
Mitsumoto A, Nakagawa Y, Takeuchi A, Okawa K, Iwamatsu A, Takanezawa Y (2001) Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic Res 35:301–310
Mochizuki H, Goto K, Mori H, Mizuno Y (1996) Histochemical detection of apoptosis in Parkinson’s disease. J Neurol Sci 137:120–123
Mochizuki H, Hayakawa H, Migita M, Shibata M, Tanaka R, Suzuki A, Shimo-Nakanishi Y, Urabe T, Yamada M, Tamayose K, Shimada T, Miura M, Mizuno Y (2001) An AAV-derived Apaf-1 dominant negative inhibitor prevents MPTP toxicity as antiapoptotic gene therapy for Parkinson’s disease. Proc Natl Acad Sci USA 98:10918–10923
Murer MG, Dziewczapolski G, Menalled LB, Garcia MC, Agid Y, Gershanik O, Raisman-Vozari R (1998) Chronic levodopa is not toxic for remaining dopamine neurons, but instead promotes their recovery in rats with moderate nigrostriatal lesions. Ann Neurol 43:561–575
Pappolla MA (1986) Lewy bodies of Parkinson’s disease. Immune electron microscopic demonstration of neurofilament antigens in constituent filaments. Arch Pathol Lab Med 110:1160–1163
Price KS, Farley IJ, Hornykiewicz O (1978) Neurochemistry of Parkinson’s disease: relation between striatal and limbic dopamine. Adv Biochem Psychopharmacol 19:293–300
Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, Carlson E, Epstein CJ, Cadet JL (1992) Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci 12:1658–1667
Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM (1996) Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci USA 93:4565–4571
Przedborski S, Chen Q, Vila M, Giasson BI, Djaldatti R, Vukosavic S, Souza JM, Jackson-Lewis V, Lee VM-Y, Ischiropoulos H (2001) Oxidative post-translational modifications of α-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J Neurochem 76:637–640
Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM (2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29:615–628
Qiu JH, Asai A, Chi S, Saito N, Hamada H, Kirino T (2000) Proteasome inhibitors induce cytochrome c-caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J Neurosci 20:259–265
Rathke-Hartlieb S, Kahle PJ, Neumann M, Ozmen L, Haid S, Okochi M, Haass C, Schulz JB (2001) Sensitivity to MPTP is not increased in Parkinson’s disease-associated mutant alpha-synuclein transgenic mice. J Neurochem 77:1181–1184
Rideout HJ, Stefanis L, Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, Citron M, Carpenter JF (2002) Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol Cell Neurosci 21:223–238
Robertson GS, Crocker SJ, Nicholson DW, Schulz JB (2000) Neuroprotection by the inhibition of apoptosis. Brain Pathol 10:283–292
Saito M, Korsmeyer SJ, Schlesinger PH (2000) BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat Cell Biol 2:553–555
Sanna MG, Duckett CS, Richter BW, Thompson CB, Ulevitch RJ (1998) Selective activation of JNK1 is necessary for the anti-apoptotic activity of hILP. Proc Natl Acad Sci USA 95:6015–6020
Sanna MG, Silva Correia J da, Ducrey O, Lee J, Nomoto K, Schrantz N, Deveraux QL, Ulevitch RJ (2002) IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Mol Cell Biol 22:1754–1766
Saporito MS, Brown EM, Miller MS, Carswell S (1999) CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J Pharmacol Exp Ther 288:421–427
Saporito MS, Thomas BA, Scott RW (2000) MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J Neurochem 75:1200–1208
Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66
Saunders PA, Chen RW, Chuang DM (1999) Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal apoptosis. J Neurochem 72:925–932
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54:823–827
Schulz JB, Dichgans J (1999) Molecular pathogenesis of movement disorders: are protein aggregates a common link in neuronal degeneration. Curr Opin Neurol 12:433–439
Schulz JB, Matthews RT, Muqit MMK, Browne SE, Beal MF (1995) Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP induced neurotoxicity in mice. J Neurochem 64:936–939
Schulz JB, Lindenau J, Seyfried J, Dichgans J (2000) Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 267:4904–4911
Shashidharan P, Chalmers-Redman RM, Carlile GW, Rodic V, Gurvich N, Yuen T, Tatton WG, Sealfon SC (1999) Nuclear translocation of GAPDH-GFP fusion protein during apoptosis. Neuroreport 10:1149–1153
Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483–487
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293:263–269
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD (1994) Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol 36:348–355
Simon DK, Pulst SM, Sutton JP, Browne SE, Beal MF, Johns DR (1999) Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology 53:1787–1793
Simons M, Beinroth S, Gleichmann M, Liston P, Korneluk RG, MacKenzie AE, Bähr M, Klockgether T, Robertson GS, Weller M, Schulz JB (1999) Adenovirus-mediated gene transfer of IAPs delays apoptosis of cerebellar granule neurons. J Neurochem 72:292–301
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Spina MB, Cohen G (1989) Dopamine turnover and glutathione oxidation: implications for Parkinson disease. Proc Natl Acad Sci USA 86:1398–1400
Swerdlow RH, Parks JK, Davis JN II, Cassarino DS, Trimmer PA, Currie LJ, Dougherty J, Bridges WS, Bennett JP Jr, Wooten GF, Parker WD (1998) Matrilineal inheritance of complex I dysfunction in a multigenerational Parkinson’s disease family. Ann Neurol 44:873–881
Teismann P, Schulz JB (2004) Cellular pathology of Parkinson’s disease—astrocytes, microglia and inflammation. Cell Tissue Res (this issue) DOI 10.1007/s00441-004-0944-0
Tompkins M, Basgall E, Zamrini E, Hill W (1997) Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am J Pathol 150:119–131
Uhl GR, Hedreen JC, Price DL (1985) Parkinson’s disease: loss of neurons from the ventral tegmental area contralateral to therapeutic surgical lesions. Neurology 35:1215–1218
Uhl GR, Walther D, Mash D, Faucheux B, Javoy-Agid F (1994) Dopamine transporter messenger RNA in Parkinson’s disease and control substantia nigra neurons. Ann Neurol 35:494–498
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease is caused by mutations in PINK1. Science 304:1158–1160
Vila M, Jackson-Lewis VV, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S (2001) Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc Natl Acad Sci USA 98:2837–2842
Viswanath V, Wu Z, Fonck C, Wei Q, Boonplueang R, Andersen JK (2000) Transgenic mice neuronally expressing baculoviral p35 are resistant to diverse types of induced apoptosis, including seizure-associated neurodegeneration. Proc Natl Acad Sci USA 97:2270–2275
Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK (2001) Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J Neurosci 21:9519–9528
Waldmeier PC, Spooren WP, Hengerer B (2000) CGP 3466 protects dopaminergic neurons in lesion models of Parkinson’s disease. Naunyn Schmiedebergs Arch Pharmacol 362:526–537
Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29:629–643
Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci USA 100:6145–6150
Wüllner U, Löschmann P-A, Schulz JB, Schmid A, Dringen R, Eblen F, Turski L, Klockgether T (1996) Glutathione depletion potentiates MPP+-toxicity in nigral dopaminergic neurons. Neuroreport 7:921–923
Wüllner U, Kornhuber J, Weller M, Schulz JB, Löschmann P-A, Riederer P, Klockgether T (1999) Cell death and apoptosis regulating proteins in Parkinson’s disease—a cautionary note. Acta Neuropathol 97:408–412
Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB (2001a) Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci USA 98:10433–10438
Xia XG, Schmidt N, Teismann P, Ferger B, Schulz JB (2001b) Dopamine mediates striatal malonate toxicity via dopamine transporter-dependent generation of reactive oxygen species and D2 but not D1 receptor activation. J Neurochem 79:63–70
Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou J-C, Penney JB Jr, Hyman BT, Beal MF (1998) MPTP neurotoxicity is attenuated in mice overexpressing Bcl-2. J Neurosci 18:8145–8152
Zhang J, Graham DG, Montine TJ, Ho YS (2000) Enhanced N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in mice deficient in CuZn-superoxide dismutase or glutathione peroxidase. J Neuropathol Exp Neurol 59:53–61
Zhou W, Freed CR (2004) Tyrosine-to-cysteine modification of human alpha-synuclein enhances protein aggregation and cellular toxicity. J Biol Chem 279:10128–10135
Zietlow R, Dunnett SB, Fawcett JW (1999) The effect of microglia on embryonic dopaminergic neuronal survival in vitro: diffusible signals from neurons and glia change microglia from neurotoxic to neuroprotective. Eur J Neurosci 11:1657–1667
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00441-005-1081-0.
Rights and permissions
About this article
Cite this article
Schulz, J.B., Falkenburger, B.H. Neuronal pathology in Parkinson’s disease. Cell Tissue Res 318, 135–147 (2004). https://doi.org/10.1007/s00441-004-0954-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-004-0954-y