
Abstract
Chromatin remodeling is a complex process shaping the nucleosome landscape, thereby regulating the accessibility of transcription factors to regulatory regions of target genes and ultimately managing gene expression. The SWI/SNF (switch/sucrose nonfermentable) complex remodels the nucleosome landscape in an ATP-dependent manner and is divided into the two major subclasses Brahma-associated factor (BAF) and Polybromo Brahma-associated factor (PBAF) complex. Somatic mutations in subunits of the SWI/SNF complex have been associated with different cancers, while germline mutations have been associated with autism spectrum disorder and the neurodevelopmental disorders Coffin–Siris (CSS) and Nicolaides–Baraitser syndromes (NCBRS). CSS is characterized by intellectual disability (ID), coarsening of the face and hypoplasia or absence of the fifth finger- and/or toenails. So far, variants in five of the SWI/SNF subunit-encoding genes ARID1B, SMARCA4, SMARCB1, ARID1A, and SMARCE1 as well as variants in the transcription factor-encoding gene SOX11 have been identified in CSS-affected individuals. ARID2 is a member of the PBAF subcomplex, which until recently had not been linked to any neurodevelopmental phenotypes. In 2015, mutations in the ARID2 gene were associated with intellectual disability. In this study, we report on two individuals with private de novo ARID2 frameshift mutations. Both individuals present with a CSS-like phenotype including ID, coarsening of facial features, other recognizable facial dysmorphisms and hypoplasia of the fifth toenails. Hence, this study identifies mutations in the ARID2 gene as a novel and rare cause for a CSS-like phenotype and enlarges the list of CSS-like genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The SWI/SNF complex (switch/sucrose nonfermentable) mobilizes nucleosomes and remodels chromatin in an ATP-dependent manner. It facilitates access of transcription factors to regulatory regions of target genes, thereby regulating gene expression, and has been implicated to play an important role in cell differentiation, cell-cycle dynamics, and DNA-repair (Hargreaves and Crabtree 2011). The SWI/SNF complex with its multiple subunits can be assembled in many different combinations and its composition can change upon cellular differentiation. Brahma-associated factor (BAF) and Polybromo Brahma-associated factor (PBAF) comprise the two major subclasses of the SWI/SNF complex and differ in their subunits.
Somatic mutations in genes encoding for subunits of the SWI/SNF complex were initially detected in a variety of cancers and growing evidence suggests a tumor suppressor role of the SWI/SNF complex (Wilson and Roberts 2011). In 2012, germline mutations in genes encoding for subunits of the SWI/SNF complex, namely ARID1B and SMARCA2, were identified as the cause of the rare intellectual disability (ID) disorders Coffin–Siris syndrome (CSS) and Nicolaides–Baraitser syndrome (NCBRS), respectively (Santen et al. 2012; Tsurusaki et al. 2012; Van Houdt et al. 2012).
CSS (MIM135900) was delineated by Coffin and Siris (1970). CSS is characterized by ID, a recognizable pattern of dysmorphic features including coarsening of the face over time and hypoplasia or absence of the fifth finger- and toenails. Approximately 150 patients with CSS have been published, of which about 100 patients carry a mutation in genes encoding for members of the SWI/SNF complex. These genes include ARID1B, ARID1A, SMARCA4, SMARCB1, and SMARCE1 (Kosho et al. 2014; Kosho and Okamoto 2014; Santen et al. 2012, 2013; Tsurusaki et al. 2012, 2014b; Wieczorek et al. 2013). ARID1B is the most frequently mutated SWI/SNF complex gene in patients with CSS (Miyake et al. 2014) and has also been reported in patients with non-syndromic ID (Hoyer et al. 2012). In addition, de novo mutations in and deletions encompassing the SOX11 gene were shown to cause a mild CSS phenotype (Hempel et al. 2016; Tsurusaki et al. 2014a). In summary, CSS-affected individuals show mutations in SWI/SNF complex subunits but a considerable number of individuals with a clinical diagnosis of CSS or a considerable phenotypic overlap remain without a mutation in these known genes.
The aim of this study is the further characterization of genetic causes of CSS-like phenotypes. In this study, we present two individuals with a CSS-like phenotype and private de novo frameshift mutations in the ARID2 gene and review previously published individuals with ARID2 mutations (Shang et al. 2015). Our study proposes de novo ARID2 mutations as a novel and rare cause of a CSS-like phenotype and expands the list of CSS-like genes.
Materials and methods
Individuals
Clinical information of the two individuals was provided by the parents and by the evaluating clinicians. Written informed consent was obtained from the families of the index individuals for participation in this study. The study was performed according to the Declaration of Helsinki protocols and was approved by the local institutional review board [ethical votum 08-3663 and 5360/13 for the Technical University Munich; The Human Research Ethics Board at the University of Alberta MS11_Pro00033138]. DNA from peripheral blood lymphocytes was obtained from the affected individuals and their parents and extracted by standard extraction procedures.
Chromosomal and array analyses
Conventional karyotyping in blood lymphocytes and microarray analyses for molecular karyotyping were normal in individuals 1 and 2 [Individual 1: CytoChip Version 3 and CytoChip Oligo 4x44k (both BlueGnome, Cambridge, UK); Individual 2: Cytoscan HD (Affymetrix, Santa Clara, CA, USA)].
Whole exome sequencing
For individual 1, trio-based whole exome sequencing was performed in a cohort of 311 patients with ID with and without associated clinical findings as described elsewhere (Bramswig et al. 2015). For individual 2, clinical WES was performed by Baylor Miraca Genetics Laboratories with parental DNA being used to test for pathogenic mutations in the index case. The variants were verified by Sanger sequencing, primer sequences will be provided upon request.
Transcript analysis
Peripheral blood of individual 1 was collected into PAXgene Blood RNA tubes (PreAnalytiX), and RNA was isolated using the PAXgene Blood RNA Kit (Qiagen). cDNA was generated with the help of the Omniscript RT Kit (Qiagen), and primers ARID2-exon15-forward 5′-GTCGAATGCAGGAGTTGGTC-3′ (NM_152641.2: c.3132-3151, exon 15) and ARID2-exon16-reverse 5′-GGTGAAGGTGGCATAGGAGT-3′ (NM_152641.2: c.4777-4796, exon 16) and the GoTaq G2 DNA polymerase (Promega) were used to amplify a 1665 bp exon–intron-boundary spanning fragment of the ARID2 cDNA. In genomic DNA, the 3′-ends of the primers are 9530 bp apart, and thus would not yield a PCR product with the used PCR protocol. The sequence at the mutation site, c.3411_3412delAG, was determined by Sanger sequencing using the ARID2-exon15-forward primer.
Results
Two individuals with de novo ARID2 frameshift variants
Individual 1 is the second child of healthy, non-consanguineous parents. The parents had two miscarriages at 25th week of gestation; one had a Truncus arteriosus, one during gestosis. The index patient was born at 36 weeks of gestation by Cesarean section with a weight of 2160 g (−1.6 SD) and a length of 45 cm (−1.6 SD). The head circumference was not recorded, but is reported as normal. At the age of 20 months, his height was 77.5 cm (−2.21 SD), weight 9 kg (−2.84 SD) and head circumference 48.5 cm (−0.14 SD). In the neonatal period he needed to undergo surgery for bilateral inguinal hernias and unilateral testicular torsion. In his first year of life, severe muscular hypotonia and delayed development were noticed. Small hands and feet were observed, as well as nail hypoplasia and delayed dental eruption with yellow–brown teeth. He showed an increased tendency for respiratory infections. Bilateral placement of tympanostomy tubes was performed. Seizures started at the age of 14 months with infantile spasms in the beginning followed by generalized tonic–clonic seizures, absences, and tonic seizures. Frequency and intensity of the seizures varied markedly over time. At the age of 8 years, antiepileptic drug therapy is still necessary and partially effective. Magnetic resonance imaging (MRI) of the brain showed wide inner and outer cerebrospinal fluid spaces, dorsal vermis hypoplasia and wide cisterna magna (Dandy–Walker formation), and a thin corpus callosum. Due to increased intracranial pressure, a shunt was implanted. Metabolic diagnostics, including testing for mucopolysaccharidosis and Morbus Niemann-Pick, were normal. At 20 months, facial dysmorphisms included slightly coarse features, tall forehead, hypertelorism, depressed nasal root and short nose, prominent and long philtrum, and thin upper lip (Fig. 1a). His latest assessment was at the age of 7 years, when he showed a height of 112 cm (−2.14 SD), weight of 17 kg (−2.81 SD). His head circumference was 53 cm (+0.68 SD). He presented with severe ID with absent speech and inability to sit alone. Facial dysmorphisms included coarsening of the face, tall forehead, hypertelorism, depressed nasal root and short nose, prominent and long philtrum, thin upper lip, and full lower lip (Fig. 1b, c). His teeth appear small, possibly due to gingival hyperplasia secondary to phenobarbital treatment. A single palmar crease, small hands with tapering fingers, small feet and nail hypoplasia of the fifth finger and toe nails were observed (Fig. 2a, b). RSK2 analysis (Coffin-Lowry syndrome) was normal. The tentative diagnosis of CSS was made, but ARID1B sequencing costs were not going to be covered by the health insurance and individual 1 was included in a WES study. Trio-based WES identified a de novo mutation in the ARID2 gene [NM_152641.2; Exon 15/21; chr12:g.46,245,317_46,245,318delAG (GRCh37/hg19); c.3411_3412delAG; p.(Gly1139Serfs*20)]. This variant is not listed in the ExAC browser. Sequencing of the cDNA from blood RNA revealed the wildtype sequence of exon 15 of ARID2 only, indicating that the mutant transcript is most likely subject to nonsense-mediated mRNA decay. In addition, a de novo mutation in the TRIM8 gene [NM_030912.2; Exon 6/6; chr10:g.104,416,788C>T; c.1333C>T; p.(Gln445*)] was identified and may be responsible for the occurrence of seizures and have an influence on the severity of ID as discussed below.

Facial phenotype of ARID2 mutation-positive individuals. a Individual 1 at the age of 20 months presenting with slightly coarse facial features, hypertelorism, flat nasal bridge, broad nose with upturned nasal tip and thin upper and thick lower vermillion. b, c Individual 1 at the age of 7 years showing coarsening of facial features, high forehead, thick eyebrows, flat nasal bridge, and an upturned nasal tip with thick, anteverted alae nasi. He has a long and prominent philtrum, a thin upper lip and thick lower lip vermillion. d Individual 2 at the age of 22 months displaying slightly coarse facial features, an exaggerated Cupid’s bow of the upper lip and a full lower lip. e, f Individual 2 at the age of 4.5 years showing further coarsening of the facial features, with a large forehead, periorbital fullness, flat nasal bridge, upturned nasal tip, prominent philtrum, large mouth with exaggerated Cupid’s bow and full lower lip vermillion

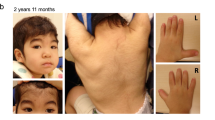
Limb phenotype of individual 1 (a, b) and individual 2 (c, d) presenting with very mild hypoplasia of the fifth fingernails and pronounced hypoplasia of the fifth toenails
Individual 2 was born at 40 weeks gestation by induced vaginal delivery to unrelated parents in good general health. At 21 weeks gestation, the fetus was found to have short femur length (3rd percentile), but no other bone-anomalies. No other congenital anomalies were identified and a maternal serum screen was negative. The mother declined amniocentesis and was followed throughout pregnancy with a consistent finding of bilateral short femur lengths and excellent biophysical profile, transient polyhydramnios, and otherwise normal fetal growth. During delivery, the patient experienced shoulder dystocia, and wet lung for which he was suctioned and received free-flow oxygen. Birth weight was 4552 g (+2.1 SD), length 51 cm (−0.7 SD), and head circumference 37 cm (+1.1 SD); Apgar scores were 5/9/9, at 1, 5, and 10 min, respectively. He was breastfed, developed some colds in the first year of life and no concerns were raised until 8 months of life when he was not yet sitting independently. He developed pincer grasp at 16 months, and walked independently at 19 months of age. He received occupational, physio- and speech therapy since 9 months of age, had normal bone age, and normal MPS and oligosaccharides screen. At the age of 18 months, he underwent an eye exam, and was found to have good fixation with no preference for one eye, no strabismus, and clear corneas with normal fundus. He exhibited difficulties tracking a moving target with jerky eye movements and repeated loss of fixation. Retinoscopy showed bilateral hyperopia. At 1 year, 8 months, the patient underwent a sensory processing evaluation, which did not show any deficits.
At the time of the first evaluation in medical genetics, he was 22 months of age, height was 79.5 cm (−2.2 SD), weight 10.6 kg (BMI 16.8 kg/m2), and head circumference 49 cm (−0.4 SD). He appeared to have a disproportionate stature, with normal built and distinctive, slightly coarse facial features (Fig. 1d). There was midface hypoplasia, normal spacing of eyes, horizontal palpebral fissures, and a small, upturned nose. He showed a Cupid’s bow and full lower lip, and there were good philtral pillars. There was generalized mild hypotonia. He was babbling but had no words; however, receptive language was much better than expressive speech. There was predisposition to constipation corrected with glycerin suppositories. He exhibited hand flapping with a screeching sound when excited. A skeletal survey completed at an outside center did not report skeletal changes. FGFR3 sequencing and microarray for copy number analysis were reported as normal.
At 4.5 years, facial features were further coarsening—there was a triangular shaped face, large forehead, horizontal palpebral fissures, periorbital fullness, somewhat flat zygomatic arches, and full cheeks (Fig. 1e, f). Nose was small and upturned with a prominent philtrum, and there was a large mouth with a Cupid’s bow, full lower lip, and crowded teeth. There were tapering fingers bilaterally, normal palm creases and small nails, and bilateral clinodactyly of the 5th finger (Fig. 2c). The X-ray image of the left hand displays hypoplastic distal phalanges (Supplemental Fig. 1). Toes showed small nails, especially on the 5th toes that also showed clinodactyly (Fig. 2d). Umbilical hernia was still present, and he maintained a tendency towards constipation. He still did not have any words, but had 12 recognizable signs. He was able to use an iPad app to order his favorite treat. Brain MRI was declined.
Initial clinical exome sequencing did not reveal a pathogenic finding explaining the individual’s phenotype (reported May 2014). However, in August 2015, a new report was issued indicating a heterozygous single base deletion in the ARID2 gene, which had recently reported to be disease associated [NM152641.2; Exon 2/21; chr12:g.46,123,890delC [GRCh37/hg19]; c.156delC, p.(Arg53Glufs*5)]. This variant is not listed in the ExAC browser.
All clinical information is summarized in Table 1. The two frameshift ARID2 mutations as well as previously published nonsense and frameshift ARID2 mutations are presented in a schematic illustration (Fig. 3).

Schematic representation of the ARID2 gene including the location of the variants of individuals 1 and 2 (top) and previously published ARID2 mutations (Shang et al. 2015) (bottom). RFX RFX-like DNA binding domain, C2H2 zinc fingers. Domains are indicated according to UniProt ID Q68CP9
Discussion
The chromatin-remodeling complex SWI/SNF is divided into the two subclasses BAF and PBAF. ARID2 (also referred to as BAF200) encodes for a protein characterized by an AT-rich interaction domain and is expressed in all tissues (Nagase et al. 2000). It was first identified as a PBAF-specific subunit regulating the expression of specific interferon-responsive genes (Yan et al. 2005). Another in vitro study determined a role for ARID2 in osteoblast differentiation (Xu et al. 2012). More recently, in vivo studies displayed heart defects and embryonic lethality (E12.5-E14.5) in Arid2 homozygously mutant mice and revealed an important role for Arid2 in heart development (He et al. 2014). The haploinsufficiency index for ARID2 is 11.01% and (https://decipher.sanger.ac.uk/) indicating a high likelihood that ARID2 exhibits haploinsuffiency. The pLI score for ARID2 is 1.00 (https://decipher.sanger.ac.uk/) illustrating that it is intolerant to loss-of-function mutations. Although little is known about the function of ARID2 these findings indicate that ARID2 and the PBAF-subclass of the SWI/SNF complex are important for tissue- and cell type-specific gene expression, as well as the sustenance of specific cellular functions, cellular identity, and organ development. In this study, we report on two individuals with de novo frameshift ARID2 mutations and a CSS-like phenotype.
Mutations in SWI/SNF complex members cause Coffin–Siris syndrome
Mutations in five different genes encoding SWI/SNF complex members, ARID1B, SMARCA4, SMARCB1, ARID1A, and SMARCE1, have been detected in CSS-affected individuals (Santen et al. 2012, 2013; Tsurusaki et al. 2012, 2014b; Wieczorek et al. 2013). However, 30.5–45.1% of these individuals do not have a mutation in the CSS candidate genes and their genetic etiology remains unknown (Kosho and Okamoto 2014; Santen et al. 2013; Tsurusaki et al. 2012; Wieczorek et al. 2013). The ARID2 gene was among the screened candidate genes in three of the CSS studies (n = 105), but no ARID2 mutations were identified (Tsurusaki et al. 2012, 2014b; Wieczorek et al. 2013). Thus, the genetic cause of about a third of CSS-affected individuals remains unsolved (Miyake et al. 2014) and screening of ARID2 in a considerable number of these individuals did not lead to the identification of pathogenic variants.
ARID2 variants in intellectual disability and CSS-like individuals
The PBAF complex had not been associated with neurodevelopmental phenotypes until recently, when Shang and colleagues reported four patients with ARID2 variants in individuals with developmental delay (DD)/ID and other similar clinical characteristics, such as short stature, dysmorphic facial features, and attention deficit hyperactivity disorder (Shang et al. 2015). In addition, a case with language delay, short stature, and a duplication of exons 1-3 of ARID2 was published very recently (Zahir et al. 2016). It was postulated that this duplication results in a loss-of-function, but there was no proof provided so that the clinical significance of these findings remains unclear.
Our data further support the interpretation of Shang and colleagues (Shang et al. 2015) that haploinsufficiency of ARID2 is the likely pathogenic cause in all six known cases with truncating ARID2 mutations. Shang and colleagues discussed that these four ARID2 mutation-positive individuals exhibit features, such as Wormian bones, frontal bossing, micro- and retrognathia, that distinguish them from individuals with other chromatin remodeling disorders. Unfortunately they did not discuss the overlap of the dysmorphic features with CSS and they did not comment on the limb phenotypes of these four individuals (i.e., the presence or absence of hypoplastic finger- and/or toenails), which presents one of the cardinal findings in CSS. Upon comparison of the available facial photographs (patient 2 and 3 from Shang et al. 2015) with our ARID2 mutation-positive individuals, we noticed remarkable similarities in their facial phenotypes. We propose that ARID2 mutations lead to a CSS-like phenotype as summarized below.
ARID2-associated CSS-like phenotype
All individuals with ARID2 mutations presented with ID (6/6), most of them exhibited muscular hypotonia (4/5) and behavioral anomalies (5/6) (Shang et al. 2015). Based on this small number of patients, birth defects do not seem to be common in ARID2 mutation carriers. One individual (patient 2 in Shang et al.) presented with an atrial septal defect and cleft palate, but she also carries a 421 kb duplication in 11q12.1 of unknown inheritance and significance. Seizures were observed in one individual (1/6) who also showed brain abnormalities, such as increased intracranial pressure, abnormal corpus callosum and Dandy–Walker anomaly. This individual also carries a nonsense mutation in the TRIM8 gene. Recently, heterozygous truncating mutations in TRIM8 were identified in two patients with early onset epileptic encephalopathy (Allen et al. 2013; Sakai et al. 2016). The case reported by Sakai and colleagues showed severe ID (similarly to individual 1); he sits in a wheelchair and has not been able to hold eye contact or speak any words (Sakai et al. 2016). Facial dysmorphic features included microcephaly, small and upslanted palpebral fissures, long-arched eyebrows, thin lips and large ear lobes (Sakai et al. 2016). Individual 1 does not exhibit microcephaly and the facial features of this case and individual 1 are difficult to compare due to different ethical backgrounds. However, the TRIM8 nonsense mutation in individual 1 might present the cause for his seizures and have an influence on the severity of his ID.
Comparison of the facial phenotypes of patients 2 and 3 published by Shang and colleagues (Shang et al. 2015) and individuals 1 and 2 of this study revealed striking similarities. ARID2 mutations are associated with coarse facial features (4/4), frontal bossing/large forehead (4/4), narrow palpebral fissures (4/4), flat nasal bridge (4/4), slightly broad nose with upturned nasal tip and thick, anteverted alae nasi (3/4), prominent philtrum (4/4), and a large mouth (3/4) with a thick lower vermillion (4/4). Individuals 1 and 2 also presented with very mild hypoplasia of the fifth fingernails (2/2) and pronounced hypoplasia of the fifth toenails (2/2), one of the cardinal findings in CSS. Although these preliminary findings need to be validated on a larger cohort of ARID2 mutation-positive individuals, we propose that ARID2 mutations lead to a CSS-like phenotype.
References
Allen AS et al (2013) De novo mutations in epileptic encephalopathies. Nature 501:217–221. doi:10.1038/nature12439
Bramswig NC et al (2015) Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of Coffin–Siris and Nicolaides–Baraitser syndromes. Hum Genet 134:553–568. doi:10.1007/s00439-015-1535-8
Coffin GS, Siris E (1970) Mental retardation with absent fifth fingernail and terminal phalanx. Am J Dis Child 119:433–439
Hargreaves DC, Crabtree GR (2011) ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21:396–420. doi:10.1038/cr.2011.32
He L et al (2014) BAF200 is required for heart morphogenesis and coronary artery development. PLoS One 9:e109493. doi:10.1371/journal.pone.0109493
Hempel A et al (2016) Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin–Siris syndrome. J Med Genet 53:152–162. doi:10.1136/jmedgenet-2015-103393
Hoyer J et al (2012) Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 90:565–572. doi:10.1016/j.ajhg.2012.02.007
Kosho T, Okamoto N (2014) Genotype-phenotype correlation of Coffin–Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet 166C:262–275. doi:10.1002/ajmg.c.31407
Kosho T, Miyake N, Carey JC (2014) Coffin–Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am J Med Genet C Semin Med Genet 166C:241–251. doi:10.1002/ajmg.c.31415
Miyake N, Tsurusaki Y, Matsumoto N (2014) Numerous BAF complex genes are mutated in Coffin–Siris syndrome. Am J Med Genet C Semin Med Genet 166C:257–261. doi:10.1002/ajmg.c.31406
Nagase T, Kikuno R, Nakayama M, Hirosawa M, Ohara O (2000) Prediction of the coding sequences of unidentified human genes. XVIII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 7:273–281
Sakai Y et al (2016) De novo truncating mutation of TRIM8 causes early-onset epileptic encephalopathy. Ann Hum Genet 80:235–240. doi:10.1111/ahg.12157
Santen GW et al (2012) Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin–Siris syndrome. Nat Genet 44:379–380. doi:10.1038/ng.2217
Santen GW et al (2013) Coffin–Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum Mutat 34:1519–1528. doi:10.1002/humu.22394
Shang L et al (2015) Mutations in ARID2 are associated with intellectual disabilities. Neurogenetics 16:307–314. doi:10.1007/s10048-015-0454-0
Tsurusaki Y et al (2012) Mutations affecting components of the SWI/SNF complex cause Coffin–Siris syndrome. Nat Genet 44:376–378. doi:10.1038/ng.2219
Tsurusaki Y et al (2014a) De novo SOX11 mutations cause Coffin–Siris syndrome. Nat Commun 5:4011. doi:10.1038/ncomms5011
Tsurusaki Y et al (2014b) Coffin–Siris syndrome is a SWI/SNF complex disorder. Clin Genet 85:548–554. doi:10.1111/cge.12225
Van Houdt JK et al (2012) Heterozygous missense mutations in SMARCA2 cause Nicolaides–Baraitser syndrome. Nat Genet 44(445–449):S441. doi:10.1038/ng.1105
Wieczorek D et al (2013) A comprehensive molecular study on Coffin–Siris and Nicolaides–Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet 22:5121–5135. doi:10.1093/hmg/ddt366
Wilson BG, Roberts CW (2011) SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11:481–492. doi:10.1038/nrc3068
Xu F, Flowers S, Moran E (2012) Essential role of ARID2 protein-containing SWI/SNF complex in tissue-specific gene expression. J Biol Chem 287:5033–5041. doi:10.1074/jbc.M111.279968
Yan Z et al (2005) PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 19:1662–1667
Zahir FR et al (2016) Intragenic CNVs for epigenetic regulatory genes in intellectual disability: survey identifies pathogenic and benign single exon changes. Am J Med Genet A 170:2916–2926. doi:10.1002/ajmg.a.37669
Acknowledgements
We are grateful to the families for participating in this study. We thank Sabine Kaya and Daniela Falkenstein for excellent technical assistance. This work was supported in part by the German Ministry of Research and Education [Grant Numbers 01GS08164 (HE), 01GS08167 (DW), 01GS08163 (TMS), German Mental Retardation Network] as part of the National Genome Research Network and 01GM1520E (DW) as part of the Chromatin-Net consortium. Exome sequencing on Individual 2 was supported by a Grant from Dart NeuroScience LLC (FVB and OC).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2017_1757_MOESM1_ESM.tif
Supplemental Fig. 1: X-ray image of the left hand of individual 2 displaying hypoplastic distal phalanges (TIFF 25738 kb)
Rights and permissions
About this article

Cite this article
Bramswig, N.C., Caluseriu, O., Lüdecke, HJ. et al. Heterozygosity for ARID2 loss-of-function mutations in individuals with a Coffin–Siris syndrome-like phenotype. Hum Genet 136, 297–305 (2017). https://doi.org/10.1007/s00439-017-1757-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1757-z