
Abstract
Microdeletions of 1q43q44 result in a recognizable clinical disorder characterized by moderate to severe intellectual disability (ID) with limited or no expressive speech, characteristic facial features, hand and foot anomalies, microcephaly (MIC), abnormalities (agenesis/hypogenesis) of the corpus callosum (ACC), and seizures (SZR). Critical regions have been proposed for some of the more prominent features of this disorder such as MIC and ACC, yet conflicting data have prevented precise determination of the causative genes. In this study, the largest of pure interstitial and terminal deletions of 1q43q44 to date, we characterized 22 individuals by high-resolution oligonucleotide microarray-based comparative genomic hybridization. We propose critical regions and candidate genes for the MIC, ACC, and SZR phenotypes associated with this microdeletion syndrome. Three cases with MIC had small overlapping or intragenic deletions of AKT3, an isoform of the protein kinase B family. The deletion of only AKT3 in two cases implicates haploinsufficiency of this gene in the MIC phenotype. Likewise, based on the smallest region of overlap among the affected individuals, we suggest a critical region for ACC that contains ZNF238, a transcriptional and chromatin regulator highly expressed in the developing and adult brain. Finally, we describe a critical region for the SZR phenotype which contains three genes (FAM36A, C1ORF199, and HNRNPU). Although ~90% of cases in this study and in the literature fit these proposed models, the existence of phenotypic variability suggests other mechanisms such as variable expressivity, incomplete penetrance, position effects, or multigenic factors could account for additional complexity in some cases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the original report of a terminal 1q43 deletion appeared in 1976 (Mankinen et al. 1976), more than 50 individuals with deletions spanning a 25 Mb interval on chromosome 1q42q44 have been reported (Boland et al. 2007; De Rosa et al. 2005; Gentile et al. 2003; Hathout et al. 1998; Hill et al. 2007; Puthuran et al. 2005; van Bever et al. 2005; van Bon et al. 2008; Zollino et al. 2003). The common clinical features include moderate to severe intellectual disability (ID), limited or no expressive speech, characteristic facial features including round face, prominent forehead, flat nasal bridge, hypertelorism, epicanthal folds, and malformed and low-set ears, hypotonia, minor heart conditions that usually resolve naturally, short stature or small size, hand and foot anomalies, microcephaly (MIC), abnormalities (agenesis/hypogenesis) of the corpus callosum (ACC), and seizures (SZR) beginning before age 3.
Historically, detection of and subsequent genotype–phenotype correlations of 1q42q44 deletions have been limited by the low resolution of techniques (e.g., G-banding, fluorescence in situ hybridization (FISH), and microsatellite marker analyses). However, the recent use of microarray analysis has allowed for the identification of numerous submicroscopic alterations and for the fine mapping of deletion sizes in individuals with distinct or variable clinical features. High-resolution microarrays have produced promising preliminary genotype–phenotype correlations, such as an association between hand and foot anomalies with more proximal 1q42 deletions (Gentile et al. 2003) and an association between MIC, ACC, and SZR with more distal 1q43q44 deletions (Boland et al. 2007; Hill et al. 2007). However, identification of the specific gene(s) responsible for the various features has remained elusive because of the conflicting reports suggesting, for at least some features, that the etiology might be more complex or additional high-resolution array data in conjunction with accurate genotype–phenotype correlations in more cases are needed.
Attempts to understand the etiology of the MIC and ACC phenotypes, and more recently the SZR phenotype, associated with 1q43q44 deletions began when Hill et al. (2007) used microsatellite and Affymetrix 50 K single nucleotide polymorphism (SNP) microarrays to characterize seven individuals with 4.9–8.3 Mb terminal deletions of 1q43q44. Their analysis identified a ~2 Mb critical region for the MIC phenotype on 1q43q44 that contains at least 11 genes including AKT3 (Fig. 1). The report by Hill et al. (2007) was followed by Boland et al. (2007), who used a bacterial artificial chromosome (BAC)-based microarray to characterize the 1q42q44 deletions in six individuals with MIC, ACC, and other features. Boland et al. (2007) established a smallest region of overlap (SRO) for MIC and/or ACC located in 1q43q44 in a ~2–3.5 Mb region encompassing AKT3 that overlapped the region reported by Hill et al. (2007) (Fig. 1).

Summary of the various proposed critical regions located within 1q43q44. Red horizontal bars indicate critical regions previously proposed in the literature. The reference and phenotypic feature(s) for which the critical region was proposed are shown to the side of each region. Yellow bars indicate RefSeq genes in the 1q43q44 interval. Because of the density of genes in some areas and the scale of the figure, gene names are listed only for the region of primary interest located between 241–245 Mb
However, a report by van Bon et al. (2008) on 13 individuals with submicroscopic pure terminal or interstitial deletions of 1q43q44 characterized primarily by multiplex ligation-dependent probe amplification (MLPA) and microarray analysis (BAC, oligonucleotide, and SNP) suggested a 360 kb critical region for ACC but not for MIC or SZR, that excluded AKT3. Furthermore, they identified a family with two affected siblings with a small 400 kb deletion disrupting both the SDCCAG8 and AKT3. Although both siblings had MIC, one sibling was considerably more affected than the other and also had ACC. Because this abnormality was inherited from the siblings’ apparently normal and healthy mother, van Bon et al. (2008) considered the deletion of AKT3 to be benign and thereby excluded AKT3 as a candidate for the phenotypic features associated with 1q43q44 deletions. However, van Bon et al. (2008) also reported one individual without ACC whose deletion overlaps their proposed 360 kb critical region for ACC, which may indicate incomplete penetrance.
In support of van Bon’s critical region, a study by Poot et al. (2007) described a single patient with MIC and ACC and a microscopically visible 4.8 Mb terminal deletion of 1q44 resulting from a paternal t(1;20)(q44;q13.33), which did not include the AKT3 or the ZNF238 genes. To complicate the phenotypic correlations further, Orellana et al. (2009) described one individual with neither MIC nor ACC and an interstitial deletion of 1q44 that overlapped the 360 kb critical region proposed by van Bon et al. (2008). Orellana et al. (2009) suggested a different critical region for MIC and ACC which is located more proximally than the region proposed by van Bon et al. (2008) that includes AKT3 and ZNF238 (Fig. 1). Finally, Caliebe et al. (2010) used microarray analysis to characterize four additional patients with deletions of the 1q43q44 region, one of whom has a balanced inv(4)(q21q35)dn in addition to the 1q44 abnormality and another has a der(1)t(1;3)(q44;q28)dn, and proposed a more distal 440 kb region containing four genes, including HNRNPU, as the critical region for ACC (Fig. 1).
We have identified over 100 rearrangements of 1q44 in individuals referred to our laboratory for microarray testing for unexplained intellectual disability and/or congenital anomalies. In this study we performed genotype–phenotype correlations for 22 individuals with pure interstitial or terminal deletions of 1q43q44 to identify critical regions and candidate genes for the MIC, ACC, and SZR phenotypes.
Materials and methods
Patient samples
Twenty-two patients with pure interstitial or terminal microdeletions of 1q43q44 were identified from >45,000 specimens submitted to our laboratory for microarray testing by searching our custom Genoglyphix Chromosome Aberration Database (GCAD; Signature Genomics, Spokane, WA). Informed consent using an IRB-approved protocol was obtained to publish photographs of patients 6, 7, 9, 11, and 17.
Oligonucleotide microarray analysis
Oligonucleotide-based microarray analysis was performed using either a custom 105 K-feature whole-genome microarray (SignatureChip® Oligo Solution™ v1.0, custom-designed by Signature Genomics, Spokane, WA; manufactured by Agilent Technologies, Santa Clara, CA) or a custom 135 K-feature whole-genome microarray (SignatureChip Oligo Solution v2.0, custom-designed by Signature Genomics; manufactured by Roche-NimbleGen, Madison, WI) using previously described methods (Ballif et al. 2008; Duker et al. 2010). Some cases in which a 1q43q44 abnormality was originally identified by SignatureChip BAC microarray analysis were reanalyzed using a 244 K-feature whole-genome catalogue microarray (Agilent Technologies) to refine the breakpoints using previously described methods (Ballif et al. 2008; Duker et al. 2010).
FISH analysis
Deletions were confirmed and visualized by metaphase FISH using BACs as previously described (Traylor et al. 2009). Parental chromosomes were also studied by metaphase FISH, when available.
Results
The microarray results for the 22 patients with pure interstitial or terminal deletions of 1q43q44 are summarized in Table 1, and the clinical features of these 22 patients relative to MIC, ACC, and SZR are summarized in Table 2. Only two patients had additional findings identified by array-based comparative genomic hybridization (array CGH) analysis of unclear clinical significance. All other CNVs detected in these patients were in known polymorphic regions and/or are predicted to be of no known clinical significance. Patient 20 had a 109 kb deletion of unclear clinical significance on 1q32.1 (chr1:202,607,836–202,716,759), and patient 21 had a paternally inherited 1.1 Mb deletion on 2q13 (chr2:108,664,793–109,784,544).
Of the 22 patients evaluated by microarray analysis in this study, 13 have MIC (Table 2 and Fig. 2). All but one of the patients with MIC had a deletion that spanned, overlapped, or disrupted the AKT3 gene including three cases with small deletions <500 kb. The deletion in patient 9 is ~403 kb in size and disrupts SDCCAG3 and the 3′ end of AKT3. The deletion in patient 11 is ~212 kb in size and disrupts only the 5′ end of AKT3. The deletion in patient 10 is ~133 kb in size and is completely contained within the AKT3 gene, resulting in a loss of exons 2–5. Parental FISH analysis indicated that patient 10 inherited the deletion from his mother who also has MIC (Table 2).

Microcephaly (MIC) phenotypes present in 22 patients with microdeletions of 1q43q44. Blue horizontal bars indicate the size of the deletion in individuals with MIC. White bars indicate deletions in individuals who do not have MIC based on their clinical evaluation. Dark gray bars indicate deletions in individuals for whom we do not have clinical information on this particular phenotypic feature. Yellow bars indicate RefSeq genes in the 1q43q44 interval. The vertical region shaded in gray indicates the proposed critical region for this phenotypic feature
Seven of the 22 patients have abnormalities of the corpus callosum (Table 2 and Fig. 3). The SRO for patients with ACC is defined by the proximal deletion breakpoint in patient 13 and the distal deletion breakpoint in patient 2. This ~75 kb interval (chr1:242,241,072–242,315,604) contains only the ZNF238 gene. However, three other patients (5, 7 and 8) have deletions spanning this interval but apparently do not have ACC.

Abnormalities of the corpus callosum (ACC) phenotypes present in 22 patients with microdeletions of 1q43q44. Purple horizontal bars indicate the size of the deletion in individuals with ACC. White bars indicate deletions in individuals who do not have ACC based on their clinical evaluation. Dark gray bars indicate deletions in individuals for whom we do not have clinical information on this particular phenotypic feature. Yellow bars indicate RefSeq genes in the 1q43q44 interval. The vertical region shaded in gray indicates the proposed critical region for this phenotypic feature
Nine patients have clinically recognized seizure phenotypes or indications for study relating to seizure disorders. Two more patients may have seizure disorders but as of their last clinic visits have had just one febrile convulsion which was associated with an illness (Table 2 and Fig. 4). The SRO for the SZR phenotype is defined by the ~100 kb interval corresponding to the proximal deletion breakpoint of patient 20 and the distal deletion breakpoint of patient 6 (chr1:243,052,583–243,153,373). This interval contains three genes, FAM36A, C1ORF199, and HNRNPU. However, two patients (12 and 16) whose deletions span this interval have not had seizures reported by 5 and 1.5 years of age, respectively.

Seizure (SZR) phenotypes present in 22 patients with microdeletions of 1q43q44. Red horizontal bars indicate the size of the deletion in individuals with SZR. Pink bars indicate deletions in individuals with only a single febrile seizure reported. White bars indicate deletions in individuals who do not have SZR based on their clinical evaluation. Yellow bars indicate RefSeq genes in the 1q43q44 interval. The vertical region shaded in gray indicates the proposed critical region for this phenotypic feature
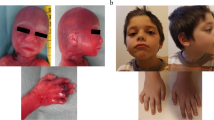
The facial features of five patients with microdeletions of 1q43q44 are shown in Fig. 5.

Facial features of five patients with microdeletions of 1q43q44. a Patient 11 at 2.5 years of age. Patient 11 has a ~212 kb deletion overlapping AKT3. b Patient 9 at 4 years of age. Patient 9 has a ~403 kb deletion overlapping AKT3. c Patient 6 at 14 years of age. Patient 6 has a ~2.4 Mb deletion overlapping the proposed critical regions for MIC, ACC, and SZR. d Patient 7 at 1.5 years of age. Patient 7 has a ~1.5 Mb deletion overlapping the proposed critical regions for MIC and ACC. e Patient 17 at 13 years of age. Patient 17 has a ~1.4 Mb deletion overlapping the proposed critical regions for SZR but not for MIC and ACC. Note the microcephaly and other characteristic facial features of individuals with microdeletions of 1q43q44 present in patients 6, 7, 9, and 11 (but less evident in patient 17) including rounded facies, prominent forehead, hypertelorism, flat nasal bridge, and low-set and malformed ears
Discussion
We report here the clinical features of 22 individuals with microdeletions of 1q43q44 identified by microarray analysis. By correlating the variable deletion sizes in these individuals with the presence or absence of MIC, ACC, and SZR, we have attempted to delineate SROs for these features. In spite of the controversy over the critical regions associated with the features of 1q43q44 microdeletions, it appears that a ~2 Mb genomic interval between 241.5 and 243.5 Mb (hg 18 build of the human genome) from the 1p telomere is the critical region for at least the MIC, ACC, and SZR phenotypes. In relative terms, deletion of the most proximal portion of this critical region results in MIC; deletion of the central portion results in ACC; and deletion of the more distal portion of this interval results in SZR. Within this 2 Mb critical region there are at least 12 genes.
MIC is described in a majority of the individuals reported in the literature with microdeletions of 1q42q44. By comparing the deletion sizes of 13 individuals with microcephaly, we have narrowed the gene causing MIC to AKT3 in all but one of the patients (Fig. 2). The presence of MIC in one patient whose deletion does not encompass AKT3 may represent a position effect on AKT3, haploinsufficiency of a gene distal to AKT3, or the disruption of a gene elsewhere in the genome. The subset of individuals with microcephaly includes one patient with a 133 kb intragenic deletion of AKT3 inherited from his similarly affected mother and two patients with 213 and 403 kb deletions overlapping the 5′ and 3′ ends of the AKT3 gene, respectively, which further implicates this gene in microcephaly. These results are in agreement with studies by Boland et al. (2007) and Hill et al. (2007), which suggested, based on the study of individuals with microcephaly and overlapping deletions, that an SRO encompassed AKT3. In addition, several other individuals in the literature with deletions of 1q42q44 encompassing AKT3 have microcephaly (Gentile et al. 2003; van Bever et al. 2005), whereas patients with deletions distal to AKT3 have normal head circumference. Therefore, it is likely that haploinsufficiency of AKT3 causes microcephaly in individuals with deletions of 1q44.
AKT3 is one of three closely related isoforms of the protein kinase B (PKB/Akt) family. All three members contain a highly conserved N-terminal pleckstrin homology (PH) domain, a central catalytic domain and a short C-terminal regulatory domain (reviewed in Bellacosa et al. 2004). AKT kinases phosphorylate a number of substrates involved in stimulation of cell proliferation, survival, intermediary metabolism and cell growth (Bellacosa et al. 2004). Like Akt family member Akt1, Akt3 plays a role in the attainment of normal organ size in mouse. However, in contrast to Akt1 −/− mice, which show a proportional decrease in the size of all organs, Akt3 −/− mice have a selective 20% decrease in brain size (Easton et al. 2005). Brains from adult nullizygotes are 25% smaller than those of wild-type littermates. Akt1 −/− and Akt2 −/− mice have increased perinatal lethality, growth retardation and altered glucose homeostasis, whereas Akt3 −/− mice brains have substantial thinning of the white matter fiber connections of the corpus callosum. All other organs are normally sized. In humans, AKT3 is most highly expressed in fetal hearts, livers, and brains and adult brains, lungs, and kidneys (Brodbeck et al. 1999). Thus, the mouse literature and our data support the original proposals of Boland et al. (2007) and Hill et al. (2007) and suggest that AKT3 remains an intriguing candidate gene for a role in brain growth in humans.
We also refined the critical region for the ACC phenotype based on seven affected individuals within our cohort (Fig. 3). The SRO is only 75 kb and contains one gene, ZNF238, which codes for a C2H2-type zinc-finger protein that functions as a transcriptional repressor. In mouse, Znf238 is widely expressed during embryogenesis with particularly high expression in the dorsal forebrain and developing cortex (Boland et al. 2007). In the adult mouse, Znf238 appears to be exclusively expressed in the brain (Becker et al. 1997). Additional studies of spatial and temporal expression of ZNF238 suggest that it plays a crucial role in neuronal proliferation, migration, and differentiation in the developing cortex (Ohtaka-Maruyama et al. 2007). Once again, our data refine the original proposals of Boland et al. (2007) and Hill et al. (2007) and suggest a potential role of ZNF238 in the ACC phenotype.
Finally, we have also refined the critical region for the seizure phenotype present in approximately half of individuals with microdeletions of 1q44. This critical region is located ~243.1 Mb from the 1p telomere and spans a ~100 kb interval that contains three genes, FAM36A, C1ORF199, and HNRNPU (Fig. 4). Little is known about FAM36A and C1ORF199. However, studies of Hnrnpu deficient mice suggest that Hnrnpu, the largest component of the heterogeneous ribonucleoprotein complex which binds to nascent transcripts, is involved in the regulation of embryonic brain development (Caliebe et al. 2010; Roshon and Ruley 2005) and is therefore a candidate for the features of 1q44 deletions. Furthermore, based on functional, genomic and evolutionary characteristics, HNRNPU has been predicted to have a high probability of being haploinsufficient when deleted, whereas FAM36A and C1ORF199 have not (Huang et al. 2010). However, the mechanism by which haploinsufficiency of HNRNPU results in seizures remains to be determined.
In our cohort the two individuals with inherited deletions in the most terminal 3.5 Mb of 1q44 lack MIC, ACC, or SZR. This is concordant with the hypothesis by van Bever et al. (2005) that, because the most distal 1.5 Mb of 1q44 consists of pseudogenes, two copies are not required for normal development. Based on our results, at least some deletions within the terminal 3.5 Mb of 1q44 might represent benign copy-number variants. However, further studies of additional cases with distal 1q44 deletions are required.
Figure 6 summarizes our proposed model for the refinement of the critical regions associated with MIC, ACC, and SZR phenotypes in patients with deletions of 1q43q44. Combining the data from this and five previously published studies indicates that 42/45 (93%) of cases for which clinical information is available on the presence or absence of MIC fit the proposed model (Table 3). Likewise, 37/43 (86%) of cases with 1q43q44 deletions and an ACC phenotype fit the model, and 40/46 (87%) of cases with 1q43q44 deletions and a SZR phenotype fit the proposed model (Table 3). Thus, our results refine the critical regions for features associated with microdeletions of 1q43q44.

Summary of proposed critical regions located within 1q43q44 based on this study. Regions shaded with vertical gray background indicate the proposed critical regions for microcephaly (MIC), abnormalities of the corpus callosum (ACC), and seizures (SZR)
For the proposed ACC and SZR critical regions, several cases in our and/or previous studies had a deletion over the critical region but lacked the associated feature (Table 3), which suggest incomplete penetrance or variable expressivity associated with disruption of a gene(s) in the critical region. For example, of the 43 cases in this and previous studies for whom clinical information was available regarding ACC, three had a deletion of the proposed ACC critical region but no ACC (Table 3). Furthermore, the absence of a consistent phenotype in a small proportion of cases suggests incomplete penetrance and variable expressivity. For example van Bon et al. (2008) describe a pair of affected sisters who inherited a small deletion over AKT3 from their unaffected carrier mother. Thus, the MIC phenotype may be incompletely penetrant in the mother based on her genetic background. The two sisters had MIC but were not equally affected, supporting variable expressivity. In contrast, in our study, a similar deletion over AKT3 was found in an affected mother and affected child, which would support complete penetrance for disruption of AKT3. Together, there is a preponderance of data that suggest incomplete penetrance and/or variable expressivity might play a role in the MIC feature of the disorder.
In addition, several cases in our and/or previous studies had a characteristic clinical feature but had a deletion that did not encompass the proposed critical region (Table 3), which may suggest a position effect on a gene(s) within the critical region or a secondary locus outside of the proposed critical region. For example, of the 43 cases for whom clinical information regarding ACC was available, three had the feature and a deletion that did not encompass the proposed critical region (Table 3). Although we were unable to perform expression studies, a large terminal deletion, such as the one in patient 13 in this study or in the study by Poot et al. (2007), could effectively silence the nearby genes, in this case the AKT3 gene, by a telomeric position effect on AKT3 as a result of bringing AKT3 into closer proximity to the subtelomeric heterochromatin (Ottaviani et al. 2008; van Karnebeek et al. 2002).
Genotype–phenotype correlations of additional pure interstitial and terminal deletions of 1q43q44 will continue to be critical to elucidate the underlying genetic causes of this rare subtelomeric disorder. The ascertainment of individuals with overlapping deletions and varying clinical features may allow researchers to narrow the region in which to search for candidate genes. Because microarray analysis can precisely refine the breakpoints for microscopic and submicroscopic deletions, the technique will continue to be instrumental in the characterization of additional abnormalities that refine the critical regions for these and other features of microdeletions of 1q43q44.
References
Ballif BC, Theisen A, McDonald-McGinn DM, Zackai EH, Hersh JH, Bejjani BA, Shaffer LG (2008) Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin Genet 74:469–475
Becker KG, Canning RD, Nagle JW, Dehejia AM, Polymeropoulos MH, Lee IJ, Gado AM, Biddison WE, Drew PD (1997) Molecular cloning and mapping of a novel developmentally regulated human C2H2-type zinc finger. Mamm Genome 8:287–289
Bellacosa A, Testa JR, Moore R, Larue L (2004) A portrait of AKT kinases: human cancer and animal models depict a family with strong individualities. Cancer Biol Ther 3:268–275
Boland E, Clayton-Smith J, Woo VG, McKee S, Manson FD, Medne L, Zackai E, Swanson EA, Fitzpatrick D, Millen KJ, Sherr EH, Dobyns WB, Black GC (2007) Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet 81:292–303
Brodbeck D, Cron P, Hemmings BA (1999) A human protein kinase Bgamma with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J Biol Chem 274:9133–9136
Caliebe A, Kroes HY, van der Smagt JJ, Martin-Subero JI, Tonnies H, Van’t Slot R, Nievelstein RA, Muhle H, Stephani U, Alfke K, Stefanova I, Hellenbroich Y, Gillessen-Kaesbach G, Hochstenbach R, Siebert R, Poot M (2010) Four patients with speech delay, seizures and variable corpus callosum thickness sharing a 0.440 Mb deletion in region 1q44 containing the HNRPU gene. Eur J Med Genet 53:179–185
De Rosa G, Pardeo M, Bria S, Caresta E, Vasta I, Zampino G, Zollino M, Zuppa AA, Piastra M (2005) Isolated myocardial non-compaction in an infant with distal 4q trisomy and distal 1q monosomy. Eur J Pediatr 164:255–256
Duker AL, Ballif BC, Bawle EV, Person RE, Mahadevan S, Alliman S, Thompson R, Traylor R, Bejjani BA, Shaffer LG, Rosenfeld JA, Lamb AN, Sahoo T (2010) Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader–Willi syndrome. Eur J Hum Genet 18(11):1196–1201
Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ (2005) Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol 25:1869–1878
Gentile M, Di Carlo A, Volpe P, Pansini A, Nanna P, Valenzano MC, Buonadonna AL (2003) FISH and cytogenetic characterization of a terminal chromosome 1q deletion: clinical case report and phenotypic implications. Am J Med Genet A 117A:251–254
Hathout EH, Thompson K, Baum M, Dumars KW (1998) Association of terminal chromosome 1 deletion with sertoli-cell-only syndrome. Am J Med Genet 80:396–398
Hill AD, Chang BS, Hill RS, Garraway LA, Bodell A, Sellers WR, Walsh CA (2007) A 2-Mb critical region implicated in the microcephaly associated with terminal 1q deletion syndrome. Am J Med Genet A 143A:1692–1698
Huang N, Lee I, Marcotte EM, Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6:e1001154
Mankinen CB, Sears JW, Alvarez VR (1976) Terminal (1)(q43) long-arm deletion of chromosome no. 1 in a three-year-old female. Birth Defects Orig Artic Ser 12:131–136
Ohtaka-Maruyama C, Miwa A, Kawano H, Kasai M, Okado H (2007) Spatial and temporal expression of RP58, a novel zinc finger transcriptional repressor, in mouse brain. J Comp Neurol 502:1098–1108
Orellana C, Rosello M, Monfort S, Oltra S, Quiroga R, Ferrer I, Martinez F (2009) Corpus callosum abnormalities and the controversy about the candidate genes located in 1q44. Cytogenet Genome Res 127:5–8
Ottaviani A, Gilson E, Magdinier F (2008) Telomeric position effect: from the yeast paradigm to human pathologies? Biochimie 90:93–107
Poot M, Kroes HY, SE VDW, Eleveld MJ, Rooms L, Nievelstein RA, Olde Weghuis D, Vreuls RC, Hageman G, Kooy F, Hochstenbach R (2007) Dandy-Walker complex in a boy with a 5 Mb deletion of region 1q44 due to a paternal t(1;20)(q44;q13.33). Am J Med Genet A 143A:1038–1044
Puthuran MJ, Rowland-Hill CA, Simpson J, Pairaudeau PW, Mabbott JL, Morris SM, Crow YJ (2005) Chromosome 1q42 deletion and agenesis of the corpus callosum. Am J Med Genet A 138:68–69
Roshon MJ, Ruley HE (2005) Hypomorphic mutation in hnRNP U results in post-implantation lethality. Transgenic Res 14:179–192
Traylor RN, Fan Z, Hudson B, Rosenfeld JA, Shaffer LG, Torchia BS, Ballif BC (2009) Microdeletion of 6q16.1 encompassing EPHA7 in a child with mild neurological abnormalities and dysmorphic features: case report. Mol Cytogenet 2:17
van Bever Y, Rooms L, Laridon A, Reyniers E, van Luijk R, Scheers S, Wauters J, Kooy RF (2005) Clinical report of a pure subtelomeric 1qter deletion in a boy with mental retardation and multiple anomalies adds further evidence for a specific phenotype. Am J Med Genet A 135:91–95
van Bon BW, Koolen DA, Borgatti R, Magee A, Garcia-Minaur S, Rooms L, Reardon W, Zollino M, Bonaglia MC, De Gregori M, Novara F, Grasso R, Ciccone R, van Duyvenvoorde HA, Aalbers AM, Guerrini R, Fazzi E, Nillesen WM, McCullough S, Kant SG, Marcelis CL, Pfundt R, de Leeuw N, Smeets D, Sistermans EA, Wit JM, Hamel BC, Brunner HG, Kooy F, Zuffardi O, de Vries BB (2008) Clinical and molecular characteristics of 1qter microdeletion syndrome: delineating a critical region for corpus callosum agenesis/hypogenesis. J Med Genet 45:346–354
van Karnebeek CD, Quik S, Sluijter S, Hulsbeek MM, Hoovers JM, Hennekam RC (2002) Further delineation of the chromosome 14q terminal deletion syndrome. Am J Med Genet 110:65–72
Zollino M, Colosimo C, Zuffardi O, Rossi E, Tosolini A, Walsh CA, Neri G (2003) Cryptic t(1;12)(q44;p13.3) translocation in a previously described syndrome with polymicrogyria, segregating as an apparently X-linked trait. Am J Med Genet A 117A:65–71
Acknowledgments
We are grateful to many clinicians, counselors, and laboratory directors for their assistance with collecting the information in this report, including Dr. Miriam Kalichman and Dr. David Aughton. We are also very grateful to the patients and their families for supporting this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ballif, B.C., Rosenfeld, J.A., Traylor, R. et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet 131, 145–156 (2012). https://doi.org/10.1007/s00439-011-1073-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1073-y