
Abstract
The etiology of intellectual disabilities (ID) remains unknown for the majority of patients. Due to reduced reproductive fitness in many individuals with ID, de novo mutations account for a significant portion of severe ID. The ATP-dependent SWI/SNF chromatin modifier has been linked with neurodevelopmental disorders including ID and autism. ARID2 is an intrinsic component of polybromo-associated BAF (PBAF), the SWI/SNF subcomplex. In this study, we used clinical whole exome sequencing (WES) in proband-parent-trios to identify the etiology of ID. We identified four independent, novel, loss of function variants in ARID2 gene in four patients, three of which were confirmed to be de novo. The patients all have ID and share other clinical characteristics including attention deficit hyperactivity disorder, short stature, dysmorphic facial features, and Wormian bones. All four novel variants are predicted to lead to a premature termination with the loss of the two conservative zinc finger motifs. This is the first report of mutations in ARID2 associated with developmental delay and ID.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Intellectual disabilities (ID) have an estimated prevalence of approximately 3 % in the general population [1, 2]. IDs are genetically extremely heterogeneous, and etiologies include aneuploidies, segmental deletions/duplications, and numerous monogenic causes. It is likely that many causes of ID have yet to be identified [1, 2]. Due to the genetic heterogeneity and small percentage of monogenic ID attributable to any one gene, as well as the number of genes involved in ID that remain to be discovered, whole exome sequencing (WES) is a useful genomic tool to identify the etiology of ID in clinical diagnostic testing. In cases of severe ID, as a result of the decreased reproductive fitness, a significant portion of genetic causes are due to de novo mutations that can be readily identified by exome sequencing using a trio comparison of the proband and the biological parents [3, 4].
Among the genes identified from clinical exome sequencing, the most frequently identified gene from one large series was ARID1B [5]. Chromatin modification is an important regulator of gene expression. Among three major epigenetic chromatin regulatory mechanisms, the ATP-dependent SWI/SNF chromatin modifier has been linked with neurodevelopmental disorders including ID and autism. The SWI/SNF complexes are evolutionarily highly conserved and alter the accessibility of DNA to nuclear factors through modification of DNA-nucleosome topology in an ATPase-dependent manner. They play essential roles in regulation of gene expression, embryogenesis, organ development and tumorigenesis [6, 7]. Based on the subunit composition, SWI/SNF can be grouped into two subcomplexes, BRG1- or hBRM-associated factors (BAF) and polybromo-associated BAF (PBAF) [8, 9]. Both subcomplexes contain ARID protein family members. BAF complexes contain ARID1A (BAF250a) or ARID1B (BAF250b) [10], while ARID2 (p200; BAF200) is a subunit of the PBAF complexes [11]. Haploinsufficiency of ARID1B has been identified as a frequent cause of intellectual disability, agenesis of the corpus callosum [12, 13], and autism [14, 15]; and mutations in ARID1A and ARID1B cause Coffin-Siris syndrome [16, 17]. Studies also have identified other core catalytic units of the SWI/SNF complexes as causes of neurological diseases, including SMARCA2 in Nicolaides-Baraitser syndrome (NBS) [18] and SMARCB1, SMARCA4, and SMARCE1 in Coffin-Siris syndrome [19].
In the present study, we performed clinical whole exome sequencing (WES) in patients with ID and identified four independent, likely gene-damaging novel variants in ARID2. Despite the well-established association between BAF subunits and neurodevelopmental disorders, little is known about PBAF function in human diseases, and this is the first report of mutations in ARID2 as a cause of intellectual disabilities.
Materials and methods
Informed consent was obtained from all individual participants included in the study. Additional informed consent was obtained from all individual participants for whom identifying information is included in this article. This study was approved by the Institutional Review Board of Columbia University.
Whole exome sequencing
Genomic DNA was extracted from whole blood from affected children and their parents. Exome sequencing was performed on exon targets captured using the Agilent SureSelect Human All Exon V4 (50 Mb) kit (Agilent Technologies, Santa Clara, CA). One microgram of DNA from blood specimen was sheared into 350–400 bp fragments, which were then repaired, ligated to adaptors, and purified for subsequent PCR amplification. Amplified products were then captured by biotinylated RNA library baits in solution following the manufacturer’s instructions. Bound DNA was isolated with streptavidin-coated beads and re-amplified. The final isolated products were sequenced using the Illumina HiSeq 2000 sequencing system with 100-bp paired-end reads (Illumina, San Diego, CA). DNA sequence was mapped to the published human genome build UCSC hg19/GRCh37 reference sequence using BWA with the latest internally validated version at the time of sequencing, progressing from BWA v0.5.8 through BWA-Mem v0.7.8 [20]. Targeted coding exons and splice junctions of known protein-coding RefSeq genes were assessed for average depth of coverage with a minimum depth of 10× required for inclusion in downstream analysis. Local realignment around insertion-deletion sites was performed using the Genome Analysis Toolkit v1.6 [21]. Variant calls were generated simultaneously on all sequenced family members using SAMtools v0.1.18 [20]. All coding exons and surrounding intron/exon boundaries were analyzed. Automated filtering removed common sequence changes (defined as >10 % frequency present in 1000 Genomes database). The targeted coding exons and splice junctions of the known protein-coding RefSeq genes were assessed for the average depth of coverage and data quality threshold values. Whole exome sequence data for all sequenced family members was analyzed using GeneDx’s XomeAnalyzer (a variant annotation, filtering, and viewing interface for WES data), which includes nucleotide and amino acid annotations, population frequencies (NHLBI Exome Variant Server and 1000 Genomes databases), in silico prediction tools, amino acid conservation scores, and mutation references. Variants were filtered based on inheritance patterns, gene lists of interest, phenotype, and population frequencies, as appropriate. Resources including the Human Gene Mutation Database (HGMD), 1000 Genomes database, NHLBI Exome Variant Server, OMIM, PubMed, ClinVar [22–25], and Exome Aggregation Consortium (ExAC; Cambridge, MA; URL: http://exac.broadinstitute.org; accessed June 2015) were used to evaluate genes and sequence changes of interest. Additional searches were performed using specific gene lists related to ID. Identified sequence changes of interest were confirmed in all family members by di-deoxy Sanger sequence analysis using an ABI3730 (Life Technologies, Carlsbad, CA) and standard protocols with a new DNA preparation.
Results
Exome sequencing was performed in 970 probands with neurodevelopmental disorders, most of whom had previous nondiagnostic genetic testing including chromosome microarrays. WES produced an average of ~13 GB of sequence per sample. Mean coverage of captured regions was ~150× per sample, with >98 % covered with at least 10× coverage, an average of >92 % of base call quality of Q30 or greater, and an overall average mean quality score of >Q36. Filtering of common SNPs (>10 % frequency present in 1000 Genomes database) resulted in ~4400 variants per proband sample. After automated filtering of variants with a minor allele frequency (MAF) of >10 %, manual curation was performed to filter less common variants with MAF of 1–10 %, single variants in genes inherited from unaffected parents, evaluate predicted effects of rare variants and known function of the genes and associated human conditions, and examine overlapping phenotypes of individuals with de novo variants in the same gene. Among the 970 probands with neurodevelopmental disorders, there were four probands with predicted loss of function mutations in ARID2 with similar phenotypes. The NHLBI Exome Variant Server did not detect any truncating variants in ARID2 in approximately 6000 individuals of European and African-American ancestry, in the Database of Single Nucleotide Polymorphisms (dbSNP), or in a local database of over 15,000 exomes. The ExAC database contained eight loss of function (LoF) variants (URL: http://exac.broadinstitute.org; accessed June 2015), of which, three were annotated in non-canonical transcripts, and their annotations in the canonical transcript (ENST00000334344) are missense and intronic variants, which are unlikely to cause LoF. Four variants have abnormal allele balance, likely due to somatic mosaicism. Additionally, one stop-gain variant is within 5 % of C-terminal and therefore unlikely to result in nonsense-mediated decay. One apparent LoF variant is part of a multinucleotide polymorphism (MNP). Based on the information above, we believe that among the eight LoF variants listed in ExAC database, five are unlikely to be LoF variants, and the remaining three are potential somatic mosaic LoF variants. Therefore, the presence of these variants in ExAC does not contradict our suggestion that germ line de novo mutations in ARID2 gene are responsible for the phenotype in our patients.
Four probands from four families were identified with novel variants in ARID2 that are predicted to result in loss of function. Among the four novel variants, three were de novo, while the origin of the fourth variant could not be determined because parental samples were not available. Two of the variants (p.H1481fs*4 and p.V846fs*3) cause frameshifts, and the other two (p.L343* and p.Q1440*) are nonsense variants in the 1835 amino acid protein. All four variants are novel and were not present in Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/), the Database of Single Nucleotide Polymorphisms (dbSNP; http://www.ncbi.nlm.nih. gov/ SNP/), 1000 Genomes (1000G; http://www.1000genomes.org/), or Exome Variant Server (ESP; http://evs.gs.washington.edu/EVS/), and all are predicted to disrupt ARID2 proximal to the two highly conservative zinc finger motifs (Fig. 1).

Location of ARID2 mutations. Germ line mutations identified in ARID2 gene in four probands diagnosed with intellectual disabilities. Figure shows ARID2 protein and the deleterious variants indicated by red arrows. p.L343* and p.Q1440* are nonsense mutations; p.V846fs*3 and p.H1481fs*4 cause frameshifts. ARID domain AT-rich interaction domain; RFX RFX-like DNA binding motif, GLN glutamine-rich region, Zinc Fingers C2H2 zinc fingers [27]
These four patients ranged in age from 6 to 15 years, and all have some degree of neurodevelopmental delay (Table 1). All of them have gross motor delays and began walking at 20–24 months of age. Hypotonia was frequently reported. They also demonstrate global developmental delay including speech and fine motor delays. Developmental quotient/intelligence quotient ranges from 50 to 89. All of the children also have behavioral issues although there is a range of reported behavioral issues including attention deficit hyperactivity disorder, tics, anxiety, obsessions, repetitive behaviors, and sensitivity to loud noises and certain food textures. Seizures were not observed.
All of the patients had a history of short stature or failure to thrive, although all had normal birth parameters. Gastroesophageal reflux disease and constipation are common.
Two of the patients have Wormian bones of the skull, one of whom also had plagiocephaly. A third patient has plagiocephaly that did not require treatment. One patient has a mild pectus excavatum, and one child has kyphoscoliosis and conductive hearing loss.
MRI of the brain was performed in two patients and demonstrated mild periventricular leukomalacia in one patient and a small arachnoid cyst with prominent lateral ventricles in the other patient.
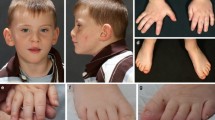
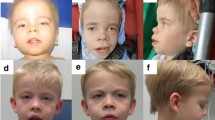
All patients have dysmorphic facial features including micrognathia or retrognathia, low set or posteriorly rotated ears, epicanthal folds, down slanting palpebral fissures, highly arched palate, and frontal bossing (Fig. 2).

Facial characteristics of individuals with ARID2 mutations
One patient also had a cleft palate, diaphragmatic eventration, and atrial septal defect. Birth defects were not observed in the other three patients.
Discussion
We have identified ARID2 (HGNC ID: 18037) as a likely novel genetic cause of ID through WES. Clinical WES revealed four novel, likely gene damaging variants in ARID2 out of 970 ID patients evaluated. Each of our four patients has global developmental delay with notable problems with speech, as well as behavioral problems including ADHD, anxiety, and obsessions that are common among patients with ID and patients with Coffin-Siris syndrome or Nicolaides Baraitser syndrome, two other SWI/SNF-A chromatin-remodeling complex disorders. Notably, none of the children have had seizures, and there are no major brain malformations. Wormian bones, frontal bossing, micrognathia, and retrognathia are distinguishing features in these ARID2 patients not observed with other chromatin remodeling disorders.
We believe it is likely that the clinical features shared by these patients are due to ARID2 mutations. Patient 2 has birth defects including an atrial septal defect, cleft palate, diaphragmatic eventration, and also has a 421 kb 11q12.1 duplication. The parents were not tested for the 11q21.1 duplication, so inheritance could not be determined, but this duplication is small and we do not believe is likely to account for the majority of the clinical presentation. Whether these birth defects are low frequency manifestations of ARID2 mutations or are due to a second genetic contribution will await the characterization of more patients with ARID2 mutations. Patient 4 also carries a novel, de novo Y629N variant in EP300 that is predicted to be deleterious by SIFT, PolyPhen2, GERP, and CADD. Mutations in EP300 cause Rubinstein-Taybi syndrome-2 [26] (Table 1). The patient clinically does not resemble individuals with Rubinstein-Taybi syndrome, so it is unclear whether the EP300 variant is contributing to the patient’s neurodevelopmental delays.
ARID2 (NP_689854.2) contains a consensus AT-rich DNA interaction domain located at the N-terminus, followed by RFX, GLN (glutamine-rich region) and two classic C2H2 zinc fingers at the C-terminus, which directly bind to DNA or interact with proteins [8, 27]. All four ARID2 mutations in our cases (p.L343*, p.Q1440*, p.H1481fs*4, and p.V846fs*3) are predicted to be deleterious since they result in frameshift/truncated proteins, and all cause the loss of the two conservative zinc finger motifs, located at amino acids 1634–1690 [27] (Fig. 1). On the basis of the predicted deleterious effect of these variants on the protein and the fact that all three cases with parents available were de novo, we conclude that haploinsufficiency of ARID2 is a likely novel cause for ID in these four patients.
ARID2 belongs to the ARID family of proteins, characterized by the AT-rich DNA interaction domain [28], and plays an important role in development, tissue-specific gene expression and control of cell proliferation [29, 30]. ARID2 is ubiquitously expressed throughout the developing spinal cord, brain and other embryonic tissues such as heart and liver in mouse [31, 32]. It is one of the three ARID proteins in SWI/SNF subunits and is an intrinsic component of the PBAF complex [11].
PBAF and BAF are related, yet distinct chromatin remodeling complexes which share multiple core complex components including the catalytic subunit BRG1, and have unique components including ARID1A (BAF250a) and ARID1B (BAF250b) in the BAF complex, and polybromo (PB1, BAF180), ARID2 (BAF200), and BRD7 in the PBAF complex [11, 33]. Through combinatorial assembly of the subunits, SWI/SNF complexes play distinct and essential roles in cell fate specificity and lineage conversion during development. BAF complexes play essential roles in controlling cell renewal, neural progenitor cell division, and neuronal maturation. A switch in subunit composition of SWI/SNF complexes BAF45a/53a and BAF45b, c/53b has been identified in the transition from neural stem/progenitors to post-mitotic neurons [31], with ARID2 physically interacting with BAF complexes in different developmental stages [34]. The importance of the BAF complex in human brain development has emerged through recent discoveries that mutations in subunits including SMARCA2, SMARCB1, SMARCA4, ARID1A, and ARID1B have all been implicated in ID and related neurobehavioral disorders including Nicolaides-Baraitser syndrome [18], Coffin-Siris syndrome [17], autism [2], and schizophrenia [35]. The high frequency of BAF subunit mutations in neurological disorders underscores the fundamental importance of BAF in the development and function of the central nervous system [36].
Beyond the role of ARID2 in brain development and function, it also appears to have a role in skeletal and cardiac development. A recent study using comprehensive single cell-resolution analysis in Caenorhabditis elegans revealed that inhibition of ARID2 (swsn-7 ortholog) caused a severe delay in cell cycle progression, suggesting an important role as chromatin regulator in embryogenesis [37]. ARID2 plays an essential role in osteoblast differentiation using MC3T3-E1 pre-osteoblasts as a model. ARID2-depletion severely impaired the mature mineralization of osteoblasts [38]. ARID2 is found in the multiple protein complex that participates in SRF-dependent gene regulation, and affects the promoter activity of cardiac genes in NIH3T3 cells [27]. SRF regulates cell survival during murine embryonic development [39], is required for PI3-kinase-regulated cell proliferation [40], and plays an important role in the regulation of immediate-early genes and muscle-specific genes [41]. Arid2 (BAF200) homozygous mutant mice died between E12.5-E14.5, and Arid2 homozygous mutant mouse embryos exhibited multiple cardiac defects [32]. These data suggest that ARID2 is required for the regulation of cardiomyocyte proliferation. ARID2 interacts with CHD7 (chromodomain helicase DNA-binding domain, member 7, an ATP-dependent chromatin remodeler mutated in CHARGE syndrome) to promote neural crest formation and cell migration in human neural crest cells [42]. CHARGE syndrome is characterized by congenital anomalies including heart and craniofacial malformations, growth retardation, and developmental delay [43, 44]. Some of those features are seen in our ARID2 patients (Table 1).
Recent sequencing studies have identified frequent mutations in subunits encoding genes in SWI/SNF chromatin remodeling complexes in a wide spectrum of human cancers, and BAF complex subunit gene mutations occur in 20 % of all human cancers. SWI/SNF complexes are wildly involved in the regulation of gene expression through interactions with various transcription factors, recruiting coactivators and corepressors at target promoters; and they play important role in modulating DNA repair [45–47]. A recent study also shows that the PBAF remodeling complex is important for double-strand breaks (DSBs)-induced transcriptional silencing and promotes repair of a subset of DNA DSBs at early time points [48]. ARID2 has been recently identified as a key cancer gene for hepatocellular carcinoma [49, 50], melanoma [51, 52], and lung cancer [53]. Recently, a missense mutation R314C in ARID2 was reported in an individual with autism [54]. R314C is the second most frequent somatic missense mutation in COSMIC in ARID2 gene, suggesting that this missense mutation leads to loss-of-function of ARID2. These cancer data further strengthen our prediction that haploinsufficiency of ARID2 is responsible for the intellectual disabilities of our patients.
To our knowledge, this is the first report of mutations in ARID2 associated with developmental delay and intellectual disabilities. Additional functional studies and evaluation of this gene in a larger patient population will be needed to definitely prove causality and fully characterize the spectrum of associated clinical features.
References
van Bokhoven H (2011) Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet 45:81–104. doi:10.1146/annurev-genet-110410-132512
Srivastava AK, Schwartz CE (2014) Intellectual disability and autism spectrum disorders: causal genes and molecular mechanisms. Neurosci Biobehav Rev 46P2:161–174. doi:10.1016/j.neubiorev.2014.02.015
Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, de Vries BB, Brunner HG, Veltman JA (2010) A de novo paradigm for mental retardation. Nat Genet 42(12):1109–1112. doi:10.1038/ng.712
Ku CS, Polychronakos C, Tan EK, Naidoo N, Pawitan Y, Roukos DH, Mort M, Cooper DN (2013) A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry 18(2):141–153. doi:10.1038/mp.2012.58
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM (2014) Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312(18):1870–1879. doi:10.1001/jama.2014.14601
Vignali M, Hassan AH, Neely KE, Workman JL (2000) ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 20(6):1899–1910
Levine M, Tjian R (2003) Transcription regulation and animal diversity. Nature 424(6945):147–151. doi:10.1038/nature01763
Mohrmann L, Langenberg K, Krijgsveld J, Kal AJ, Heck AJ, Verrijzer CP (2004) Differential targeting of two distinct SWI/SNF-related Drosophila chromatin-remodeling complexes. Mol Cell Biol 24(8):3077–3088
Nie Z, Xue Y, Yang D, Zhou S, Deroo BJ, Archer TK, Wang W (2000) A specificity and targeting subunit of a human SWI/SNF family-related chromatin-remodeling complex. Mol Cell Biol 20(23):8879–8888
Wang X, Nagl NG, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P, Dallas PB, Moran E (2004) Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J 383(Pt 2):319–325. doi:10.1042/BJ20040524
Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, Parsons R, Zhao K, Wang W (2005) PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 19(14):1662–1667. doi:10.1101/gad.1323805
Backx L, Seuntjens E, Devriendt K, Vermeesch J, Van Esch H (2011) A balanced translocation t(6;14)(q25.3;q13.2) leading to reciprocal fusion transcripts in a patient with intellectual disability and agenesis of corpus callosum. Cytogenet Genome Res 132(3):135–143. doi:10.1159/000321577
Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, Zweier M, Gohring I, Zink AM, Rappold G, Schrock E, Wieczorek D, Riess O, Engels H, Rauch A, Reis A (2012) Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 90(3):565–572. doi:10.1016/j.ajhg.2012.02.007
Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM, Henriksen KF, Hjalgrim H, Kirchhoff M, Bijlsma EK, Nielsen M, den Hollander NS, Ruivenkamp CA, Isidor B, Le Caignec C, Zannolli R, Mucciolo M, Renieri A, Mari F, Anderlid BM, Andrieux J, Dieux A, Tommerup N, Bache I (2012) Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet 82(3):248–255. doi:10.1111/j.1399-0004.2011.01755.x
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485(7397):246–250. doi:10.1038/nature10989
Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CA, van Ommen GJ, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M (2012) Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet 44(4):379–380. doi:10.1038/ng.2217
Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N (2012) Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44(4):376–378. doi:10.1038/ng.2219
Van Houdt JK, Nowakowska BA, Sousa SB, van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard MJ, Bottani A, Castori M, Cormier-Daire V, Deardorff MA, Filges I, Fryer A, Fryns JP, Gana S, Garavelli L, Gillessen-Kaesbach G, Hall BD, Horn D, Huylebroeck D, Klapecki J, Krajewska-Walasek M, Kuechler A, Lines MA, Maas S, Macdermot KD, McKee S, Magee A, de Man SA, Moreau Y, Morice-Picard F, Obersztyn E, Pilch J, Rosser E, Shannon N, Stolte-Dijkstra I, Van Dijck P, Vilain C, Vogels A, Wakeling E, Wieczorek D, Wilson L, Zuffardi O, van Kampen AH, Devriendt K, Hennekam R, Vermeesch JR (2012) Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 44(4):445–449. doi:10.1038/ng.1105, S441
Kosho T, Okamoto N, Coffin-Siris Syndrome International C (2014) Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C: Semin Med Genet 166C(3):262–275. doi:10.1002/ajmg.c.31407
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. doi:10.1093/bioinformatics/btp324
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. doi:10.1038/ng.806
Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA (2012) An integrated map of genetic variation from 1092 human genomes. Nature 491(7422):56–65. doi:10.1038/nature11632
Serve EV, NHLBI GO (2015) NHLBI GO exome sequencing project (ESP). Seattle, WA
OMIM Online Mendelian Inheritance in Man. In: McKusick-Nathans Institute of Genetic Medicine JHM, National Human Genome Research Institute, editor. An online catalog of human genes and genetic disorders. Baltimore, MA
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR (2014) ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42(Database issue):D980–D985. doi:10.1093/nar/gkt1113
Bartholdi D, Roelfsema JH, Papadia F, Breuning MH, Niedrist D, Hennekam RC, Schinzel A, Peters DJ (2007) Genetic heterogeneity in Rubinstein-Taybi syndrome: delineation of the phenotype of the first patients carrying mutations in EP300. J Med Genet 44(5):327–333. doi:10.1136/jmg.2006.046698
Zhang X, Azhar G, Zhong Y, Wei JY (2006) Zipzap/p200 is a novel zinc finger protein contributing to cardiac gene regulation. Biochem Biophys Res Commun 346(3):794–801. doi:10.1016/j.bbrc.2006.05.211
Patsialou A, Wilsker D, Moran E (2005) DNA-binding properties of ARID family proteins. Nucleic Acids Res 33(1):66–80. doi:10.1093/nar/gki145
Wilsker D, Patsialou A, Dallas PB, Moran E (2002) ARID proteins: a diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ 13(3):95–106
Kortschak RD, Tucker PW, Saint R (2000) ARID proteins come in from the desert. Trends Biochem Sci 25(6):294–299
Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR (2007) An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55(2):201–215. doi:10.1016/j.neuron.2007.06.019
He L, Tian X, Zhang H, Hu T, Huang X, Zhang L, Wang Z, Zhou B (2014) BAF200 is required for heart morphogenesis and coronary artery development. PLoS One 9(10), e109493. doi:10.1371/journal.pone.0109493
Xue Y, Canman JC, Lee CS, Nie Z, Yang D, Moreno GT, Young MK, Salmon ED, Wang W (2000) The human SWI/SNF-B chromatin-remodeling complex is related to yeast rsc and localizes at kinetochores of mitotic chromosomes. Proc Natl Acad Sci U S A 97(24):13015–13020. doi:10.1073/pnas.240208597
Yoo AS, Crabtree GR (2009) ATP-dependent chromatin remodeling in neural development. Curr Opin Neurobiol 19(2):120–126. doi:10.1016/j.conb.2009.04.006
Loe-Mie Y, Lepagnol-Bestel AM, Maussion G, Doron-Faigenboim A, Imbeaud S, Delacroix H, Aggerbeck L, Pupko T, Gorwood P, Simonneau M, Moalic JM (2010) SMARCA2 and other genome-wide supported schizophrenia-associated genes: regulation by REST/NRSF, network organization and primate-specific evolution. Hum Mol Genet 19(14):2841–2857. doi:10.1093/hmg/ddq184
Son EY, Crabtree GR (2014) The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am J Med Genet C: Semin Med Genet 166C(3):333–349. doi:10.1002/ajmg.c.31416
Kruger AV, Jelier R, Dzyubachyk O, Zimmerman T, Meijering E, Lehner B (2014) Comprehensive single cell-resolution analysis of the role of chromatin regulators in early C. elegans embryogenesis. Dev Biol. doi:10.1016/j.ydbio.2014.10.014
Xu F, Flowers S, Moran E (2012) Essential role of ARID2 protein-containing SWI/SNF complex in tissue-specific gene expression. J Biol Chem 287(7):5033–5041. doi:10.1074/jbc.M111.279968
Schratt G, Philippar U, Hockemeyer D, Schwarz H, Alberti S, Nordheim A (2004) SRF regulates Bcl-2 expression and promotes cell survival during murine embryonic development. EMBO J 23(8):1834–1844. doi:10.1038/sj.emboj.7600188
Poser S, Impey S, Trinh K, Xia Z, Storm DR (2000) SRF-dependent gene expression is required for PI3-kinase-regulated cell proliferation. EMBO J 19(18):4955–4966. doi:10.1093/emboj/19.18.4955
Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428(6979):185–189. doi:10.1038/nature02382
Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, Wysocka J (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463(7283):958–962. doi:10.1038/nature08733
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 36(9):955–957. doi:10.1038/ng1407
Sanlaville D, Verloes A (2007) CHARGE syndrome: an update. Eur J Hum Genet 15(4):389–399. doi:10.1038/sj.ejhg.5201778
Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T, Yasui A (2014) SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res 74(9):2465–2475. doi:10.1158/0008-5472.CAN-13-3608
Tolstorukov MY, Sansam CG, Lu P, Koellhoffer EC, Helming KC, Alver BH, Tillman EJ, Evans JA, Wilson BG, Park PJ, Roberts CW (2013) Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci U S A 110(25):10165–10170. doi:10.1073/pnas.1302209110
Helming KC, Wang X, Roberts CW (2014) Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 26(3):309–317. doi:10.1016/j.ccr.2014.07.018
Kakarougkas A, Ismail A, Chambers AL, Riballo E, Herbert AD, Kunzel J, Lobrich M, Jeggo PA, Downs JA (2014) Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol Cell 55(5):723–732. doi:10.1016/j.molcel.2014.06.028
Shiraishi Y, Fujimoto A, Furuta M, Tanaka H, Chiba K, Boroevich KA, Abe T, Kawakami Y, Ueno M, Gotoh K, Ariizumi S, Shibuya T, Nakano K, Sasaki A, Maejima K, Kitada R, Hayami S, Shigekawa Y, Marubashi S, Yamada T, Kubo M, Ishikawa O, Aikata H, Arihiro K, Ohdan H, Yamamoto M, Yamaue H, Chayama K, Tsunoda T, Miyano S, Nakagawa H (2014) Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS One 9(12), e114263. doi:10.1371/journal.pone.0114263
Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, Calvo F, Zucman-Rossi J (2012) Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 44(6):694–698. doi:10.1038/ng.2256
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L (2012) A landscape of driver mutations in melanoma. Cell 150(2):251–263. doi:10.1016/j.cell.2012.06.024
Ding L, Kim M, Kanchi KL, Dees ND, Lu C, Griffith M, Fenstermacher D, Sung H, Miller CA, Goetz B, Wendl MC, Griffith O, Cornelius LA, Linette GP, McMichael JF, Sondak VK, Fields RC, Ley TJ, Mule JJ, Wilson RK, Weber JS (2014) Clonal architectures and driver mutations in metastatic melanomas. PLoS One 9(11), e111153. doi:10.1371/journal.pone.0111153
Huang HT, Chen SM, Pan LB, Yao J, Ma HT (2015) Loss of function of SWI/SNF chromatin remodeling genes leads to genome instability of human lung cancer. Oncol Rep 33(1):283–291. doi:10.3892/or.2014.3584
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, Sullivan CA, Walker MF, Waqar Z, Wei L, Willsey AJ, Yamrom B, Lee YH, Grabowska E, Dalkic E, Wang Z, Marks S, Andrews P, Leotta A, Kendall J, Hakker I, Rosenbaum J, Ma B, Rodgers L, Troge J, Narzisi G, Yoon S, Schatz MC, Ye K, McCombie WR, Shendure J, Eichler EE, State MW, Wigler M (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515(7526):216–221. doi:10.1038/nature13908
Acknowledgements
We thank the families for their generous contributions. We thank the data review support from ExAC consortium. This work was supported in part by a grant from the Simons Foundation.
Conflicts of interest
Megan Cho, Kyle Retterer, Leandra Folk, Patrik Vitazka, and Jane Juusola are employees of GeneDx. Wendy Chung is a consultant to BioReference Laboratories. The other authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shang, L., Cho, M.T., Retterer, K. et al. Mutations in ARID2 are associated with intellectual disabilities. Neurogenetics 16, 307–314 (2015). https://doi.org/10.1007/s10048-015-0454-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-015-0454-0