
Abstract
Proper epigenetic regulation processes are crucial in the normal development of the human brain. An ever-increasing group of neurodevelopmental disorders due to derangements of epigenetic regulation involve both microdeletion and monogenic syndromes. Some of these syndromes have overlapping clinical phenotypes due to haploinsufficiency-sensitive genes involved in microdeletions. It was shown recently that the ZMYND11 gene has important functions in epigenetic regulation as an unconventional transcription co-repressor of highly expressed genes, possibly acting in the repression of cryptic transcription from gene bodies. The aim of our study was to compare the clinical phenotypes of patients with 10p15.3 deletions with the phenotypes of patients with loss-of-function ZMYND11 mutations. The results of our study further confirm that the ZMYND11 gene is the critical gene for the clinical phenotype of 10p15.3 microdeletion involving the terminal ~4 Mb of chromosome 10p. In addition, accumulating clinical data allow for further characterisation of this syndrome, including neurodevelopmental disorder, characteristic dysmorphic features and some other more frequent symptoms, such as behavioural disturbances, hypotonia, seizures, low birth weight, short stature in those older than 10 years of age, genitourinary malformations and recurrent infections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proper epigenetic regulation processes are crucial in the normal development of the human brain. Numerous derangements of major systems for epigenetic gene expression regulation were recognised as causes of various inherited neurodevelopmental disorders, including abnormalities of DNA methylation or binding to methylated DNA (e.g. Angelman, Prader–Willi, Rett and fragile X syndromes), abnormalities of histone modifications (e.g. Rubinstein–Taybi, Sotos, Kleefstra and Coffin–Lowry syndromes) and abnormalities of chromatin-modelling proteins involved in the organisation, composition, localisation and movement of nucleosomes (Millan 2013). It was recently shown that the ZMYND11 gene has important functions in epigenetic regulation as an unconventional transcription co-repressor of highly expressed genes, possibly acting in the repression of cryptic transcription from gene bodies (Wen et al. 2014a, b). The clinical phenotype resulting from the haploinsufficiency of the ZMYND11 gene could be attributed to neurodevelopmental disorders due to derangements of epigenetic regulation.
Terminal 10p15.3 deletion is a clinically recognisable syndrome with invariable neurodevelopmental disorder. More than 25 patients with deletions involving the terminal 4 Mb of chromosome 10p have been described to date, with the largest cohort (19 patients) being presented by DeScipio et al. (2012). Interestingly, the clinical phenotype of these patients having deletions ranging in size from 155 kb to 4 Mb was not substantially different and seemingly unrelated to deletion size. The minimal critical region of the overlap of the deletions pointed to two genes, ZMYND11 and DIP2C, as the main candidate genes determining the clinical phenotype of 10p15.3 microdeletion syndrome. A patient with a de novo mutation in ZMYND11 resulting in a phenotype closely resembling 10p15.3 microdeletion syndrome was recently described (Cobben et al. 2014). Furthermore, six more cases of heterozygous loss-of-function mutations in the ZMYND11 gene were found in a large cohort of 4716 patients with developmental delay/autism (Coe et al. 2014). Of major importance, no mutations in the DIP2C gene were found in this large cohort of patients, while truncating mutations were found in two of 2193 controls. Therefore, ZMYND11 haploinsufficiency was confirmed as a major cause of 10p15.3 microdeletion syndrome.
The aim of our study was to compare the clinical phenotypes of 14 patients with terminal 10p15.3 deletions, involving terminal ~800 kb or ~4 Mb of chromosome 10p, and eight patients with loss-of-function ZMYND11 mutations. Further characterisation of the syndrome due to presumable ZMYND11 haploinsufficiency is also provided.
Materials and methods
Case reports
Patient 1 (patient 9 in Tables 1, 2 and the supplementary table)
This 34-month-old male patient was born at 42 weeks of gestation from the second pregnancy of a healthy Lithuanian non-consanguineous couple, both aged 30 years, and there is one healthy older male sibling in the family. Length at birth was 45 cm (<< 3rd centile) and weight at birth was 2.7 kg (< 3rd centile). Failure to thrive, muscular hypotonia and psychomotor retardation was observed from the first several months. Increase in weight gain occurred with the introduction of solid foods, but his weight was still below the 3rd centile, while his height reached the 10th percentile at the age of 34 months. Lack of spontaneous active movements was noted by the parents from the first several months, and left hemiparesis with spasticity was diagnosed at the age of 5 months. There was global psychomotor delay: independent sitting was achieved at the age of 30 months, and no independent walking was observed at 34 months. The child was examined at the age of 7 months by a pulmonologist due to consistently loud breathing and frequent respiratory infections, and laryngotracheomalacia was diagnosed after bronchoscopic evaluation. A normalising breathing pattern with less frequent respiratory infections was observed with age. Unilateral cryptorchidism was also diagnosed.
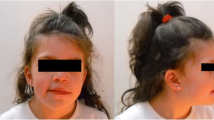
At the age of 34 months, the child actively communicated and was characterised by a quiet but cheerful disposition and frequent laughing. There was global psychomotor retardation with muscular hypotonia and left hemiparesis, and his vocabulary consists of several syllables. Although his arms are the same size, facial asymmetry with a hypotrophic left side and leg length asymmetry, with the left leg being 2 cm shorter than the right one, has been observed. Dysmorphic features include a metopic ridge, high forehead, low-set ears, epicanthus inversus with upslanting palpebral fissures, broad nasal bridge, anteverted nostrils, smooth philtrum, high palate and slightly hypertrophic gums (Fig. 1).

Patient 1 at the age of 9 months (a) and 3 years (b). Note facial asymmetry, metopic ridge, high forehead, low-set ears, epicanthus inversus with upslanting palpebral fissures, broad nasal bridge, anteverted nostrils and smooth philtrum
Slight hyperlactaciduria till 4 mmol/L (n.r. 0.6–2.4 mmol/L) and hyperammonaemia till 111 micromol/L (n.r. < 40 micromol/L), slight increase of liver transaminases (AST till 72 IU/L, n.r. < 40 IU/L; ALT 63 IU/L, n.r. < 40 IU/L) were observed on several occasions. Also, 3-methylglutaconic, 3-methylglutaric and succinic aciduria were found on two occasions in urinary organic acid analysis, pointing to possible derangements of mitochondrial functions. However, the phenotype of the patient was deemed non-compatible with any type of methylglutaconic aciduria.
Patient 2 (patient 10 in Tables 1, 2 and the supplementary table)
This 30-year-old patient (see also Ciuladaite et al. 2014) is the third child of healthy, non-consanguineous parents. There was one female sibling who died in infancy after being diagnosed with congenital heart disease and one healthy female sibling. She was born with breech presentation. Her weight at birth was 2.9 kg (the 3rd centile) and her length at birth was 50 cm (25th centile). The neonatal period was complicated by sepsis and encephalopathy with seizures. Recurrent respiratory infections occurred throughout childhood. Psychomotor retardation was evident from the first months of life, with head lag until 6 months of age, sitting alone from the age of 18 months and walking unaided from the age of 24 months. Talipes was noted from infancy, and scoliosis and a waddling gait due to hip dysplasia developed with age. There was delayed pubertal development, with menarche at 15 years and secondary amenorrhoea afterwards. At the age of 30 years, the patient is obese (weight 90 kg, height 170 cm, BMI 31) and microcephalic (OFC 54 cm). She has a waddling gait and moderately severe scoliosis. Dysmorphic features include arched eyebrows, low-set ears, short philtrum, high and narrow palate, dental crowding and clinodactyly of her 2nd toes (Fig. 2). She has moderate intellectual disability and has been able to talk in short, 2–3-word sentences since the age of 4 years, but incomprehensiveness of speech is noticeable, and although she is able to write some letters, she is not able to read or calculate. Her hearing and sight are normal. Behavioural problems developed with age and included temper tantrums, episodes of mild self-mutilation (biting of palms) and impatience, but autistic traits were not apparent. There was also occasional diurnal and constant nocturnal enuresis.

Pedigrees of patient 1 (a) and patient 2 (b). The father of patient 2 (unavailable for studies) is presumably an inversion carrier (rec(10)dup(10q)inv(10)(p15.3q26.12)
Methods
Karyotyping was performed on metaphase spreads prepared from PHA stimulated cultured peripheral blood lymphocytes using standard methods.
Array CGH for patient 1 and patient 2 was performed by using 180 K and 60 K arrays (Agilent Technologies, Santa Clara, CA, USA), respectively, according to the manufacturer’s recommendations. Image analysis, normalisation and annotation were based on Agilent Feature Extraction 10.7.1. Data were analysed with CytoGenomics 3.0.0.27 software for microarrays (Agilent Technologies). All parameter thresholds were set to default values (segment mean log2 ratio of > 0.5 or − 0.8 were called gain or loss events, respectively). The array data were analysed using the annotation GRCh37/hg19. Fluorescent in situ hybridisation (FISH) analyses of patient 1 and his parents, and patient 2, her sibling and mother were performed by using two probes from chromosome 10 (10pTEL006, D10S2290) (Vysis, Downers Grove, IL, USA). All FISH procedures were done according to the manufacturer’s recommendations. Fluorescent signals were analysed using a Nikon Eclipse 80i fluorescent microscope with CytoVision version 3.6 (Applied Imaging, UK).
Results
G-banded chromosome analysis did not reveal abnormalities in either patient.
Array CGH revealed a 4.3-Mb deletion of 10p15.3pter (patient 1: chr10:102,539–4,440,292; patient 2: chr10:1–4,396,320, NCBI Build GRCh37) and a 13.4-Mb duplication of 10q26.12qter (patient 1: chr10:122,032,434–135,434,178; patient 2: chr10:122,014,670–135,404,523, NCBI build GRCh37). FISH analysis confirmed the array CGH results by showing an unbalanced derivative chromosome 10 in both patient 1 and patient 2: rec(10)dup(10q)inv(10)(p15.3q26.12). The healthy father of patient 1 and healthy female sibling of patient 2 were found to be inversion carriers: ish inv(10)(p15.3)(10pTEL006−,D10S2290+)(q26.3)(D10S2290−,10pTEL006+) (Fig. 2). The paternal inversions detected by FISH proves that the recombinant chromosome originated from a crossing-over between a displaced segment in the patients: rec(10)dup(10q)inv(10)(p15.3q26.12).
Discussion
Copy number variation (CNV) is an important part of human genetic pathology. The pathogenicity of microdeletions is largely determined by genes with inherent sensitiveness to haploinsufficiency. Mutations in these genes usually lead to autosomal dominant syndromes with mostly de novo mutations. Indeed, recent large exome sequencing studies involving from 500 to 2000 patients show that de novo heterozygous mutations are responsible for a large part of genetic human pathology and usually amount to slightly more than 40% of all detected causal variants in exomes (Farwell et al. 2015; Yang et al. 2014). Comparisons of clinical phenotypes arising from microdeletions versus those determined by mutations in genes contained in relevant microdeletions can give important insights into the aetiology of genetic syndromes.
As was noted in two recent publications (Coe et al. 2014; Cobben et al. 2014), comparison of the clinical phenotypes of 10p15.3 microdeletion patients and of those with heterozygous ZMYND11 mutations reveals striking phenotypic similarities, leading to a definition of a ‘syndrome due to presumable ZMYND11 gene haploinsufficiency’. Besides intellectual disability/developmental delay, behavioural disturbances, hypotonia, mild craniofacial dysmorphism, brain anomalies and seizures, as described by DeScipio et al. (2012) and Coe et al. (2014), low birth weight (10/17 patients with birth weight indicated, 59%), microcephaly (5/22 patients, 23%) or macrocephaly (4/22 patients, 18%), and short stature in those older than 10 years of age (3/9 patients, 30%) were observed in a substantial portion of patients in this study to be associated with the syndrome. Also, recurrent infections, laryngomalacia, hemiparesis and movement disorders, including myoclonus, tremor, choreoathetosis and progressive dystonia, were observed in some of them (Tables 1 and 2). Genitourinary malformations, including cryptorchidism, hypospadia and hydronephrosis, were seen in 10p15.3 microdeletion patients only.
Somewhat dysmorphic features of the syndrome may be recognised (Table 2), including prominent forehead/metopic ridge (5/22 patients), hypertelorism/broad or depressed nasal bridge (9/22 patients), upslanting or downslanting palpebral fissures, epicanthus (8/22 patients) and smooth/broad philtrum (6/22). Other features, like facial asymmetry, ptosis, synophrys/arched eyebrows, wide mouth and high palate, were reported less frequently (DeScipio et al. 2012; Ciuladaite et al. 2014; Lindstrand et al. 2010; Vargiami et al. 2014; Cobben et al. 2014; Coe et al. 2014). Although similarity of craniofacial dysmorphic features is more pronounced in patients with smaller terminal 10p15.3 microdeletions (up to 1 Mb from 10pter, ‘group A’ patients according to DeScipio et al. 2012), there is a high variability of facial dysmorphism in some patients having larger microdeletions but showing no dysmorphic features (e.g. patient 11 (5) in Table 2), while other patients with ZMYND11 mutations have more pronounced dysmorphism (e.g. patient 16 in Table 2). Moreover, there is a high variability in head circumference, ranging from microcephaly to macrocephaly. Of note, the phenotype of a patient with 10p15.3 microdeletion not including ZMYND11 is somewhat different (patient 8 in Tables 1 and 2). Phenotypic variability is a feature of multiple genetic syndromes, including those resulting from aberrations in genes for epigenetic regulation (Grillo et al. 2013; Bartsch et al. 2010). Although downstream targets of ZMYND11 are currently largely unknown, genes regulated by ZMYND11 potentially interact with multiple other genes with functions in craniofacial morphogenesis, creating variability in clinical phenotypes.
Intellectual disability/developmental delay was observed as an almost constant feature of the syndrome. Furthermore, it seems that the severity of neurodevelopmental disorder is not different between patients with the 10p15.3 microdeletion involving the terminal ~4 Mb of 10p and patients with loss-of-function heterozygous ZMYND11 mutations. It should be noted, however, that a de novo substitution at codon 239 G>A in the ZMYND11 gene was described in a patient with autism spectrum disorder without intellectual disability (Iossifov et al. 2012). Also, behavioural problems, including autism/autistic traits, attention deficit and hyperactivity disorder (ADHD), aggressiveness/temper tantrums/self-mutilation, bipolar disorder, psychosis, and alcohol and drug abuse (10/17 patients), are observed in the majority of patients. Interestingly, a sociable and affectionate disposition was mentioned in four children from 2 to 9 years old. Hypotonia is a remarkable feature of the syndrome (13/22 patients, 59%) and unaided walking was achieved at the age of 1.5 to 4 years (9 patients). Epilepsy or seizures were observed in five of 22 patients (23%).
There were some limitations to our study. Both our patients had concomitant 10q26.12 duplications, hindering phenotype comparisons. According to available data based on the analysis of publications and the DECIPHER database, terminal 10q duplications involving only the 10q26 chromosomal band are associated with mild skeletal symptoms only, and all cases with definitely pathogenic 10q terminal duplications involve bands 10q25 and/or 10q24 (supplementary table). Both duplications include 79 RefSeq genes, four of them are associated with autosomal dominant diseases (FGFR2, HTRA1, EBF3, WDR11). Recently, point mutations of the EBF3 gene were identified in several patients with neurodevelopmental disorder (Blackburn et al. 2017; Chao et al. 2017; Harms et al. 2017; Sleven et al. 2017). Although these patients have some overlapping features, including intellectual disability, muscular hypotonia, seizures, behavioural problems and urogenital anomalies, there are currently no data indicating that whole-gene duplications of the EBF3 gene could be associated with this phenotype. The phenotypes of other autosomal dominant diseases are not compatible with the phenotypes of our patients.
According to available data, ZMYND11 functions as an unconventional transcription co-repressor of these highly expressed genes, possibly acting in the repression of cryptic transcription from gene bodies (Wen et al. 2014a). Since a knockdown of ZMYND11 was associated with only moderate changes in gene expression, it is also proposed that this protein acts in fine-tuning gene expression (Wen et al. 2014b). An ever-increasing group of neurodevelopmental disorders caused by derangements of epigenetic regulation involve both microdeletion and monogenic syndromes. Some of these syndromes have overlapping clinical phenotypes due to haploinsufficiency-sensitive genes involved in microdeletions, with Kleefstra syndrome being caused by both 9q34.3 deletion and EHMT1 gene mutation, and Angelman syndrome being caused by both maternal 15q11–13 deletion and UBE3A gene mutation, among others. These findings point to a possible inherent sensitivity to the haploinsufficiency of some genes involved in epigenetic regulation (Millan 2013).
In conclusion, comparison of the clinical phenotypes of 14 patients with 10p15.3 microdeletion involving the terminal ~4 Mb of 10p and eight patients with loss-of-function ZMYND11 mutations allowed us to further confirm an earlier established concept that the ZMYND11 gene is the critical gene for the 10p15.3 microdeletion syndrome involving the terminal ~4 Mb of 10p. Accumulating clinical data also allow for further characterisation of a syndrome that includes neurodevelopmental disorder, characteristic dysmorphic features and other clinical signs and symptoms, and is due to presumable ZMYND11 haploinsufficiency. This syndrome could be included into an ever-increasing group of neurodevelopmental disorders caused by derangements of epigenetic regulation.
References
Bartsch O, Kress W, Kempf O, Lechno S, Haaf T, Zechner U (2010) Inheritance and variable expression in Rubinstein–Taybi syndrome. Am J Med Genet A 152A(9):2254–2261
Blackburn PR, Barnett SS, Zimmermann MT, Cousin MA, Kaiwar C, Pinto E Vairo F, Niu Z, Ferber MJ, Urrutia RA, Selcen D, Klee EW, Pichurin PN (2017) Novel de novo variant in EBF3 is likely to impact DNA binding in a patient with a neurodevelopmental disorder and expanded phenotypes: patient report, in silico functional assessment, and review of published cases. Cold Spring Harb Mol Case Stud 3(3):a001743
Chao HT, Davids M, Burke E, Pappas JG, Rosenfeld JA, McCarty AJ, Davis T, Wolfe L, Toro C, Tifft C, Xia F, Stong N, Johnson TK, Warr CG; Undiagnosed Diseases Network, Yamamoto S, Adams DR, Markello TC, Gahl WA, Bellen HJ, Wangler MF, Malicdan MC (2017) A syndromic neurodevelopmental disorder caused by de novo variants in EBF3. Am J Hum Genet 100(1):128–137
Ciuladaite Z, Preiksaitiene E, Utkus A, Kučinskas V (2014) Relatives with opposite chromosome constitutions, rec(10)dup(10p)inv(10)(p15.1q26.12) and rec(10)dup(10q)inv(10)(p15.1q26.12), due to a familial pericentric inversion. Cytogenet Genome Res 144(2):109–113
Cobben JM, Weiss MM, van Dijk FS, De Reuver R, de Kruiff C, Pondaag W, Hennekam RC, Yntema HG (2014) A de novo mutation in ZMYND11, a candidate gene for 10p15.3 deletion syndrome, is associated with syndromic intellectual disability. Eur J Med Genet 57(11–12):636–638
Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, Schuurs-Hoeijmakers JH, Hoischen A, Pfundt R, Krumm N, Carvill GL, Li D, Amaral D, Brown N, Lockhart PJ, Scheffer IE, Alberti A, Shaw M, Pettinato R, Tervo R, de Leeuw N, Reijnders MR, Torchia BS, Peeters H, O’Roak BJ, Fichera M, Hehir-Kwa JY, Shendure J, Mefford HC, Haan E, Gécz J, de Vries BB, Romano C, Eichler EE (2014) Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46(10):1063–1071
DeScipio C, Conlin L, Rosenfeld J, Tepperberg J, Pasion R, Patel A, McDonald MT, Aradhya S, Ho D, Goldstein J, McGuire M, Mulchandani S, Medne L, Rupps R, Serrano AH, Thorland EC, Tsai AC, Hilhorst-Hofstee Y, Ruivenkamp CA, Van Esch H, Addor MC, Martinet D, Mason TB, Clark D, Spinner NB, Krantz ID (2012) Subtelomeric deletion of chromosome 10p15.3: Clinical findings and molecular cytogenetic characterization. Am J Med Genet A 158A(9):2152–2161
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC, Gandomi SK, Lu I, Li X, Lu H, Lu HM, Salvador D, Ruble D, Lao M, Fischbach S, Wen J, Lee S, Elliott A, Dunlop CL, Tang S (2015) Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 17(7):578–586
Grillo E, Lo Rizzo C, Bianciardi L, Bizzarri V, Baldassarri M, Spiga O, Furini S, De Felice C, Signorini C, Leoncini S, Pecorelli A, Ciccoli L, Mencarelli MA, Hayek J, Meloni I, Ariani F, Mari F, Renieri A (2013) Revealing the complexity of a monogenic disease: rett syndrome exome sequencing. PLoS One 8(2):e56599
Harms FL, Girisha KM, Hardigan AA, Kortüm F, Shukla A, Alawi M, Dalal A, Brady L, Tarnopolsky M, Bird LM, Ceulemans S, Bebin M, Bowling KM, Hiatt SM, Lose EJ, Primiano M, Chung WK, Juusola J, Akdemir ZC, Bainbridge M, Charng WL, Drummond-Borg M, Eldomery MK, El-Hattab AW, Saleh MA, Bézieau S, Cogné B, Isidor B, Küry S, Lupski JR, Myers RM, Cooper GM, Kutsche K (2017) Mutations in EBF3 disturb transcriptional profiles and cause intellectual disability, ataxia, and facial dysmorphism. Am J Hum Genet 100(1):117–127
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M (2012) De novo gene disruptions in children on the autistic spectrum. Neuron 74(2):285–299
Lindstrand A, Malmgren H, Verri A, Benetti E, Eriksson M, Nordgren A, Anderlid BM, Golovleva I, Schoumans J, Blennow E (2010) Molecular and clinical characterization of patients with overlapping 10p deletions. Am J Med Genet A 152A(5):1233–1243
Millan MJ (2013) An epigenetic framework for neurodevelopmental disorders: from pathogenesis to potential therapy. Neuropharmacology 68:2–82
Sleven H, Welsh SJ, Yu J, Churchill ME, Wright CF, Henderson A, Horvath R, Rankin J, Vogt J, Magee A, McConnell V, Green A, King MD, Cox H, Armstrong L, Lehman A, Nelson TN; Deciphering Developmental Disorders study; CAUSES study, Williams J, Clouston P, Hagman J, Németh AH (2017) De novo mutations in EBF3 cause a neurodevelopmental syndrome. Am J Hum Genet 100(1):138–150
Vargiami E, Ververi A, Kyriazi M, Papathanasiou E, Gioula G, Gerou S, Al-Mutawa H, Kambouris M, Zafeiriou DI (2014) Severe clinical presentation in monozygotic twins with 10p15.3 microdeletion syndrome. Am J Med Genet A 164A(3):764–768
Wen H, Li Y, Li H, Shi X (2014a) ZMYND11: An H3.3-specific reader of H3K36me3. Cell Cycle 13(14):2153–2154
Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, Li B, Barton MC, Li W, Li H, Shi X (2014b) ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508(7495):263–268
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM (2014) Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312(18):1870–1879
Acknowledgements
We are very grateful to our patients and their families for their contribution.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The results of this study were in part obtained from the Lithuanian–Swiss cooperation program to reduce economic and social disparities within the enlarged European Union under project agreement no. CH-3-ŠMM-01/04, the UNIGENE project. The study was approved by the Vilnius Region Bioethics Committee.
Conflict of interest
The authors declare that they have no financial or other conflicts of interest in relation to this research and its publication.
Additional information
Communicated by: Michal Witt
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 29 kb)
Rights and permissions
About this article

Cite this article
Tumiene, B., Čiuladaitė, Ž., Preikšaitienė, E. et al. Phenotype comparison confirms ZMYND11 as a critical gene for 10p15.3 microdeletion syndrome. J Appl Genetics 58, 467–474 (2017). https://doi.org/10.1007/s13353-017-0408-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-017-0408-3