
Abstract
Microdeletion syndromes are frequent causes of neuropsychiatric disorders leading to intellectual disability as well as autistic features accompanied by epilepsy and craniofacial anomalies. From comparative deletion mapping of the smallest microdeletion to date at 12q24.31, found in a patient with overlapping clinical features of 12q24.31 microdeletion syndrome, we narrowed the putative critical region to 445 kb containing seven genes, one microRNA, and one non-coding RNA. Zebrafish in situ hybridization and comprehensive transcript analysis of annotated genes in the panels of human organ and brain suggest that these are all candidates for neurological phenotypes excluding the gene HPD. This is also corroborated by synteny analysis revealing the conservation of the order of these six candidate genes between humans and zebrafish. Among them, we propose histone demethylase KDM2B and histone methyltransferase SETD1B as the two most plausible candidate genes involved in intellectual disability, autism, epilepsy, and craniofacial anomalies. These two chromatin modifiers located approximately 224 kb apart were both commonly deleted in six patients, while two additional patients had either KDM2B or SETD1B deleted. The four additional candidate genes (ORAI1, MORN3, TMEM120B, RHOF), a microRNA MIR548AQ, and a non-coding RNA LINC01089 are localized between KDM2B and SETD1B. The 12q24.31 microdeletion syndrome with syndromic intellectual disability extends the growing list of microdeletion syndromes and underscores the causative roles of chromatin modifiers in cognitive and craniofacial development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the prevalent application of high-resolution microarray in medical genetics, a large number of new microdeletion and microduplication syndromes involving many chromosomal regions are emerging as disorders of syndromic intellectual disability (Nevado et al. 2014). We report a genomic delineation of eight unrelated patients with non-recurrent 12q24.31 microdeletion syndrome to map the size, extent, and gene content. The deleted regions in seven cases significantly overlap with a microdeletion we identified, the smallest one described to date, enabling the refinement of the candidate gene region for syndromic intellectual disability. Four cases of microdeletions spanning this region have been reported with similar phenotypes (Baple et al. 2010; Chouery et al. 2013; Palumbo et al. 2015; Qiao et al. 2013) and we found three additional microdeletion cases in the DECIPHER database (Firth et al. 2009).
Our clinical delineation of the 12q24.31 interval suggests that intellectual disability, autism, epilepsy, and craniofacial anomalies constitute core clinical features of the 12q24.31 microdeletion syndrome. We refined the smallest overlapping region to 445 kb encompassing seven genes, one microRNA, and one non-coding RNA by qPCR. Among them, we propose two chromatin regulators, KDM2B (MIM 609078) and SETD1B (MIM 611055), as the most plausible two candidate genes for syndromic intellectual disability along with four additional candidate genes (ORAI1, MORN3, TMEM120B, and RHOF). The perturbation of a chromatin regulatory mechanism is a well-known underlying cause of neurodevelopmental and psychiatric disorders (Ronan et al. 2013). Whether the two chromatin modifiers, KDM2B as a demethylase and SETD1B as a methyltransferase, act independently or work in concert to coordinate histone modifications remains to be seen.
In the current study, we have analyzed putative candidate genes within this chromosomal region. Zebrafish in situ hybridization and RT-qPCR in different human organ and brain regions were performed to validate individual candidate genes. We determined the level of transcript reduction for all six candidate genes in our microdeletion.
Materials and methods
Genomic DNA extraction
Blood samples were obtained from patient DGDP343, his parents as well as his younger brother. Genomic DNA was extracted from blood using a standard phenol–chloroform protocol.
Microarray
Genomic DNA derived from the patient was assayed on a 4 × 180 K oligonucleotide array (Agilent Technologies) to identify pathogenic copy number variations (CNVs).
Cell culture
Lymphoblastoid cell lines (LCLs) were established from the patient, his brother and father using the protocol described in Nishimoto et al. (2014).
Real-time PCR (qPCR and RT-qPCR)
Primers for qPCR were designed across the chromosomal regions containing the proximal and distal deletion breakpoints. We also designed RT-qPCR primers against exonic sequences of genes including KDM2B and SETD1B (Table S1). Total RNA was extracted from LCLs using the RNeasy Plus Mini kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Total RNA from whole human brain, fetal brain and from an additional 11 different regions of the brain (Clontech, Mountain View, CA, USA) were also used to investigate the transcript levels of KDM2B, ORAI1, MORN3, TMEM120B, RHOF, and SETD1B. Samples of total RNA used were derived from several patients aged between 16 and 89, except for hippocampus and kidney where the total RNA was obtained from single individuals aged 27 and 40, respectively. Fetal brain total RNA was pooled from 21 spontaneously aborted 26–33-week-old Caucasians. cDNA was synthesized from 1 µg of total RNA using the RevertAid First cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). Real-time PCR was carried out using 2 µl cDNA, 2.5 µM primer and 10 µl FastStart DNA Green Master (Roche, Indianapolis, IN, USA) in a 20 µl reaction volume. The ∆∆ct method was used to determine relative transcript levels of genes, as well as copy number of the loci of interest, and beta-2-microglobulin (MIM 109700) was used as the endogenous control. Samples were run in triplicates and standard deviations were calculated from 2–3 independent experiments.
Cloning and RNA probe preparation from zebrafish
Total RNA was extracted from adult zebrafish brains using TRIzol® (Ambion). Total RNA was reverse transcribed to cDNA using the SuperScript III First-Strand synthesis kit (Invitrogen™). From the sequences at NCBI (National Center for Biotechnology Information) and ZFIN (The Zebrafish Model Organism Database) primers were designed for cloning of the following parts of 7 genes in zebrafish. kdm2bb (XM_009305661.1) primer pair: forward 5′-GAGGACGATGAGGAGTACGAGGAGC-3′, reverse 5′-GCTCCCATTTAGGCCCCAGCCAATC- 3′ for a 439 bp fragment; orai1b (NM_205600.1) primer pair : forward 5′-GTGGAGGTCCAGCTGGACACCAATC-3′, reverse 5′-GGCGGCCACTCCGGCAGACACAGT-3′ for a 342 bp fragment; morn3 (NM_001017666.1) primer pair : forward 5′-GTGAAAAGACCCCCGACCGCAGAAC-3′, reverse 5′-TTCTCATACTGCATTCGTCCCCATCC-3′ for a 389 bp fragment; tmem120b (NM_001045230.2) primer pair: forward 5′-GGCCTGAAAGACCTCAAGCAGAGCC-3′, reverse 5′-TTGAGACGTAGTGATGTGACACCCAC-3′ for a 433 bp fragment; rhof (NM_001020642.1) primer pair : forward 5′-TCGTGATCGTCGGGGATGGAGGATG-3′, reverse 5′-AGCTCTGGCTGCTAGCGCTCGCTTT-3′ for a 527 bp fragment; setd1ba (NM_001045134.2) primer pair : 5′-CCCTAGAACCCCTGGTCATGAAGGC-3′, reverse 5′-GAGGAATGCCAGCACTGAGCACAGG-3′ for a 383 bp fragment; hpda (NM_201167.1) primer pair : forward 5′-GCCCGAGAGAGGAAAGTTTCTAAACTT-3′, reverse 5′-TAAGAGAGGGTCTCTGAACAGCGGCT-3′ for a 484 bp fragment. The RT-PCR products were subcloned into the pGEM-T easy vector (Promega) and digoxigenin (DIG)-labeled sense and antisense RNAs were produced using a DIG-RNA labeling kit (Roche) according to the manufacturer’s instructions.
Whole mount in situ hybridization
At least ten embryos at a time were used for each probe, and whole mount in situ hybridization was repeated three times for each probe. Two developmental stages (48 h and 5 day) of embryos were used. Thus, at least 60 embryos (10 × 3 × 2) were used for each probe. Control sense probes were also tested as negative controls.
Fixation and storage
Whole mount in situ hybridization was performed with digoxigenin-labeled probes, essentially as described in Thisse et al. (1993). 48-hpf (hour post-fertilization) embryos were fixed overnight in 4 % paraformaldehyde in PBS at 4 °C and then transferred to methanol before being stored at −20 °C until use.
Proteinase K treatment and hybridization
48-hpf embryos were rehydrated with PBST, incubated in proteinase K (10 µg/ml, Roche) for 20 min, fixed for 20 min in 4 % paraformaldehyde, and then washed with PBST. Embryos were prehybridized in HYB [hybridization solution: 50 % formamide (Bioneer), 5X SSC, 50 µg/ml heparin (Sigma-Aldrich), 500 µg/ml torula RNA (Sigma-Aldrich), 46 mM citric acid, pH 6.0 and 0.1 % Tween-20] for 1 h at 70 °C. After prehybridization, RNA probes were added (100 ng per 10 embryos), and embryos hybridized at 70 °C overnight.
Probe washing and blocking
After the hybridization, embryos were washed at 70 °C for 30 min with 50 % HYB in 2X SSC, once with 100 % 2X SSC for 15 min, and three times with 0.2X SSC for 15 min. Through a series of 4 washes with increasing percentages of PBST, embryos were transferred into PBST at room temperature, and washed three times with PBST for 10 min each wash. For staining, embryos were protected with 5 % horse serum (Sigma-Aldrich)/PBST blocking solution for 30 min.
Staining
Embryos were incubated with anti-digoxigenin Fab fragment conjugated with alkaline phosphatase (Roche) at a concentration of 1:4000 overnight in 4 °C. After extensive washing with PBST, labeling was visualized using NBT/BCIP (nitroblue tetrazolium/5-bromo 4-chloro 3-indolyl phosphate, Roche) as alkaline phosphatase substrate. After a sufficient intensity of staining was reached, the reaction was stopped by washing in stop solution (1 mM EDTA in PBST, pH 5.5). The whole mount in situ hybridization results were examined with a differential interference contrast microscope (Leica MX16FA).
Synteny analysis
The chromosomal location of kdm2bb, setd1ba, orai1b, morn3, tmem120b, rhof, setd1ba and hpda genes in zebrafish was compared with that of human counterpart genes based on the Synteny database (http://teleost.cs.uoregon.edu/acos/synteny_db).
Clinical report
The patient, now age 3, is a white male born at 415/7 weeks by vaginal delivery from a 35-year-old mother after an uneventful pregnancy. At ~6 months, he displayed significant torticollis and developmental delay. He sat at 10-1/2 months and exhibited command crawling at 15 months. However, he often threw himself backwards onto the floor from a sitting position.
At 16/12 years, myoclonic seizures began. He soon developed screaming seizures during which his face displayed a terrified look. His screaming seizures ranged from mild panic to loud, horrified screaming. He was placed on valproic acid treatment, but he became extremely passive and did not engage with people, so it was discontinued. He was then placed on a ketogenic diet. When he was 17/12 years, an electroencephalogram (EEG) showed a generalized epileptiform activity classified as E1, generalized.
At 110/12 years, he crawled and maintained a standing position using the wall for support. He preferentially used his right hand with a raking grasp. He imitated some of his parents’ speech, and learned to make a few consonant sounds. He had seizures several times a day, but the duration lessened with clonazepam treatment. At age 2, the boy underwent Occupational Therapy (OT), Physiotherapy (PT), and Speech–Language–Pathology (SLP) assessments. He was followed by infant development services, and genetics and neurology specialists. The child enjoyed banging toys together, spinning toys, and clapping. At age 2, his EEG showed a very active epileptiform disturbance classifying his epilepsy as E3, generalized.
At 23/12 years, he was diagnosed with mild to moderate hypotonia. He displayed poor head righting bilaterally, mild to moderate head lag, significant tightness in motion, and did not fully develop protective reactions. Early stages of four-point crawling were observed. His sensory seeking and arching into full extension impacted his gross motor skill development as he often fell out of position. An evaluation placed his gross motor skills at a ~7-month-old level, and he received physiotherapy. Interestingly, he had a high pain tolerance. He frequently curled his fingers into his palms, but had full active and passive range of motion in his fingers and upper extremities. The boy also often sucked on his fingers. He enjoyed car rides, played on the exercise ball, swing, and walker.
At 23/12 years, the child was evaluated for fine motor, language, pragmatic and social skills. He had difficulty processing touch input and particularly with vestibular/movement in space input. He seemed to require repetition to understand, and he did not point to items or wave to express himself. He displayed attention to pictures, sometimes responded to the ‘give me’ command, and understood some new words. When asked questions, he vocalized in response. He did not understand words referring to body parts, prepositions, or the word “no”. The patient showed developmental delay in fine motor skills and his pre-language skills were severely delayed for his age. His developing pragmatic skills were at the 3–6-month age level and developing interaction–attachment skills at the 9–12-month age level on the Rossetti Infant-Toddler Scale. His social pragmatic skills were also severely delayed for his age. He exhibited less than half of the language comprehension skills at the 12–15-month age level on the Rossetti and half of the skills at the 9–12-month age level. His receptive and expressive language skills were severely delayed for his age.
At 26/12 years, his epilepsy was categorized as E3 generalized, E3 multifocal, D1 generalized indicating a change in epilepsy type over time. When 29/12 years, the myoclonic seizures were replaced by tonic seizures. He appeared happy and rarely cried except during a seizure. He was fed small pieces of food and often sucked on his finger, probably to help with swallowing. He tended to hold onto objects and transferred them from hand to hand and into his mouth. At 211/12 years, he showed an interest in numbers and letters, music, lights, and fans. He enjoyed looking at books, turning pages, and listening to someone reading. He often rubbed the knuckles of his hands against each other in a rhythmic way. The child attended school 4 days a week where he was monitored by a one-on-one worker.
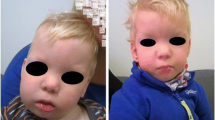
At 211/12 years, he drank thin liquids, but occasionally coughed while drinking. He also frequently had a cough. He had a narrow face, a prominent forehead, and hypertelorism (Fig. 1a). At this age he was diagnosed having a mild plagiocephaly (Fig. 1b), and mild tapering fingers (Fig. 1e, f). He enjoyed exaggerated sensations such as loud music. He loved being in a swing and listening to music, so he was enrolled in music therapy. His social engagement was very limited. He was interested in other children, and sometimes tried to touch them, but in general, he was very passive with other people. The child did not engage in any creative or pretend play. He displayed some unusual finger movements and repetitive scratching of one arm. At that time, the family was informed that the boy’s behaviors were probably compatible with an autism spectrum diagnosis.

Composite figure showing the facial and hand pictures of patient DGDP343, comparative deletion mapping of CNVs at 12q24.31 and whole mount in situ hybridization. The patient has a narrow face and also displays a prominent forehead and hypertelorism (a). He displays a mild plagiocephaly with flattened occiput in the side view (b) as well as mild tapering fingers (e and f) at the age of 211/12. g Genomic delineation at 12q24.31 showing the size of the deletion in our patient DGDP343 relative to other microdeletion cases. DGDP343 has the smallest microdeletion reported in this region. The deleted chromosomal region in DGDP343 is completely contained within the microdeletions reported in Baple et al. (2010), Palumbo et al. (2015), as well as the three DECIPHER (DCP) cases. Blue dotted lines show the proximal region of DGDP343’s microdeletion that overlaps distal end of the deleted region reported in Chouery et al. (2013), while dotted lines in purple indicate the distal part of the microdeletion partially overlapping with the proximal deleted region in Qiao et al. (2013). h–q Whole mount in situ hybridization analysis in zebrafish showing tissue-specific expression levels of 7 candidate genes involved in the 12q24.31 microdeletion. All are shown as side view of head region, except for i, j and l (dorsal view). h, i kdm2bb is extensively expressed in central nervous system and retinal photoreceptor cell layer at 48 hpf. j No signals are obtained with kdm2bb sense control probe (k, l) setd1ba is strongly expressed in brain but weakly expressed in retinal cell layer. m tmem120b is ubiquitously expressed. n orai1b is expressed in brain. o morn3 is expressed in ependymal cilia cells and brain region faintly. p rhof produced little or no signal at 48 hpf. q hpda is only expressed in liver. Scale bar 100 µm
At 42/12 years, his head circumference was 19”, being at the 4th percentile. This study was approved by the Institutional Review Board of Augusta University and a written informed consent was obtained from the mother of the patient DGDP343 for the publication of this report and accompanying images.
Results
Microarray analysis
Microarray analysis in DGDP343 revealed a minimal 360 kb interstitial microdeletion at 12q24.31 (chr12:121908905-122269437, hg19) involving at least six genes namely, KDM2B, ORAI1 (MIM 610277), MORN3, TMEM120B, RHOF, SETD1B, a microRNA MIR548AQ, as well as a non-coding RNA LINC01089 (Table 1; Fig. 2a).

Gene organization at 12q24.31, synteny analysis and delineation of deletion breakpoints. a Schematic representation of genes involved in the 12q24.31 microdeletion. The minimal deletion determined by microarray is displayed as well as the location of the deletion breakpoints defined by qPCR. Genes ORAI1, MORN3, TMEM120B, RHOF, SETD1B, microRNA MIR548AQ, and non-coding RNA LOC338799 are fully deleted heterozygously while KDM2B and HPD are both truncated. b Shared synteny of candidate genes between human chromosome 12q24.31 and zebrafish chromosome 10. Among the seven genes of interest on human chromosome 12q24.31, six genes show preserved synteny with the corresponding orthologous genes on chromosome 10 of zebrafish. The conserved order and organization might be evolutionarily advantageous. Interestingly, there is a synteny break between the genes SETD1B and HPD. It is important to note that KDM2B and SETD1B are within shared synteny. c Heterozygous deletion of KDM2B exon 8 showing that the 12q24.31 microdeletion in present only in the patient. d The proximal deletion breakpoint is located between KDM2B exon 13 and 14, while the distal deletion breakpoint resides between HPD 5′-UTR and intron 10. A value close to 1 indicates no deletion whereas a value close to 0.5 indicates heterozygous deletion
Additionally, one 453 kb microdeletion (chr3: 110,462,714-110,915,623/hg19) at 3q13.13 was detected that encompasses the gene PVRL3 (poliovirus receptor-related 3 or Nectin-3). This gene is predominantly expressed in the testis and placenta and a knockout resulted in defects in the later stages of sperm morphogenesis in mice (Inagaki et al. 2006). Since this was inherited from his healthy mother and since several CNVs including partial PVRL3 were reported in the DGV (Database of Genomic Variant), it was considered a benign polymorphism.
qPCR
qPCR assays showed that the microdeletion is present only in the patient. No 12q24.31 deletion was found in DGDP343’s mother, father or his brother (Fig. 2c) indicating a de novo chromosomal event. Refinement of the deletion breakpoints showed that the proximal breakpoint lies in KDM2B intron 13 (Fig. 2a, d). The real distal breakpoint was found to extend beyond SETD1B, residing between intron 10 and the 5′-UTR of the truncated HPD gene (NM_001171993) (Fig. 2a, d).
Real-time qPCR
The transcript levels of KDM2B, ORAI1, MORN3, TMEM120B, RHOF, and SETD1B were lower in DGDP343 compared to his brother in the same age group (2–4 years) (Fig. 3a–f). A similar reduction was observed in the father, although not to the same extent as the patient (Fig. 3a, e). The transcript levels of KDM2B (Fig. 3g), SETD1B (Fig. 3h) MORN3 (Fig. 3i), RHOF (Fig. 3l) were higher in the brain, and significantly lower in other tissues, including heart, kidney, and liver. Notably, MORN3 transcript levels were more than 100-fold higher in the brain relative to lymphocytes (Fig. 3i). The TMEM120B gene was expressed at low levels in all organs tested including the brain (Fig. 3j). The expression level of ORAI1 was ~85-fold higher in skeletal muscle, but was expressed at low levels in the brain and other tissues (Fig. 3k). A twofold higher expression of RHOF was detected in heart and skeletal muscle compared to lymphocytes (Fig. 3l).

Transcripts levels of genes residing in the 12q24.31 interval. Transcript levels of KDM2B (a), SETD1B (b) and MORN3 (c) TMEM120B (d) ORAI1 (e) RHOF (f) were highest in patient’s brother and lowest as expected in DGDP343. The transcript levels in DGDP343’s were set at 1 because he is only ~3 years younger than the patient. The patient’s father also showed reduced transcript levels for all six genes, although not to the same extent as DGDP343. KDM2B (g), SETD1B (h), MORN3 (i) and RHOF (l) transcript levels were higher in the brain compared to other tissues such as heart, liver and lung. Interestingly, the expression in human brain was highest for SETD1B (h) and MORN3 (i) where ~14-fold and 100-fold higher transcript levels were detected, respectively, relative to lymphocyte. Transcripts of TMEM120B (j) are expressed at very low levels in all tissues including whole human brain. The ORAI1 gene (k) is expressed at high levels in skeletal muscle, while RHOF (l) transcripts were moderately expressed in all tissues assayed
A comprehensive transcript analysis of KDM2B, SETD1B, MORN3, TMEM120B, ORAI1 and RHOF in the various human brain regions revealed that they are expressed at different levels throughout the brain (Fig. 4). Interestingly, the transcript levels of SETD1B (Fig. 4b) and MORN3 (Fig. 4c) in fetal brain were ~250- and 600-fold higher, respectively, relative to lymphocytes. The MORN3 gene is expressed more than 100-fold higher in the cerebral cortex, temporal lobe, insula, parietal lobe and post-central gyrus compared to lymphocytes (Fig. 4c). Moreover, the transcript levels of MORN3 were 200-fold higher in the occipital lobe, and ~50-fold higher in the hippocampus relative to lymphocytes (Fig. 4c). Transcripts of SETD1B were ~50-fold higher in the cerebellum and at least 30-fold higher in the occipital lobe and post-central gyrus compared to lymphocytes (Fig. 4b). While KDM2B transcripts were low in the dorsal root ganglion, spinal cord, and substantia nigra, ~eightfold and sixfold higher expressions were detected in the cerebellum and occipital lobe, compared to lymphocytes (Fig. 4a). The transcript levels of TMEM120B were low in all regions of the brain except in the cerebellum where a ~fivefold higher expression was observed relative to lymphocytes (Fig. 4d). The ORAI1 gene was expressed at low levels in all regions of the brain assayed, but ~fivefold and threefold higher transcript levels were recorded in fetal brain and cerebral cortex (Fig. 4e). An ~tenfold higher expression of RHOF was observed in the cerebellum and fetal brain relative to lymphocytes, while its transcript levels were lower in other brain regions (Fig. 4f).

Expression profile of genes at 12q24.31 in various regions of the human brain. The transcript levels of KDM2B (a) and SETD1B (b) were highest in the cerebellum, occipital lobe and post-central gyrus. b SETD1B is highly expressed in the fetal brain. c MORN3 is expressed abundantly throughout the brain with the highest transcript levels detected in fetal brain and occipital lobe, respectively. d TMEM120B is expressed at low levels in the brain, except in cerebellum where ~fivefold higher expression was observed. e ORAI1 is expressed at a higher level in the cerebral cortex (threefold) and fetal brain (fivefold). Other brain regions show low level of ORAI1 transcripts. f RHOF transcripts were highest in cerebellum (~11-fold) and fetal brain (tenfold) relative to lymphocytes. The lowest transcripts were detected in dorsal root ganglion and spinal cord, respectively
Synteny analysis
Synteny analysis confirmed that the two chromatin modifiers and additional four annotated genes deleted in six human patients were conserved in human chromosome 12q24.31 and zebrafish chromosome 10, except for HPD on zebrafish chromosome 5 (Fig. 2b). KDM2B, ORAI1, MORN3, TMEM120B, RHOF and SETD1B were organized in the same order in both the human and zebrafish.
Whole mount in situ hybridization in zebrafish
Whole mount in situ hybridization analysis showed the expression of seven candidate genes in 48 hpf zebrafish embryos (Fig. 1h–q). KDM2B, SETD1B, TMEM120B, and ORAI1 mRNA transcripts were all detected in the neural tissues during early development of the zebrafish brain (Fig. 1H–N). MORN3 transcripts were moderately expressed in neural tissue (Fig. 1o); however, the other two annotated genes, RHOF and HPD, were not expressed (Fig. 1p, q). The zebrafish HPD homologue showed a tissue-specific expression pattern in the liver (Fig. 1q). These results along with the human results suggest that the regulation of gene expression for KDM2B and SETD1B is well conserved during vertebrate evolution.
Comparative deletion mapping
We compared the size and phenotypes of the eight microdeletion cases at 12q24.31 (Fig. 1g). Our patient DGDP343 turned out to be the most informative due to the microdeletion’s smallest size and the patient’s detailed phenotypes. The proximal end of the microdeletion in DGDP343 overlapped with the distal end of the microdeletion reported in Chouery et al., as well as the proximal end of the microdeletion reported in Palumbo et al. (two vertical blue dotted lines in Fig. 1g). The distal end of the genomic region deleted in DGDP343 overlapped with the proximal end of a microdeletion case in Qiao et al. (vertical purple lines). In addition, the microdeletion in DGDP343 was fully contained in four additional microdeletion cases at 12q24.31 including Baple et al., and three DECIPHER cases (Firth et al. 2009). Furthermore, the genomic deletion in DGDP343 almost completely resides within the microdeletion in Palumbo et al.
Discussion
Among the seven genes (KDM2B, ORAI1, MORN3, TMEM120B, RHOF, SETD1B, and HPD) within the refined 12q24.31 microdeletion syndrome region in DGDP343, four genes (KDM2B, SETD1B, TMEM120B, and ORAI1) demonstrated relatively high expression and one (MORN3) showed moderated expression in the zebrafish head region (Fig. 1h–q). Moreover, synteny analysis revealed that six genes found within a 445 kb genomic region at 12q24.31 are also conserved in the zebrafish chromosome 10 region (Fig. 2b). Zebrafish hpda located on chromosome 5 is not included in the synteny, suggesting a synteny break in this region of zebrafish.
KDM2B and SETD1B are chromatin modifiers, and mutations in chromatin remodeling proteins, such as members of the SWI/SNF complex and transcriptional activators, cause syndromic intellectual disability as seen in Coffin–Siris syndrome (Tsurusaki et al. 2012) and autism (De Rubeis et al. 2014). The high level of expression of kdm2bb observed in zebrafish midbrain, hindbrain, forebrain and retinal neurons suggests that the functional role of kdm2bb may be conserved between zebrafish and humans. Whole mount in situ hybridization also showed expression of setd1ba in the brain region. The SETD1B gene is highly expressed in the human fetal brain (Fig. 4b), as well as in mouse embryos (Bledau et al. 2014).
KDM2B (aka JHDM1B/FBXL10, MIM 609078) is a histone demethylase, demethylating histones H3K36me1/2 (He et al. 2008) and H3K4me3 (Janzer et al. 2012). Apart from a CXXC domain, a PHD domain, and an Fbox domain, it contains a JmjC domain, which is the catalytic core for lysine demethylation (Shi and Whetstine 2007). The JmjC domain is present in a wide range of species including yeast and drosophila, and has important roles in chromatin remodeling and transcription (Klose et al. 2006; Takeuchi et al. 2006). Several genes containing JmjC domains have been associated with neurological disorders. Mutations in KDM5C are responsible for autism, X-linked intellectual disability and seizures (Abidi et al. 2008; Adegbola et al. 2008; Jensen et al. 2005). Similarly, mutations in KDM6A (aka UTX, MIM 300128) were identified in three individuals manifesting Kabuki syndrome characterized by peculiar facies, developmental delay, and mild to moderate intellectual disability (Lederer et al. 2012). The gene PHF8 (MIM 300560) encoding another JmjC domain-containing protein, PHD finger protein 8, has been identified to cause X-linked intellectual disability (Laumonnier et al. 2005; Loenarz et al. 2010). Heterozygous Kdm2b +/− mice show no discernible phenotype (Fukuda et al. 2011). However, homozygous Kdm2b knockout mice display failure of neural tube closure during embryonic neural development due to excessive apoptosis in neuroepithelium and mesenchyme, resulting in exencephaly and death shortly after birth (Boulard et al. 2015; Fukuda et al. 2011). Interestingly, in humans a heterozygous mutation was sufficient to manifest the phenotype, whereas heterozygous KO mice of a specific gene do not show any phenotype as evidenced in the PHF21A gene (Iwase et al. 2006). This could be explained by compensation of the detrimental effect of heterozygous KO by another gene in mice. Alternatively, it could be explained by differential penetrance and expressivity in mice and humans.
Kdm2b is required for normal closure of the neural tube and optic fissure. It has an essential role in neural development, and Kdm2b deficiency causes increased cell proliferation and cell death in neural progenitor cells (Fukuda et al. 2011).
Among the genes residing in the putatively critical region of the 12q24.31 microdeletion, five genes have residual variation intolerance scores (RVIS) (SETD1B: +0.202, ORAI1: +0.105, MORN3: −0.1156, TMEM120B: −0.1138, RHOF: +0.35) higher than average RVIS for a developmental disorder (−0.56) which argues against haploinsufficiency of these genes (Petrovski et al. 2013). KDM2B, however, had the lowest RVIS (−2.17) suggesting that a heterozygous deletion of KDM2B could be pathogenic. Moreover, KDM2B had the highest haploinsufficiency score (HI) (0.302) followed by SETD1B (0.189) (Huang et al. 2010) supporting their pathogenicity in the 12q24.31 microdeletion syndrome.
The SETD1B gene is a subunit of a histone methyltransferase complex, which creates H3K4me3 (Lee et al. 2007). SETD1B is a member of SET domain-containing proteins which use cofactor S-adenosyl-l-methionine (SAM) during methylation of their substrate (Herz et al. 2013). The SET domain is evolutionarily conserved and is made up of a polypeptide of ~130 amino acids in length (Xiao et al. 2003). In mice, the histone methyltransferase Setd1b has been shown to be essential in the differentiation of neural progenitor cells. Ablation of Setd1b leads to severe brain defects and early lethality (Tan et al. 2012). It has also been shown that mono-allelic loss of Setd1b is associated with tumors (Rusiniak et al. 2012).
Recently, Palumbo et al. (2015) reported a female patient with a 1.66 Mb microdeletion at 12q24.31 encompassing 31 genes including KDM2B, SETD1B and microRNA MIR4304. The authors suggested SETD1B and MIR4304 as the two candidate genes possibly contributing to intellectual disability, seizures, and facial dysmorphisms seen in their patient (Fig. 1g) (Palumbo et al. 2015). Previous studies have also shown that histone methyltransferases are involved in a number of neurological disorders. Mutations of SET domain-containing methyltransferase SETD5 (MIM 615743) caused intellectual disability in the 3p25 microdeletion syndrome (Grozeva et al. 2014) and this gene is also linked to autism spectrum disorder (Pinto et al. 2014). Loss of function mutations in EHMT1 (MIM 607005) are causative for Kleefstra syndrome (MIM 610253) characterized by intellectual disability, craniofacial dysmorphisms, brachycephaly, heart defects, and seizures (Kleefstra et al. 2006). Furthermore, rare missense variants in the EHMT1 gene have been found to be associated with autism (Balan et al. 2014). Similarly, disruption of the methyltransferase METTL23 (MIM 615262) is responsible for mild non-syndromic autosomal recessive intellectual disability (Bernkopf et al. 2014).
TMEM120B and ORAI1 showed widespread expression in the zebrafish head (Fig. 1m, n) and in the human brain (http://www.gtexportal.org/home/gene/TMEM120B and http://www.gtexportal.org/home/gene/ORAI1). TMEM120B transcript levels were higher in human cerebellum (Fig. 4d), whereas ORAI1 was highly expressed (85-fold) in human skeletal muscle (Fig. 3k). A ~fivefold and threefold higher expression of ORAI1 was detected in fetal brain and cerebral cortex, respectively (Fig. 4e). Although rhof appears not to be expressed at 48-hpf zebrafish embryos (Fig. 1p), RHOF transcripts were expressed at moderate levels in many regions of the human brain (Fig. 4f). Therefore, we cannot exclude the candidacy of TMEM120B, ORAI1 and RHOF in the neurological phenotypes.
Weak or no expression of hpda in zebrafish indicates that it is unlikely to play an important role in brain development and function (Fig. 1q). Interestingly, hpda was specifically expressed in the liver, suggesting its possible role in liver development and function. While the role of MORN3 is yet to be elucidated in humans, a recent study in mice has shown that Morn3 is abundantly expressed in the testis, and together with Meig1 it plays a role in regulating spermatogenesis (Zhang et al. 2015). The MORN motif consists of 14 highly conserved residues present in many species (Takeshima et al. 2000; Nishi et al. 2000). Interestingly, MORN motif-containing proteins have been shown to be involved in neurological disorders. The JPH1 (MIM 605266) with eight MORN motifs (Nishi et al. 2000) has been shown to act as a genetic modifier gene (Pla-Martin et al. 2015) in GDAP1-associated Charcot–Marie–Tooth disease axonal type 2 k (CMT2K) (MIM 607831), which is characterized by motor impairment and demyelination (Chung et al. 2008; Zimon et al. 2011). Notably, JHP3 (MIM 605268), which also has eight MORN motifs, is specifically expressed in the human brain (Nishi et al. 2000) and is associated with Huntington disease-like 2 (MIM 606438) (Holmes et al. 2001; Todd and Paulson, 2010) defined by progressive decline in cognitive and motor function (Margolis et al. 2001). Furthermore, ALS2 (MIM 606352), another MORN motif-containing protein, is involved in the neurodegenerative disorder amyotrophic lateral sclerosis (MIM 205100). Overall, these findings suggest that MORN3 may have an important role in cognitive development. The higher expression of morn3 in ependymal cells also suggests that it may play a role in brain function in zebrafish (Bruni 1998; Lindsey et al. 2012), but not to the same extent as in humans.
Conspicuously, the transcript levels of all six genes (KDM2B, SETD1B, MORN3, ORAI1, TMEM120B and RHOF) were reduced in DGDP343 compared to his normal brother (Fig. 3a–f). Among all genes assayed, the KDM2B transcript levels in the patient were lowest compared to his two healthy family members. The transcript levels of six genes in the unaffected brother (2 years old) and unaffected father (41 years old) are significantly different (Fig. 3a–f). More reduction of six transcript levels seen in the healthy father might be age related or due to inter-personal variation. The two chromatin modifiers (KDM2B and SETD1B) were expressed in various parts of the human brain, with the highest transcript levels detected in the cerebellum, occipital lobe, and post-central gyrus (Fig. 4a, b). Interestingly, MORN3 transcripts were expressed at high levels in most regions of the brain including cerebral cortex and occipital lobe (Fig. 4c). In addition, SETD1B and MORN3 transcript levels were exceptionally high in the fetal brain aged 20–33 weeks indicating that they may play an important role in early brain development. The cerebellum contributes not only to motor coordination and sensorimotor integration, (Allen and Courchesne 1998; Miall et al. 2001; Proville et al. 2014; Reeber et al. 2013) but also to cognitive processing and emotional control (Schmahmann and Caplan 2006). The severe delay in gross motor skills, autistic behavior and language delay observed in DGDP343 are consistent with a possible dysregulation in the expression of both chromatin modifiers and MORN3 in the cerebellum as a consequence of the genomic deletion at 12q24.31. Furthermore, the occipital lobe is a brain region that can give rise to epilepsy often accompanied by visual hallucinations (Pfaender et al. 2004). Epilepsy was a major clinical feature of our patient, and he also experienced hallucinations. It is interesting to note that SETD1B and MORN3 are expressed at ~tenfold and 50-fold higher levels relative to lymphocytes, respectively, in the hippocampus, a region known to be important in learning and memory (Bliss and Collingridge 1993).
Autism, intellectual disability, epilepsy, and craniofacial anomalies constitute the principal clinical features seen in the 12q24.31 microdeletion syndrome (Table 2). These phenotypes often occur singularly or together in various combinations, sharing sometimes the same genetic etiology (Gecz et al. 2006). For example, mutations of NRXN1cause either autism (Kim et al. 2008), intellectual disability (Zweier et al. 2009), epilepsy (Moller et al. 2013), or craniofacial anomalies (Ching et al. 2010). Furthermore, a regulatory element of a specific gene located outside of the microdeletion might be dysregulated, exerting a position effect (Zweier et al. 2010; Kim et al. 2010; Kleinjan and van Heyningen 2005). Alternatively, parental imprinting at 12q24.31 might be involved (Chouery et al. 2013), which will complicate the comparative deletion mapping. In this case, a 1 Mb interstitial deletion was inherited from a father to his daughter. Although the 2.3-year-old daughter is affected with developmental delay, infantile spasms, hypotonia, and craniofacial anomalies, her father with the same deletion had only insulin-dependent diabetes mellitus. The clinical features in the girl are likely caused by the absence of paternally expressed and maternally silenced genes at 12q24.31 or alternatively, the lack of phenotypes in the father can be explained by incomplete penetrance. Given that both genes KDM2B and HPD, with the same transcription direction, are truncated in our microdeletion patient, it is theoretically possible that a fusion gene using the promoter of HPD could be generated. The possibility that this aberrant gene with a dominant-negative function is a cause of the principal phenotypes can be excluded, because of the overlapping phenotypes seen across many patients sharing the deletion region (Table 2). Although we assumed that a single gene contributes to a constellation of core phenotypes in 12q24.31 microdeletion syndrome, alternatively, 1) the main phenotypes, especially craniofacial anomalies, could be caused by an epistatic effect of dosage imbalance of multiple genes within the overlapping deletion region as seen in 17p13.1 microdeletion syndrome (Carvalho et al. 2014); or 2) a single gene contributes to a partial component of the syndromic phenotype. Therefore, it is difficult to establish a genotype/phenotype relationship and thus, all six genes (KDM2B, ORAI1, MORN3, TMEM120B, RHOF, SETD1B), microRNA MIR548AQ, and non-coding RNA LINC01089 deleted in our DGDP343 patient are potential candidates for neurological and craniofacial phenotypes. Point mutations in single genes would be invaluable in understanding the individual contributions of each gene in the clinical phenotypes in 12q24.31microdeletion syndrome.
References
Abidi FE, Holloway L, Moore CA, Weaver DD, Simensen RJ, Stevenson RE, Rogers RC, Schwartz CE (2008) Mutations in JARID1C are associated with X-linked mental retardation, short stature and hyperreflexia. J Med Genet 45:787–793
Adegbola A, Gao H, Sommer S, Browning M (2008) A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD). Am J Med Genet A 146A:505–511
Allen G, Courchesne E (1998) The cerebellum and non-motor function: clinical implications. Mol Psychiatry 3:207–210
Balan S, Iwayama Y, Maekawa M, Toyota T, Ohnishi T, Toyoshima M, Shimamoto C, Esaki K, Yamada K, Iwata Y, Suzuki K, Ide M, Ota M, Fukuchi S, Tsujii M, Mori N, Shinkai Y, Yoshikawa T (2014) Exon resequencing of H3K9 methyltransferase complex genes, EHMT1, EHTM2 and WIZ, in Japanese autism subjects. Mol Autism 5:49
Baple E, Palmer R, Hennekam RC (2010) A microdeletion at 12q24.31 can mimic Beckwith-Wiedemann syndrome neonatally. Mol Syndromol 1:42–45
Bernkopf M, Webersinke G, Tongsook C, Koyani CN, Rafiq MA, Ayaz M, Muller D, Enzinger C, Aslam M, Naeem F, Schmidt K, Gruber K, Speicher MR, Malle E, Macheroux P, Ayub M, Vincent JB, Windpassinger C, Duba HC (2014) Disruption of the methyltransferase-like 23 gene METTL23 causes mild autosomal recessive intellectual disability. Hum Mol Genet 23:4015–4023
Bledau AS, Schmidt K, Neumann K, Hill U, Ciotta G, Gupta A, Torres DC, Fu J, Kranz A, Stewart AF, Anastassiadis K (2014) The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 141:1022–1035
Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39
Boulard M, Edwards JR, Bestor TH (2015) FBXL10 protects Polycomb-bound genes from hypermethylation. Nat Genet 47:479–485
Bruni JE (1998) Ependymal development, proliferation, and functions: a review. Microsc Res Tech 41:2–13
Carvalho CM, Vasanth S, Shinawi M, Russell C, Ramocki MB, Brown CW, Graakjaer J, Skytte AB, Vianna-Morgante AM, Krepischi AC, Patel GS, Immken L, Aleck K, Lim C, Cheung SW, Rosenberg C, Katsanis N, Lupski JR (2014) Dosage changes of a segment at 17p13.1 lead to intellectual disability and microcephaly as a result of complex genetic interaction of multiple genes. Am J Hum Genet 95:565–578
Ching MS, Shen Y, Tan WH, Jeste SS, Morrow EM, Chen X, Mukaddes NM, Yoo SY, Hanson E, Hundley R, Austin C, Becker RE, Berry GT, Driscoll K, Engle EC, Friedman S, Gusella JF, Hisama FM, Irons MB, Lafiosca T, LeClair E, Miller DT, Neessen M, Picker JD, Rappaport L, Rooney CM, Sarco DP, Stoler JM, Walsh CA, Wolff RR, Zhang T, Nasir RH, Wu BL (2010) Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet 153B:937–947
Chouery E, Choucair N, Abou Ghoch J, El Sabbagh S, Corbani S, Megarbane A (2013) Report on a patient with a 12q24.31 microdeletion inherited from an insulin-dependent diabetes mellitus father. Mol Syndromol 4:136–142
Chung KW, Kim SM, Sunwoo IN, Cho SY, Hwang SJ, Kim J, Kang SH, Park KD, Choi KG, Choi IS, Choi BO (2008) A novel GDAP1 Q218E mutation in autosomal dominant Charcot-Marie-Tooth disease. J Hum Genet 53:360–364
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita-Laza J, Jimenz Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimaki T, Lin CF, Ma’ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnstrom K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Ruther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Li-San W, Weiss LA, Willsey AJ, Yu TW, Yuen RK, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, Buxbaum JD (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 84:524–533
Frescas D, Guardavaccaro D, Bassermann F, Koyama-Nasu R, Pagano M (2007) JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature 450:309–313
Fukuda T, Tokunaga A, Sakamoto R, Yoshida N (2011) Fbxl10/Kdm2b deficiency accelerates neural progenitor cell death and leads to exencephaly. Mol Cell Neurosci 46:614–624
Fuller CM (2012) Time for TMEM? J Physiol 590:5931–5932
Gecz J, Cloosterman D, Partington M (2006) ARX: a gene for all seasons. Curr Opin Genet Dev 16:308–316
Grozeva D, Carss K, Spasic-Boskovic O, Parker MJ, Archer H, Firth HV, Park SM, Canham N, Holder SE, Wilson M, Hackett A, Field M, Floyd JA, Hurles M, Raymond FL (2014) De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am J Hum Genet 94:618–624
Hager SE, Gregerman RI, Knox WE (1957) p-Hydroxyphenylpyruvate oxidase of liver. J Biol Chem 225:935–947
He J, Kallin EM, Tsukada Y, Zhang Y (2008) The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat Struct Mol Biol 15:1169–1175
Herz HM, Garruss A, Shilatifard A (2013) SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci 38:621–639
Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL (2001) A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29:377–378
Huang N, Lee I, Marcotte EM, Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6:e1001154
Inagaki M, Irie K, Ishizaki H, Tanaka-Okamoto M, Miyoshi J, Takai Y (2006) Role of cell adhesion molecule nectin-3 in spermatid development. Genes Cells 11:1125–1132
Iwase S, Shono N, Honda A, Nakanishi T, Kashiwabara S, Takahashi S, Baba T (2006) A component of BRAF-HDAC complex, BHC80, is required for neonatal survival in mice. FEBS Lett 580:3129–3135
Janzer A, Stamm K, Becker A, Zimmer A, Buettner R, Kirfel J (2012) The H3K4me3 histone demethylase Fbxl10 is a regulator of chemokine expression, cellular morphology, and the metabolome of fibroblasts. J Biol Chem 287:30984–30992
Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns JP, Van Esch H, Kleefstra T, Hamel B, Moraine C, Gecz J, Turner G, Reinhardt R, Kalscheuer VM, Ropers HH, Lenzner S (2005) Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet 76:227–236
Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, Descartes M, Holt L, Braddock S, Troxell R, Kaplan L, Volkmar F, Klin A, Tsatsanis K, Harris DJ, Noens I, Pauls DL, Daly MJ, MacDonald ME, Morton CC, Quade BJ, Gusella JF (2008) Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet 82:199–207
Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, Ha KS, Itokawa Y, Meliciani I, Wenzel W, Lee D, Rosenberger G, Ozata M, Bick DP, Sherins RJ, Nagase T, Tekin M, Kim SH, Kim CH, Ropers HH, Gusella JF, Kalscheuer V, Choi CY, Layman LC (2010) WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 87:465–479
Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, Genevieve D, Cormier-Daire V, van Esch H, Fryns JP, Hamel BC, Sistermans EA, de Vries BB, van Bokhoven H (2006) Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet 79:370–377
Kleinjan DA, van Heyningen V (2005) Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 76:8–32
Klose RJ, Kallin EM, Zhang Y (2006) JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 7:715–727
Koyama-Nasu R, David G, Tanese N (2007) The F-box protein Fbl10 is a novel transcriptional repressor of c-Jun. Nat Cell Biol 9:1074–1080
Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, Schwartz CE, Lespinasse J, Van Esch H, Lacombe D, Goizet C, Phan-Dinh Tuy F, van Bokhoven H, Fryns JP, Chelly J, Ropers HH, Moraine C, Hamel BC, Briault S (2005) Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J Med Genet 42:780–786
Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B, Verellen-Dumoulin C (2012) Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet 90:119–124
Lee JH, Tate CM, You JS, Skalnik DG (2007) Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem 282:13419–13428
Lindsey BW, Darabie A, Tropepe V (2012) The cellular composition of neurogenic periventricular zones in the adult zebrafish forebrain. J Comp Neurol 520:2275–2316
Loenarz C, Ge W, Coleman ML, Rose NR, Cooper CD, Klose RJ, Ratcliffe PJ, Schofield CJ (2010) PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nepsilon-dimethyl lysine demethylase. Hum Mol Genet 19:217–222
Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285:895–898
Margolis RL, O’Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML, Callahan C, Hwang HS, Troncoso JC, Ross CA (2001) A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann Neurol 50:373–380
Miall RC, Reckess GZ, Imamizu H (2001) The cerebellum coordinates eye and hand tracking movements. Nat Neurosci 4:638–644
Moller RS, Weber YG, Klitten LL, Trucks H, Muhle H, Kunz WS, Mefford HC, Franke A, Kautza M, Wolf P, Dennig D, Schreiber S, Ruckert IM, Wichmann HE, Ernst JP, Schurmann C, Grabe HJ, Tommerup N, Stephani U, Lerche H, Hjalgrim H, Helbig I, Sander T (2013) Exon-disrupting deletions of NRXN1 in idiopathic generalized epilepsy. Epilepsia 54:256–264
Nevado J, Mergener R, Palomares-Bralo M, Souza KR, Vallespin E, Mena R, Martinez-Glez V, Mori MA, Santos F, Garcia-Minaur S, Garcia-Santiago F, Mansilla E, Fernandez L, de Torres ML, Riegel M, Lapunzina P (2014) New microdeletion and microduplication syndromes: a comprehensive review. Genet Mol Biol 37:210–219
Nishi M, Mizushima A, Nakagawara K, Takeshima H (2000) Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun 273:920–927
Nishimoto HK, Ha K, Jones JR, Dwivedi A, Cho HM, Layman LC, Kim HG (2014) The historical Coffin-Lowry syndrome family revisited: identification of two novel mutations of RPS6KA3 in three male patients. Am J Med Genet A 164A:2172–2179
Palumbo O, Palumbo P, Delvecchio M, Palladino T, Stallone R, Crisetti M, Zelante L, Carella M (2015) Microdeletion of 12q24.31: report of a girl with intellectual disability, stereotypies, seizures and facial dysmorphisms. Am J Med Genet A 167A:438–444
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB (2013) Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9:e1003709
Pfaender M, D’Souza WJ, Trost N, Litewka L, Paine M, Cook M (2004) Visual disturbances representing occipital lobe epilepsy in patients with cerebral calcifications and coeliac disease: a case series. J Neurol Neurosurg Psychiatry 75:1623–1625
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, Vorstman JA, Thompson A, Regan R, Pilorge M, Pellecchia G, Pagnamenta AT, Oliveira B, Marshall CR, Magalhaes TR, Lowe JK, Howe JL, Griswold AJ, Gilbert J, Duketis E, Dombroski BA, De Jonge MV, Cuccaro M, Crawford EL, Correia CT, Conroy J, Conceicao IC, Chiocchetti AG, Casey JP, Cai G, Cabrol C, Bolshakova N, Bacchelli E, Anney R, Gallinger S, Cotterchio M, Casey G, Zwaigenbaum L, Wittemeyer K, Wing K, Wallace S, van Engeland H, Tryfon A, Thomson S, Soorya L, Roge B, Roberts W, Poustka F, Mouga S, Minshew N, McInnes LA, McGrew SG, Lord C, Leboyer M, Le Couteur AS, Kolevzon A, Jimenez Gonzalez P, Jacob S, Holt R, Guter S, Green J, Green A, Gillberg C, Fernandez BA, Duque F, Delorme R, Dawson G, Chaste P, Cafe C, Brennan S, Bourgeron T, Bolton PF, Bolte S, Bernier R, Baird G, Bailey AJ, Anagnostou E, Almeida J, Wijsman EM, Vieland VJ, Vicente AM, Schellenberg GD, Pericak-Vance M, Paterson AD, Parr JR, Oliveira G, Nurnberger JI, Monaco AP, Maestrini E, Klauck SM, Hakonarson H, Haines JL, Geschwind DH, Freitag CM, Folstein SE, Ennis S et al (2014) Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94:677–694. doi:10.1016/j.ajhg.2014.03.018
Pla-Martin D, Calpena E, Lupo V, Marquez C, Rivas E, Sivera R, Sevilla T, Palau F, Espinos C (2015) Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum Mol Genet 24:213–229
Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443:230–233
Proville RD, Spolidoro M, Guyon N, Dugue GP, Selimi F, Isope P, Popa D, Lena C (2014) Cerebellum involvement in cortical sensorimotor circuits for the control of voluntary movements. Nat Neurosci 17:1233–1239
Qiao Y, Tyson C, Hrynchak M, Lopez-Rangel E, Hildebrand J, Martell S, Fawcett C, Kasmara L, Calli K, Harvard C, Liu X, Holden JJ, Lewis SM, Rajcan-Separovic E (2013) Clinical application of 2.7 M Cytogenetics array for CNV detection in subjects with idiopathic autism and/or intellectual disability. Clin Genet 83:145–154
Reeber SL, Otis TS, Sillitoe RV (2013) New roles for the cerebellum in health and disease. Front Syst Neurosci 7:83
Ronan JL, Wu W, Crabtree GR (2013) From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 14:347–359
Rusiniak ME, Kunnev D, Freeland A, Cady GK, Pruitt SC (2012) Mcm2 deficiency results in short deletions allowing high resolution identification of genes contributing to lymphoblastic lymphoma. Oncogene 31:4034–4044
Schmahmann JD, Caplan D (2006) Cognition, emotion and the cerebellum. Brain 129:290–292
Shi Y, Whetstine JR (2007) Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 25:1–14
Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K (2000) Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 6:11–22
Takeuchi T, Watanabe Y, Takano-Shimizu T, Kondo S (2006) Roles of jumonji and jumonji family genes in chromatin regulation and development. Dev Dyn 235:2449–2459
Tan SL, Nishi M, Ohtsuka T, Matsui T, Takemoto K, Kamio-Miura A, Aburatani H, Shinkai Y, Kageyama R (2012) Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 139:3806–3816
Thisse C, Thisse B, Schilling TF, Postlethwait JH (1993) Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119:1203–1215
Todd PK, Paulson HL (2010) RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol 67:291–300
Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N (2012) Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44:376–378
Xiao B, Wilson JR, Gamblin SJ (2003) SET domains and histone methylation. Curr Opin Struct Biol 13:699–705
Zhang L, Shang XJ, Li HF, Shi YQ, Li W, Teves ME, Wang ZQ, Jiang GF, Song SZ, Zhang ZB (2015) Characterization of membrane occupation and recognition nexus repeat containing 3, meiosis expressed gene 1 binding partner, in mouse male germ cells. Asian J Androl 17:86–93
Zimon M, Baets J, Fabrizi GM, Jaakkola E, Kabzinska D, Pilch J, Schindler AB, Cornblath DR, Fischbeck KH, Auer-Grumbach M, Guelly C, Huber N, De Vriendt E, Timmerman V, Suter U, Hausmanowa-Petrusewicz I, Niemann A, Kochanski A, De Jonghe P, Jordanova A (2011) Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77:540–548
Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MA, Ekici AB, Reis A, Schenck A, Rauch A (2009) CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet 85:655–666
Zweier M, Gregor A, Zweier C, Engels H, Sticht H, Wohlleber E, Bijlsma EK, Holder SE, Zenker M, Rossier E, Grasshoff U, Johnson DS, Robertson L, Firth HV, Ekici AB, Reis A, Rauch A (2010) Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat 31:722–733
Acknowledgments
We would like to thank the patient and family members who kindly consented to participate in our study. We wish to extend our thanks to Paul Browne. We are also thankful to Bronwyn Kerr, Alex Henderson and Joris Andrieux who provided phenotypes for DCDP251274, DCP259210, and DCP272960, respectively. This genetics study was performed following the standards established by the Institutional Review Board (IRB) at AU. S.I. has been collectively supported by NIH: NS089896 and Cooley’s Anemia Foundation. C.H.K. was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future (NRF-2015M3A9A8029261). We also gratefully wish to acknowledge the support of funding provided by Caroline Jones-Carrick and Collin Carrick.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Labonne, J.D.J., Lee, KH., Iwase, S. et al. An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum Genet 135, 757–771 (2016). https://doi.org/10.1007/s00439-016-1668-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1668-4