
Abstract
Pathogenic variants in genes encoding subunits of the spliceosome are the cause of several human diseases, such as neurodegenerative diseases. The RNA splicing process is facilitated by the spliceosome, a large RNA–protein complex consisting of small nuclear ribonucleoproteins (snRNPs), and many other proteins, such as heterogeneous nuclear ribonucleoproteins (hnRNPs). The HNRNPU gene (OMIM *602869) encodes the heterogeneous nuclear ribonucleoprotein U, which plays a crucial role in mammalian development. HNRNPU is expressed in the fetal brain and adult heart, kidney, liver, brain, and cerebellum. Microdeletions in the 1q44 region encompassing HNRNPU have been described in patients with intellectual disability (ID) and other clinical features, such as seizures, corpus callosum abnormalities (CCA), and microcephaly. Recently, pathogenic HNRNPU variants were identified in large ID and epileptic encephalopathy cohorts. In this study, we provide detailed clinical information of five novels and review two of the previously published individuals with (likely) pathogenic de novo variants in the HNRNPU gene including three non-sense and two missense variants, one small intragenic deletion, and one duplication. The phenotype in individuals with variants in HNRNPU is characterized by early onset seizures (6/7), severe ID (6/6), severe speech impairment (6/6), hypotonia (6/7), and central nervous system (CNS) (5/6), cardiac (4/6), and renal abnormalities (3/4). In this study, we broaden the clinical and mutational HNRNPU-associated spectrum, and demonstrate that heterozygous HNRNPU variants cause epilepsy, severe ID with striking speech impairment and variable CNS, cardiac, and renal anomalies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The accurate RNA splicing process is of major importance for proper cell function and disruptions of this process can lead to human diseases (Wang and Cooper 2007). In RNA splicing, nascent RNA transcripts are processed into mature messenger RNA (mRNA) by the joining of exons and the removal of introns. Pathogenic variants in genes encoding subunits of the spliceosome, or its auxiliary factors, as well as mutations in splice donor or acceptor sites are the cause of human diseases (Lehalle et al. 2015; Wang and Cooper 2007).
The RNA splicing process is facilitated by the spliceosome, which is a large RNA–protein complex (Fu and Ares 2014). It consists of five small nuclear ribonucleoproteins (snRNPs: U1, U2, U4, U5, and U6) and many other proteins, such as heterogeneous nuclear ribonucleoproteins (hnRNPs). Several hnRNPs have been identified (hnRNP A1 to hnRNP U), which bind to intronic regions to enhance or repress splicing (Fu and Ares 2014). The gene HNRNPU (OMIM *602869) is located on chromosome 1q44 within a region, in which microdeletions have been described in patients with intellectual disability (ID) and other varying clinical features, such as seizures, corpus callosum abnormalities (CCA), and microcephaly (Ballif et al. 2012; Boland et al. 2007; Caliebe et al. 2010; Hill et al. 2007; Nagamani et al. 2012; Thierry et al. 2012; van Bon et al. 2008). Different critical regions within this area have been proposed and various candidate genes for different phenotypic features, such as CCA, seizures, and microcephaly, have been discussed in detail (Ballif et al. 2012; Boland et al. 2007; Caliebe et al. 2010; Hill et al. 2007; Nagamani et al. 2012; Thierry et al. 2012; van Bon et al. 2008). However, the search for the causative gene(s) has been complicated by conflicting data and proof for the causative gene(s) was not established. Recently, individuals with HNRNPU variants were identified in large cohorts of individuals with moderate or severe ID (Hamdan et al. 2014; Monroe et al. 2016), epileptic encephalopathy (Allen et al. 2013; Carvill et al. 2013; de Kovel et al. 2016), undiagnosed conditions (Need et al. 2012), and listed in the Decipher database, but only scarce clinical information of these patients was included. While this study was in revision, Depienne and colleagues reported on seven individuals with HNRNPU variants focusing on their developmental and epilepsy phenotype (Depienne et al. 2017).
In this study, we provide detailed phenotypic analysis of five individuals with novel (likely) pathogenic variants in the HNRNPU gene identified by whole-exome sequencing (WES) and array analysis, and include comprehensive clinical information of two previously published HNRNPU-mutation-positive individuals (Allen et al. 2013; Hamdan et al. 2014). The aim of this study is the further characterization of HNRNPU-mutation-positive individuals and their comparison to previously published patients with HNRNPU variants and microdeletions in the 1q44 region to elucidate the much discussed role of HNRNPU in human disease.
Materials and methods
Individuals
Clinical information of the seven individuals was provided by the parents and the evaluating clinicians. Written informed consent was obtained from the families of the index individuals for participation in this study. The study was performed according to the Declaration of Helsinki protocols and was approved by the local institutional review board [ethical votum 08-3663 and 5360/13 for the Technical University Munich] and by the ethics committee of the University Medical Center Göttingen (individual 5). Peripheral blood lymphocytes were collected from the affected individuals and their parents, and genomic DNA was extracted by standard extraction procedures.
Chromosomal analysis
The conventional karyotyping in blood lymphocytes and microarray analyses for molecular karyotyping were normal in individuals 1–4, 6, and 7 [Individual 1:105 k-Chip (Agilent, Santa Clara, CA, USA); Individual 2: 250 k SNP array (Affymetrix, Santa Clara, CA, USA); Individual 3: 135 k-feature whole-genome microarray [SignatureChip OS2.0, Signature Genomic Laboratories (Spokane, WA, USA) by Roche Nimble-Gen, Madison, WI, USA] (Allen et al. 2013); Individual 4: no array performed; Individual 6: Agilent 180 K custom HD-CGH microarray (Agilent, Santa Clara, CA, USA). Individual 7: HumanOmniExpress-12 BeadChip (Illumina, San Diego, CA, USA).
Whole-exome sequencing
For individual 1, trio-based whole-exome sequencing (WES) was performed as described elsewhere (Bramswig et al. 2015). For individuals 2, 6, and 7, diagnostic WES was performed as described elsewhere (de Ligt et al. 2012). The molecular findings and WES methods for individual 3 [case 1464.524, (Hamdan et al. 2014)] and individual 4 [trio hv, (Allen et al. 2013)] have previously been published. The variants were verified by Sanger sequencing; primer sequences will be provided upon request.
Exome capture on the individual 5′s DNA was accomplished using the NimbleGen SeqCap EZ Human Exome Library v2.0 enrichment kit followed by sequencing on an Illumina HiSeq 2000. The Varbank pipeline of the Cologne Center for Genomics (CCG) (Hussain et al. 2014) was used to analyze the exome data as described before (Keupp et al. 2013). The data set was analyzed independently by three team members. In addition we used a Wollnik-lab in-house bioinformatic pipeline, which includes copy number variation analyses with XHMM (Fromer et al. 2012) and CODEX (Jiang et al. 2015).
qPCR analysis for HNRNPU and EFCAB2 genes in individual 5
The CNV analysis was performed by quantitative real-time PCR using SYBR® Green. The qPCR was carried out using the StepOne™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The primers for HNRNPU analysis are located in intron 1 (forward: 5′-AGAGGCTTGGCCTTTATGGG-3′) and exon 2 (reverse: 5′-GGCCATGATCTTCTCGTGGT-3′), for the EFCAB2 in exon 1 (forward: 5′-TGAGCAGGCCGGGAC-3′) and intron 1 (reverse: 5′-TCCCCTACCTTACCTTCCCTG-3′), and for the reference gene KANK1 are located in intron 11 (forward: 5′-CGACCACTTGGTGTTTTGGC-3′) and exon 12 (reverse: 5′-AGCAGCTATGTAGTCCCCCA-3′). 10 ng DNA was used for qPCR. The ddCt method was used to calculate real-time PCR results. To simulate the dCt created by one copy, two copies and three copies of HNRNPU and EFCAB2 genes, one control sample with different DNA amounts were used. Using the Ct values of the different DNA amounts (5, 10, and 15 ng) of the control sample, we calculated the expected 1, 2 and 3 copies.
Transcript analysis in individual 4
Peripheral blood of individual 4 was collected into PAXgene Blood RNA tubes (PreAnalytiX), and RNA was isolated using the PAXgene Blood RNA Kit (Qiagen). cDNA was generated with the help of the Omniscript RT Kit (Qiagen), and a forward primer that overlaps the boundaries of exon 8 and exon 9, HNRNPU-forward 5′-GGATAAGATGATGGTGGCAGG-3′ (NM_031844.2: c.1602-1622, exons 8 and 9), and a reverse primer located in exon 12, HNRNPU-reverse 5′-CCGATTCCACCACTTCCTCC-3′ (NM_031844.2: c.2230-2249, exon 12), and the GoTaq G2 DNA polymerase (Promega) were used to amplify a 648 bp exon–intron-boundary spanning fragment of the HNRNPU cDNA. Because the forward primer overlaps the exon-8-9-boundary, no PCR product can be amplified from genomic DNA. The sequence at the mutation site was determined by Sanger sequencing using the above-mentioned primers.
Results
Seven individuals with HNRNPU-variants
Individual 1 was identified in a cohort of 311 patients with ID (1/311; trio-based WES, Institute of Human Genetics, University Duisburg-Essen and Institute of Human Genetics, University Bonn). Individuals 2, 6, and 7 were identified in a cohort of 2000 patients with ID (3/2000) (diagnostic WES, Radboud University Medical Center). Individual 3 was identified in a cohort of 264 probands with epileptic encephalopathy [1/264; trio-based WES, CHU Sainte-Justine Research Center; (Hamdan et al. 2014)]. Individual 4 was identified in a cohort of 41 individuals with ID [1/41; trio-based WES, Epi4K Consortium; (Allen et al. 2013)]. The variant information and some clinical findings of individuals 3 and 4 were previously published as indicated above (Allen et al. 2013; Hamdan et al. 2014). Routine array analysis for multiple congenital anomalies identified the HNRNPU duplication in individual 5. The clinical information of individuals 1–7 is summarized in Table 1.
Individual 1 is the only child of healthy, non-consanguineous parents with an uneventful family history. He was born at 32 weeks of gestation with a weight of 1680 g (−0.47 SD), a length of 42 cm (−0.39 SD), and an OFC of 29 cm (−0.74 SD). Due to perinatal asphyxia, he was intubated and ventilated for 2 days. He showed muscular hypotonia and required tube feeding for 4 weeks. He was diagnosed with atrial septal defect (ASD II), right renal agenesis, and hypoplastic aorta abdominalis. In addition, he was diagnosed with sensorineural hearing loss and got hearing aids at the age of 3 months. At the age of 7 months, he got glasses for hyperopia. He reached head control at the age of 12 months, sat at the age of 18 months, and crawled at 31 months. At the age of 14 months, he developed epileptic spasms, showed hypsarrhythmia in the EEG, and presented with multiple (up to 100) seizures per day. The administration of valproate led to significant improvement and he did not have any seizures for two years. Brain MRI showed a thin corpus callosum, cerebral atrophy, multiple lesions including glial lesions, and atrophy of the cerebellar vermis. His first assessment was at the age of 32/12 years when his height was 90 cm (−2.03 SD), his weight was 13 kg (−1.06 SD, BMI: 16.05 kg/m2), and his OFC was 51 cm (+0.44 SD). He presented with severe ID. Facial dysmorphisms included hypertelorism, deep nasal bridge, short nose with an upturned nasal tip, long philtrum, and thin upper vermillion. When he was reexamined at the age of 10 4/12 years, his height was about 125 cm (−2.91 SD), his weight 33 kg (−0.51 SD, BMI: 21.12 kg/m2), and his OFC 54 cm (−0.02 SD). He had developed severe scoliosis. He was able to walk several steps with help. Facial dysmorphisms were similar to the previous assessment and also included downturned corners of the mouth. He had large incisors which showed a malposition due to chewing on a chain and pulling it through his front teeth. Due to the remarkable phenotypic overlap with published individuals with microdeletions in 1q44 [especially with patient 6 published by van Bon and colleagues (van Bon et al. 2008)], a microdeletion 1q44 was suspected at the age of 11 4/12 years. However, the re-evaluation of the array data could not detect any aberration in this region. Over the years, anti-epileptic treatment included different combinations of valproate with sultiame, levetiracetam, or lamotrigine. The combination of valproate and lamotrigine led to good control of his myoclonic epilepsy with six to seven seizures per year. His last assessment was at the age of 146/12 years, his height was about 151 cm (−1.86 SD), and his OFC 57 cm (+1.16 SD). He walked with the help of a walker, but was not able to walk alone. He did not speak, but communicated with gestures and sounds. He salivated and showed a similar facial phenotype to his previous assessments, but his facial features appeared coarser (Fig. 1a, b). WES revealed a heterozygous de novo missense variant in the HNRNPU gene [NM_031844.2; chr1:g.245,023,684T>C [GRCh37/hg19]; c.970A>G; p.(Arg324Gly)] and a heterozygous de novo frameshift mutation in the KDM6B gene [NM_001080424.1; chr17:g.7,756,440delG [GRCh37/hg19]; c.4733delG; p.(Cys1578Serfs*11)]. The HNRNPU variant involves an evolutionary conserved amino acid, which is located in the B30.2/SRY domain, which mediates protein–protein interactions (Fig. 2). Four in silico tools (PROVEAN, SIFT v5.2.2, PolyPhen-2, and MutationTaster) predict the HNRNPU variant as probably disease causing. It was not found in the ExAC or in the 1000 Genomes databases.

Facial phenotypes of individuals with HNRNPU variants. a, b Individual 1 at the age of 146/12 years presenting with coarse facial features, deep set eyes, depressed nasal bridge, short nose with anteverted nares, long and flat philtrum, and thin upper vermillion. c Individual 2 at the age of 32 years showing a coarse face with broad full eyebrows, hypertelorism and epicanthal folds, short broad nose, and full lips. d Individual 3 at the age of 4 years with telecanthus. e, f Individual 4 presenting with hypertelorism, deep-set eyes, and short neck. g Individual 5 at the age of 25 days with microcephaly, hypertelorism, short nose with bulbous nasal tip, thin upper vermillion, and microretrognathia. h, i Individual 6 at the age of 23 years showing synophrys, hypertelorism, depressed nasal bridge and broad nasal tip, and full lips

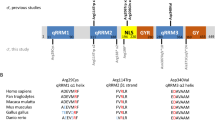
Schematic representation of HNRNPU with the SAP domain, the protein–protein interaction domain B30.2/SPRY, ATP binding and the RNA binding RGG–box (Uni-Prot Q00839), and depiction of the described HNRNPU variants (NM_031844.2/NP_114032.2)
Individual 2 is the second child of healthy, non-consanguineous parents with an unremarkable family history. She was born at term with a weight of 2780 g (−1.64 SD) and a length of 48 cm (−1.68 SD). After 2 weeks, she was admitted to the hospital because of severe oedema and failure to thrive. This was due to a Pentalogy of Fallot for which she was operated on at the age of 2 years. She had severe feeding problems that improved after the operative correction of her cor vitium. Several fever-related seizures occurred until the age of 5 years, but EEG results were normal. A cerebral brain MRI has not been performed. She was first seen at the clinical genetic center at the age of 32 years when her height was 151 cm (−2.13 SD), her weight was 64.7 kg (+0.57 SD), and her OFC was 52 cm (−1.8 SD). She was a friendly female with obsessive–compulsive behavior. She had mild webbing of neck and had a coarse face with broad full eyebrows, hypertelorism, epicanthal folds, short forehead, short broad nose, full lips, small palate, short philtrum, prominent jaw, and crowded teeth (Fig. 1c). Her extremities showed small hands and feet with short and broad fingers and toes, cutaneous syndactyly of digits 2–3 and 3–4. She had a metabolic screen and investigation of the RAI1 gene, which gave normal results. Trio-based WES was performed and revealed a heterozygous de novo non-sense mutation in the HNRNPU gene [NM_031844.2; chr1:g.245,025,823G>A [GRCh37/hg19]; c.817C>T; p.(Gln273*)].
Individual 3 (Hamdan et al. 2014) was born following an uneventful pregnancy and delivery. He was born with a weight of 3310 g (−0.95 SD). He has severe global developmental delay. He started to sit at the age of 22 months, crawl at 24 months. At the age of 4 years, he could stand with support and take a few steps with support. He did not have a pincer grasp but could reach for objects and bring them to his mouth. He does not understand simple commands. He does not speak. He has some autistic features with poor eye contact and repetitive stereotypic movements. He had febrile seizures at the age of 12 months, then unprovoked generalized tonic–clonic seizures at the age of 18 months following which he was treated with clobazam and remains well controlled, with one seizure per year on average. EEG was normal. At the age of four years, his weight was 17.7 kg (+0.69 SD), his height 96 cm (−1.75 SD), and OFC 48.6 cm (−1.65 SD). His eye contact was poorly sustained. He had a large forehead, apparent telecanthus (Fig. 1d) and bilateral single palmar creases. His tone was decreased centrally and in his limbs. Fragile X testing and metabolic studies (plasma lactate, ammonia, and very long chain fatty acids measurements; plasma amino acids and urine organic acids chromatographies; isoelectric focusing of serum transferrin; urine purines/pyrimidines measurements) were normal. Brain MRI at the age of 18 months showed broad sylvian fissures, enlarged sub-arachnoid spaces, and discrete non-specific signal anomalies in the white matter in the periventricular region and the right frontal sub-cortical region. Trio-based WES revealed a de novo non-sense mutation in HNRNPU [NM_031844.2; chr1:g.245,027,099G>A [GRCh37/hg19]; c.511C>T: p.(Gln171*)] (Hamdan et al. 2014).
Individual 4 (Allen et al. 2013) is the second child of non-consanguineous parents with no significant family medical history. She was born at 40 week gestation after an uncomplicated delivery with birth weight of 2640 g (−1.97 SD) and length 47 cm (−2.13 SD). Shortly after birth, she was noted to have low tone and difficulty feeding but did not require supplementation. Parents became concerned about her development at 6 months of age leading to evaluation and initiation of therapies at 9 months of age. She sat at 18 months and walked at 2½ years of age. First spoken words occurred at 3 years of age. Autism was diagnosed at 5 years of age. She currently speaks in 1–2 words utterances. She is able to identify 3–4 colors; she does not read or count. She has severe ID. There have been several admissions for aggressive behavior and psychosis. She continues on risperidone to manage these difficulties. At 11 years of age, she was noted to have short stature and delayed bone age. At age 15, there was an incidental finding of a right maxillary cyst with an associated tooth that required surgical intervention. Pathology demonstrated odontogenic keratocyst. Seizures began at 16 months of age with a generalized tonic–clonic seizure. Absence seizures were noted around 30 months of age and atonic seizures began at 3 years of age. Subsequent EEGs were consistent with a diagnosis of Lennox–Gastaut Syndrome. Several anti-seizure medications have been used including phenobarbital, topiramate, lamotrigine, carbamazepine, rufinamide, clobazam, and vagus nerve stimulation. Generalized tonic–clonic seizures are now rare, occurring once per year. Atonic seizures occur daily in the morning. Absence seizures occur 4–5 times a day. She has had periods of several months without seizures. MRI demonstrates several nodules of sub ependymal gray matter bilaterally in the frontal horns of the lateral ventricles. Frontal lobes contain punctate foci of T2 abnormality and there is a Chiari I malformation. At the age of 17 years, her height was 146.5 cm (−2.44 SD), her weight was 63.6 kg (+0.84 SD), and her OFC was 54.2 cm (−0.19 SD). Facial dysmorphisms included hypertelorism, deep-set eyes, and short neck (Fig. 1e, f). Trio-based WES revealed a small deletion variant in the HNRNPU gene overlapping the splice acceptor site of intron 9 and the beginning of exon 10 (Allen et al. 2013) [NM_031844.2; chr1:245,019,922_245,019,931delATTTGTcttt [GRCh37/hg19]; intron sequence is indicated in lower case letters]. The effect of this variant was investigated by transcript analysis. We found that the mutant allele yields a stable transcript. In the absence of the normal splice acceptor chr1:245,019,928_245,019,929, a cryptic splice acceptor within exon 9, chr1:245,019,904_245,019,905, is used to yield a mature mRNA that lacks the codons 582 through 589 of the wild-type transcript. Thus, the effect for this variant could be determined as an in frame deletion of eight codons, c.1744_1767del, p.Thr582_Gln589del. This part of the protein is not known as a specific domain (http://www.uniprot.org/uniprot/Q00839), but the in silico tool PROVEAN (http://provean.jcvi.org/index.php) predicts the deletion of eight amino acids as deleterious for protein function with a score of −34.661.
Individual 5 was born by emergency Caesarean section at the 34th week of gestation to consanguineous parents (first cousins). It was the fourth pregnancy; they have two healthy sons and have lost one girl who suffered from hydrocephaly, possible brain maldevelopment, and intracranial hemorrhage at the age of 4.5 months. The individual’s prenatal history was complicated by prematurity, intrauterine growth retardation (IUGR), fetal distress, and perinatal asphyxia. He was transferred intubated to the neonatal intensive care unit immediately after birth. His birth weight was 1815 g (−1.24 SD), and he presented with distinct microcephaly and multiple arthrogryposis. Neonatal seizures started in the first week of life and required phenobarbital treatment. Physical examination was performed at the age of 25 days when he showed the following measurements: weight 1890 g (−2.73 SD for 37th week of gestation), length 42 cm (−3.42 SD for 37th week of gestation), and OFC 27 cm (−4.93 SD for 37th week of gestation). Clinical features were distinct microcephaly, closed anterior fontanelle, sloping forehead, facial dysmorphism with hypertelorism, short nose, bulbous nasal tip, high palate, thin upper vermillion, microretrognathia, big ears, and a small notch on the left ear (Fig. 1g). He also showed cryptorchidism, hypospadia, scrotal raffe, pes equinovarus and multiple joint contractures, hypotony, clonus, and focal myoclonies particularly on lower extremities. Echocardiography revealed a PDA. He presented with pale optic discs and the thorax X-ray showed 10 ribs. Cerebral ultrasonography displayed an increased ventricular index of 0.55 and the cranial CT showed microcephaly, a cystic malformation between posterior fossa and the fourth ventricle, a Dandy–Walker malformation, and a hemorrhage on the left plexus choroideus. Abdominal ultrasonography revealed minimal bilateral renal pelvic ectasia; the left testis was in the inguinal canal. He passed away at about 2 months of age at home. We performed WES in individual 5, but the initial analysis and interpretation of WES variants did not identify likely causative variants. Therefore, we also determined copy number variants from the WES data and a duplication on chromosome 1 with a minimal size of 108 kb was predicted (chr1:g.245,025,709_245,133,797). The first three exons of the HNRNPU gene and the first exon of the EFCAB2 gene are located within this duplicated region and we confirmed the presence of the duplication by qPCR with amplicons located in the first exons of both genes.
Individual 6 was born from non-consanguineous healthy parents after a pregnancy that was complicated by polyhydramnios. He was born at term with a weight of 3400 g (+0.31 SD), a length of 50 cm (+0.13 SD), and a normal OFC (exact measurement unknown). His neonatal history was uneventful. His developmental milestones were late-normal for the first 18 months, but he has always been different in making social contact. He walked at 18 months of age, spoke several words and was able to climb the stairs. The onset of epilepsy was at 10 months of age with a convulsion during fever. After that, convulsions without fever started to occur. He was treated with valproate, but still had multiple temporal seizures per week. He started with teeth grinding and showed stereotypic hand movements. At 2 years of age, he was admitted to the hospital in status epilepticus. He had received a vaccination (measles, mumps, and rubella) 1 week prior to this event. He had a fever and a viral infection was presumed, but not detected. After this event, he lost the ability to walk and stopped speaking. The epilepsy was first deduced as temporal epilepsy, but evolved into West syndrome at 2.5 years, based on hypsarrythmia in the EEG and epileptic spasms. Vigabatrin was added to his medication and later changed to lamictal, but this seemed to cause more frequent seizures. He had multiple (up to 40) seizures a day, occurring mostly in the morning after awaking and before bedtime. His hands turned blue-purple frequently, sometimes related to the seizures, but sometimes without clear cause. His epilepsy evolved into a Lennox–Gastaut-like phenotype with a matching EEG and tonic and atonic insults and absences. At 4 years of age, he could walk several steps independently again. The epilepsy remained difficult to control; he showed frequent temporal seizures, tonic seizures, and infrequent atonic seizures, sometimes provoked by fever, but mostly occurring spontaneously and most frequently in the mornings. Several anti-eplieptic drugs were tried, but none induced seizure control. He became obese at 10 years of age. At 12 years of age, he became somnolent, caused by several subdural hematomas after frequent falls caused by atonic seizures. The patient has used a ketogenic diet for several years with good effect in the beginning. Neurological examination showed a spastic quadriplegia at first, but later on, he became more hypotonic. He can walk several steps with help, but because of the danger of falling, he uses a wheelchair. He has a severe intellectual disability and signs of an autism spectrum disorder. His sleeping pattern is normal. He was diagnosed with cortical visual impairment, with a vision of 0.2 in both eyes. He has a divergent strabismus, rotatory nystagmus, myopia, and astigmatism. He has osteoporosis, probably because of immobility combined with anti-epileptic drugs. He has an extra tooth that came through in his palate, and seemingly exists of several fused elements. His central incisors are thickened and long (Fig. 1h). MRI brain at 2 years of age showed wide ventricles frontotemporal and retarded frontal myelinisation. MRI brain at 12 years of age showed wide ventricles, subdural hematomas and a glandula pinealis cyst. Physical examination at 23 years of age showed a height of 175 cm (−1.24 SD), a weight of 69 kg (−0.55 SD), and an OFC of 61 cm (+1.81SD). He has an accessory whorl in his hair. Facial dysmorphisms included a synophrys, mild hypertelorism, a depressed nasal bridge, a broad nasal tip, and full lips with eversion of the lower lip (Fig. 1h, i). The ears showed slightly overfolded helices laterally. He has tapering fingers. His facial features appear coarser over the years. Array analysis revealed two variants inherited from his healthy mother of normal intellect [arr 8p23.1(10,066,633–10,780,057) × 3 mat (713 kb), arr 13q31.3(90,863,461–91,085,378) × 1 mat (222 kb)]. WES revealed a heterozygous de novo missense variant in the HNRNPU gene [NM_031844.2; chr1:g.245,022,129A>G [GRCh37/hg19]; c.1132T>C; p.(Ser378Pro)]. The variant involves an evolutionary conserved amino acid, and four in silico tools (PROVEAN, SIFT v5.2.2, PolyPhen-2, and MutationTaster) predict it as probably disease causing. It is located in B30.2/SRY domain (Fig. 2). The variant was absent in the ExAC and the 1000 genomes databases.
Individual 7 is the only child of healthy, non-related parents. Her father and her paternal aunt have had learning problems. The patient was born at 38 weeks of gestation with a weight of 2835 g (−1.08 SD), a length of 48 cm (−1.34 SD), and an OFC of 32.5 cm (−1.71 SD). The mother had a cerebral vascular incident 5 days after delivery. At the age of 11 months, she had her first febrile tonic–clonic seizure. In the following months, the seizures, absences, occurred also without fever. She was treated with valproate. Her development was delayed; she could walk at the age of 18 months with a broad based gait. At the age of 3 years and 8 months, a new assessment was done; she had a normal weight, height, and OFC. Facial dysmorphisms included bulbous nasal tip with fleshy alae nasi, a thick columella, and a thin upper vermillion. She had hypertrophic gums and short broad thumbs. Her gaze is intermittently exotropic (strabismus divergens). She could speak some words and sometimes short sentences. The estimated level was of a child of 19 months. Brain MRI showed no abnormalities. WES revealed a heterozygous de novo non-sense mutation in the HNRNPU gene [NM_031844.2; chr1: g.245027087G>A [GRCh37/hg19]; c.523C>T; p.(Gln175*)].
A schematic representation of HNRNPU with its functional domains and the location of the described variants are depicted in Fig. 2. The phenotypes of the seven individuals with HNRNPU variants are compared to the seven recently published individuals with HNRNPU variants and the phenotypes of individuals with 1q44 deletions including the HNRNPU gene (Table 2; Ballif et al. 2012; Caliebe et al. 2010; Depienne et al. 2017; Nagamani et al. 2012; Thierry et al. 2012; van Bon et al. 2008).
Discussion
Hn RNP U plays an important role in mammalian development
The large family of hnRNPs plays a crucial role in the splicing process and affects cellular homeostasis in health and several diseases, such as cancer and neurodegenerative diseases (Geuens et al. 2016). Different studies have demonstrated that the family member hnRNP U plays a crucial role in mammalian development. A hypomorphic mutation of Hnrnpu causes early embryonic lethality in mice (Roshon and Ruley 2005). A conditional knockout mouse, in which Hnrnpu is only deleted in the heart, revealed that hnRNP U is necessary for postnatal heart development and function in mice (Ye et al. 2015). RNA-sequencing data of Hnrnpu mutant hearts showed defective alternative splicing (Ye et al. 2015). More specifically, in vitro experiments displayed that hnRNP U is involved in the control of alternative splicing by the regulation of U2 snRNP maturation (Xiao et al. 2012). In addition, hnRNP U has also been implicated in many other processes of RNA metabolism, such as RNA transport and stability control (Xiao et al. 2012). HNRNPU is expressed at low levels in the fetal brain and adult heart, kidney, liver, brain and cerebellum (Thierry et al. 2012). The haploinsufficiency index for HNRNPU is 7.65% and the pLI score is 1.00 indicating a high likelihood that HNRNPU exhibits haploinsufficiency and is intolerant to loss-of-function (LoF) mutations (https://decipher.sanger.ac.uk/). A haploinsufficiency index of 0–10% indicates that a gene is more likely to exhibit haploinsufficiency, while a haploinsufficiency index of 90–100% indicates that a gene is more likely to not exhibit haploinsufficiency (https://decipher.sanger.ac.uk/). Genes with pLI ≥ 0.9 are extremely intolerant to LoF mutations, whereas genes with pLI ≤ 0.1 are tolerant to LoF mutations (https://decipher.sanger.ac.uk/). Due to the above-mentioned murine developmental phenotypes, the expression pattern, and the 1q44 microdeletion phenotypes (as discussed below), HNRNPU presents a promising candidate gene for neurodevelopmental phenotypes.
The 1q44 microdeletion phenotypes
Many studies describing the phenotypes of individuals with 1q43q44 deletions have been published and it has been discussed in great detail which gene could be causative for the main clinical features such as ID, speech delay, seizures, CCA, and microcephaly (Ballif et al. 2012; Boland et al. 2007; Caliebe et al. 2010; Hill et al. 2007; Nagamani et al. 2012; Poot 2013; Thierry et al. 2012; van Bon et al. 2008). However, there were some discrepancies between these studies with regard to the deleted critical regions and the associated phenotypes. In addition, sequencing of the AKT3 gene in a cohort with 45 patients with agenesis of the corpus callosum (Boland et al. 2007) and sequencing of the HNRNPU gene in a cohort with 191 ID patients (Thierry et al. 2012) did not reveal any pathogenic variants. Caliebe and colleagues concluded that other genes and other mechanisms besides hemizygosity, e.g., for AKT3 or HNRNPU may be involved in the phenotypic variability of these individuals, but HNRNPU sequencing might be worthwhile in patients with CCA (Caliebe et al. 2010). Other studies suggested that HNRNPU, FAM36A, and HNRNPU-AS1 (also referred to as NCRNA00201) are promising candidate genes for ID and seizures (Ballif et al. 2012; Thierry et al. 2012). In summary, conflicting data and phenotypic variability among the 1q44 deletion-positive individuals did not allow for the identification of a single causative gene for selected clinical features, but there was strong support for HNRNPU being a candidate gene for ID and seizures. Recently, Depienne and colleagues reported on seven individuals with HNRNPU mutations presenting with epilepsy and developmental delay (DD)/ID (Depienne et al. 2017), which will be discussed in more detail below.
Mutational spectrum of HNRNPU variants
In this study, we present detailed clinical descriptions of seven individuals with HNRNPU variants including three non-sense and two missense variants, one intragenic deletion, and one duplication. Based on these findings, we suspect haploinsufficiency to be the major pathogenic mechanism. Depienne et al. reported on one non-sense, four frameshift, and two splice site HNRNPU mutations (Depienne et al. 2017). The two missense variants identified in our study, p.(Arg324Gly) and p.(Ser378Pro), result in the substitution of the affected amino acids to amino acids with different chemical and steric properties, and are located within the B30.2/SPRY domain of the protein (Fig. 2). B30.2/SPRY domains, which mediate protein–protein interactions, have been found in many protein families involved in a variety of functions in different biological processes (Perfetto et al. 2013). The 198aa-domain of HNRNPU is conserved to almost 100% identity in all mammalian HNRNPU orthologues, and even its coding DNA sequence is highly conserved. Although no specific function of the two affected amino acids is known, it is well conceivable that the predicted amino-acid substitutions in this extremely invariable domain influence protein function.
Individual 1 also carries a heterozygous de novo frameshift mutation in the KDM6B gene. The KDM6B gene (also known as JMJD3) encodes a H3K27 demethylase. Its deletion in mice leads to impaired neuronal differentiation (Park et al. 2014). To date, there are no relevant variants listed in the Decipher database, and to the best of our knowledge, KDM6B has not been associated with a clinical phenotype, so that it remains unclear whether the KDM6B variant influences the individual’s phenotype. However, the clinical overlap of individual 1 to the HNRNPU- and 1q44-associated spectrum (as discussed below) supports the notion that the HNRNPU variant explains at least part of his phenotype.
Individual 5 carries a duplication with a minimal size of 108 kb including the first three exons of HNRNPU and the first exon of EFCAB2 (no OMIM reference). Unfortunately, we are not able to prove the location of the duplicated fragment as were unable to collect additional material from the deceased individual to determine the effect of this variant on HNRNPU function. There were no other likely causative variants identified in WES and individual 5 showed features at the severe end of the HNRNPU-associated spectrum. Thus, this duplication presents an important finding and calls for further evaluation of a larger number of patients with similar duplications.
In summary, our study broadens the clinically relevant mutational spectrum of the HNRNPU gene.
HNRNPU variants cause epilepsy and severe ID
Our phenotypic analysis yielded the following HNRNPU-associated phenotype. All individuals presented with severe ID (6/6) with the exception of the newborn individual 5, who passed away at the age of 2 months. Speech impairment was severe in all individuals (6/6). More specifically, three individuals (1, 3, and 6) were non-verbal, two individuals (2 and 4) showed hardly any speech, and individual 7 spoke words and short sentences. Most individuals displayed early onset seizures (6/7, age of onset: neonatal to 16 months) except individual 2, who showed fever-related seizures only at a young age with a normal EEG. In individual 5, we cannot exclude that the neonatal seizures could possibly be due to structural malformations and ventricular hemorrhage. The epilepsy-associated findings are similar to those reported by Depienne et al. with 6/7 individuals with HNRNPU variants showing early onset epilepsy and 7/7 individuals presenting with moderate to severe ID with severe speech impairment (Depienne et al. 2017). In addition, most individuals in our study (5/6) demonstrated structural CNS abnormalities as detected by brain MRI, such as cerebral atrophy, heterotopia, wide ventricles, but with no apparent pattern among the affected individuals. Teeth abnormalities, such as large incisors or crowded teeth, were another common feature (5/6) as well as hypotonia (6/7). Teeth abnormalities were not mentioned by Depienne and colleagues, CNS abvnormalities was reported in 3/6, and hypotonia was noted in 3/5 individuals (Depienne et al. 2017).
Only individual 1 displayed an abnormal corpus callosum (1/6), similar to the numbers reported by Depienne et al. (2/6) (Depienne et al. 2017). Only two individuals in our cohort presented with microcephaly (2/7) as well as one individual in the cohort published by Depienne et al. (1/6) indicating that HNRNPU does not play a crucial role in corpus callosum development or in the pathogenesis of microcephaly. Depienne and colleagues provide a detailed discussion of the role of ZBTB18 in CC development and the role of AKT3 in microcephaly (Depienne et al. 2017).
Although the facial phenotypes of some individuals, e.g., individuals 1 and 2 show resemblance to previously published individuals [patient 6 published by van Bon et al. (2008); patient 3 published by Thierry et al. (2012); patient 17 published by Ballif et al. (2012)]. Individual 1 presented with a HNRNPU missense mutation, while individual 2 presented with a HNRNPU non-sense mutations, so that there does not seem to be a correlation between the type of mutation and the occurrence of the facial phenotype. Overall, phenotypic comparison of our HNRNPU-mutation-positive individuals does not lead to a recognizable facial phenotype (Fig. 1; Table 1). Interestingly, 4/6 individuals in our cohort displayed cardiac abnormalities and 3/4 individuals showed renal abnormalities, while only 1/6 individuals reported by Depienne et al. presented with cardiac abnormalities and no renal abnormalities were described (Depienne et al. 2017). The reason for the discrepant numbers between the two cohorts and for the low number of patients with 1q44 microdeletions displaying cardiac or renal abnormalities remains unclear. Additional studies and the investigation of additional individuals with HNRNPU variants are needed to elucidate the role of HNRNPU in cardiac and renal diseases.
In summary, we broaden the clinical and mutational spectrum of heterozygous HNRNPU variants, which lead to early onset epilepsy and severe ID with speech impairment and variable CNS, cardiac, and renal abnormalities.
References
Allen AS et al (2013) De novo mutations in epileptic encephalopathies. Nature 501:217–221. doi:10.1038/nature12439
Ballif BC et al (2012) High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet 131:145–156. doi:10.1007/s00439-011-1073-y
Boland E et al (2007) Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet 81:292–303
Bramswig NC et al (2015) Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of Coffin-Siris and Nicolaides-Baraitser syndromes. Hum Genet 134:553–568. doi:10.1007/s00439-015-1535-8
Caliebe A et al (2010) Four patients with speech delay, seizures and variable corpus callosum thickness sharing a 0.440 Mb deletion in region 1q44 containing the HNRPU gene. Eur J Med Genet 53:179–185. doi:10.1016/j.ejmg.2010.04.001
Carvill GL et al (2013) Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45:825–830. doi:10.1038/ng.2646
de Kovel CG et al (2016) Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients Mol Genet. Genomic Med 4:568–580. doi:10.1002/mgg3.235
de Ligt J et al (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367:1921–1929. doi:10.1056/NEJMoa1206524
Depienne C et al (2017) Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Hum Genet. doi:10.1007/s00439-017-1772-0
Fromer M et al (2012) Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 91:597–607. doi:10.1016/j.ajhg.2012.08.005
Fu XD, Ares M Jr (2014) Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15:689–701. doi:10.1038/nrg3778
Geuens T, Bouhy D, Timmerman V (2016) The hnRNP family: insights into their role in health and disease. Hum Genet 135:851–867. doi:10.1007/s00439-016-1683-5
Hamdan FF et al (2014) De novo mutations in moderate or severe intellectual disability. PLoS Genet 10:e1004772. doi:10.1371/journal.pgen.1004772
Hill AD, Chang BS, Hill RS, Garraway LA, Bodell A, Sellers WR, Walsh CA (2007) A 2-Mb critical region implicated in the microcephaly associated with terminal 1q deletion syndrome. Am J Med Genet A 143A:1692–1698. doi:10.1002/ajmg.a.31776
Hussain MS et al (2014) Mutations in CKAP2L, the human homolog of the mouse Radmis gene, cause Filippi syndrome. Am J Hum Genet 95:622–632. doi:10.1016/j.ajhg.2014.10.008
Jiang Y, Oldridge DA, Diskin SJ, Zhang NR (2015) CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res 43:e39. doi:10.1093/nar/gku1363
Keupp K et al (2013) Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet 92:565–574. doi:10.1016/j.ajhg.2013.02.010
Lehalle D, Wieczorek D, Zechi-Ceide RM, Passos-Bueno MR, Lyonnet S, Amiel J, Gordon CT (2015) A review of craniofacial disorders caused by spliceosomal defects. Clin Genet 88:405–415. doi:10.1111/cge.12596
Monroe GR et al (2016) Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet Med 18:949–956. doi:10.1038/gim.2015.200
Nagamani SC et al (2012) Delineation of a deletion region critical for corpus callosal abnormalities in chromosome 1q43-q44. Eur J Hum Genet 20:176–179. doi:10.1038/ejhg.2011.171
Need AC et al (2012) Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet 49:353–361. doi:10.1136/jmedgenet-2012-100819
Park DH et al (2014) Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep 8:1290–1299. doi:10.1016/j.celrep.2014.07.060
Perfetto L, Gherardini PF, Davey NE, Diella F, Helmer-Citterich M, Cesareni G (2013) Exploring the diversity of SPRY/B30.2-mediated interactions. Trends Biochem Sci 38:38–46. doi:10.1016/j.tibs.2012.10.001
Poot MaK MJ (2013) Antisense May Make Sense of 1q44 Deletions. Seizures, and HNRNPU Am J Med Genet Part A 161A:910–912
Roshon MJ, Ruley HE (2005) Hypomorphic mutation in hnRNP U results in post-implantation lethality. Transgenic Res 14:179–192
Thierry G et al (2012) Molecular characterization of 1q44 microdeletion in 11 patients reveals three candidate genes for intellectual disability and seizures. Am J Med Genet A 158A:1633–1640. doi:10.1002/ajmg.a.35423
van Bon BW et al (2008) Clinical and molecular characteristics of 1qter microdeletion syndrome: delineating a critical region for corpus callosum agenesis/hypogenesis. J Med Genet 45:346–354. doi:10.1136/jmg.2007.055830
Wang GS, Cooper TA (2007) Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8:749–761
Xiao R et al (2012) Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol Cell 45:656–668. doi:10.1016/j.molcel.2012.01.009
Ye J et al (2015) hnRNP U protein is required for normal pre-mRNA splicing and postnatal heart development and function. Proc Natl Acad Sci USA 112:E3020–E3029. doi:10.1073/pnas.1508461112
Acknowledgements
We are grateful to the families for participating in this study and we thank Daniela Falkenstein and Sabine Kaya for excellent technical assistance. This work was in part supported by the German Ministry of Research and Education [Grant Numbers 01GS08164 (HE), 01GS08167 (DW), 01GS08163 (TMS), German Mental Retardation Network] as part of the National Genome Research Network. This work was supported by grants from the National Institute of Neurological Disorders and Stroke (The Epilepsy Phenome/Genome Project NS053998, Epi4K—Administrative Core NS077274, Epi4K—Sequencing, Biostatistics and Bioinformatics Core NS077303; Epi4K Project 1—Epileptic Encephalopathies NS077364, and Epi4K—Phenotyping and Clinical Informatics Core NS077276).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Bramswig, N.C., Lüdecke, HJ., Hamdan, F.F. et al. Heterozygous HNRNPU variants cause early onset epilepsy and severe intellectual disability. Hum Genet 136, 821–834 (2017). https://doi.org/10.1007/s00439-017-1795-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1795-6