
Abstract
A collection of 1,108 case–parent trios ascertained through an isolated, nonsyndromic cleft lip with or without cleft palate (CL/P) was used to replicate the findings from a genome-wide association study (GWAS) conducted by Beaty et al. (Nat Genet 42:525–529, 2010), where four different genes/regions were identified as influencing risk to CL/P. Tagging SNPs for 33 different genes were genotyped (1,269 SNPs). All four of the genes originally identified as showing genome-wide significance (IRF6, ABCA4 and MAF, plus the 8q24 region) were confirmed in this independent sample of trios (who were primarily of European and Southeast Asian ancestry). In addition, eight genes classified as ‘second tier’ hits in the original study (PAX7, THADA, COL8A1/FILIP1L, DCAF4L2, GADD45G, NTN1, RBFOX3 and FOXE1) showed evidence of linkage and association in this replication sample. Meta-analysis between the original GWAS trios and these replication trios showed PAX7, COL8A1/FILIP1L and NTN1 achieved genome-wide significance. Tests for gene–environment interaction between these 33 genes and maternal smoking found evidence for interaction with two additional genes: GRID2 and ELAVL2 among European mothers (who had a higher rate of smoking than Asian mothers). Formal tests for gene–gene interaction (epistasis) failed to show evidence of statistical interaction in any simple fashion. This study confirms that many different genes influence risk to CL/P.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cleft lip with/without cleft palate (CL/P) is a common and etiologically heterogeneous birth defect where both genes and environmental risk factors are known to influence risk (Dixon et al. 2011; Marazita 2012). CL/P includes cleft lip (CL) and cleft lip and palate (CLP) which share some epidemiologic features, such as a gender ratio skewed toward males and substantial differences in birth prevalence rates across racial groups (with rates generally higher in Asian populations compared to European populations, and lowest in populations of African ancestry). CL/P can occur as a phenotypic feature of several hundred malformation syndromes, including some Mendelian syndromes for which etiologic genes have been identified (e.g., Van der Woude syndrome; see Dixon et al. 2011 for a review). About 70 % of CL/P cases are classified as isolated and nonsyndromic, and it is becoming increasingly clear that multiple genes determine risk to CL/P.
Several genome-wide association studies (GWAS) have been conducted using both case–control (Birnbaum et al. 2009; Grant et al. 2009; Mangold et al. 2010) and case–parent trio study designs (Beaty et al. 2010), and different genes or specific chromosomal regions have shown strong evidence of harboring causal genes for CL/P. As a follow-up study to our case–parent trio GWAS from an international consortium (Beaty et al. 2010), we conducted an independent replication study on 1,108 case–parent trios. We genotyped a custom panel of 1,269 tagging single nucleotide polymorphic (SNP) markers in 33 selected genes/regions showing some evidence of linkage and association.
Methods
Samples
A total of 1,108 case–parent trios were drawn from several ongoing studies of oral clefts (see Table 1), and these trios included parents of various European ancestries (German and Turkish) plus trios from the multi-site EuroCran study. In addition, case–parent trios from the Philippines were included representing parents of Southeast Asian ancestry. Each CL/P case was examined by a health care provider, and cases with other congenital anomalies, recognized syndromic forms of CL/P or major development delay were excluded. Informed consent was obtained from parents of minor children and all affected individuals able to give informed consent directly. DNA was obtained from whole blood, saliva or a mouthwash sample. Questionnaire data on maternal smoking during the first trimester of pregnancy was collected from personal interview of the mother.
Genotyping
A custom SNP panel was developed to include tagging SNPs in and around genes/regions identified in the GWAS as primary or secondary “hits”, plus genes showing evidence in one of the two major racial sub-groups (European and Asian) represented in Beaty et al. (2010). Additional candidate genes were considered based on prior evidence in the literature, and 33 genes were selected for genotyping using the Illumina Golden Gate platform. A custom panel of 1,536 SNPs was selected based on high Illumina design scores, reported minor allele frequencies (MAF) in European and Asian populations, and their potential for tagging the 33 genes/regions of interest. Among these, 267 SNPs had poor genotype qualities and were dropped, leaving 1,269 SNPs. Additional quality control (QC) criteria were used to flag SNPs: (1) four SNPs were flagged for missing genotype calls >1 %; (2) SNPs with MAF <0.005 in European or Filipino parents separately (54 SNPS in Filipino parents and 1 SNP in European parents, where the one SNP flagged in Europeans also had a low MAF in both populations); (3) SNPs were flagged for deviation from Hardy–Weinberg equilibrium (HWE) in the European and Filipino parents separately at p < 1 × 10−5 (3 SNPs in Europeans and 1 SNP in Filipinos and again, this single SNP showed deviation from HWE in both groups). Supplemental Table 1 lists all SNPs in or near 33 genes/regions examined here with the physical distance (on build 36) spanned by markers and the number of SNPs typed and not flagged in each gene.
Checks for biological relationships among family members (parent–offspring and parent–parent) were run, and SNPs showing excess Mendelian errors were excluded. Three trios were discovered to have sample switches between a parent and child, and correcting the pedigree structure resolved inconsistencies. Seven trios were dropped entirely and 17 individual parents were dropped resulting in incomplete trios, which cannot contribute to the analysis.
Statistical methods
The genotypic transmission disequilibrium test (gTDT) was used to test for evidence of linkage and association (i.e., linkage disequilibrium between the observed marker allele and an unobserved causal gene). This gTDT can be formulated as a conditional logistic regression model where the odds ratio of being the observed case (as opposed to the three ‘pseudo-controls’ possible from any given parental mating type) becomes a function of marker genotype. Within the ith trio, this model is written as:
where X i represents the corresponding risk genotype(s) under an additive, dominant or recessive model (for an additive model, heterozygotes for the risk allele are coded as 1 and homozygotes for the risk allele are coded as 2; for a dominant model both heterozygotes and homozygotes for the risk allele are coded as 1; for a recessive model only homozygotes for the risk allele are coded as 1). The association of a marker with CL/P can be assessed using the estimated odds ratio as (\( {\text{OR}}\left( {{\text{CL}}/{\text{P}}} \right) = { \exp }\hat{\beta }_{\text{G}} \)) with 95 %CI calculated from estimated standard errors about \( \hat{\beta }_{\text{G}} \) (\( \hat{\beta }_{\text{G}} \) is an unbiased estimator of the log relative risk; Schaid 1996). The gTDT has an advantage in terms of statistical power over other TDT models and allows for different underlying genetic models (Schaid 1999; Fallin et al. 2002).
This conditional logistic regression model can also be extended to incorporate gene–environment (G×E) interaction (Cordell 2009a) and even gene–gene (G×G) interaction (Cordell 2002, 2009b; Schwender et al. 2012). Under an additive model, the extension to consider G×E interaction requires two regression coefficients, one for the effect of genotype (β G) and one for the interaction term itself (β G×E). The test statistic for G×E interaction alone is a 1 df test of β G×E = 0. These two regression coefficients can then be used to calculate the odds ratio of having a CL/P for unexposed carriers, \( {\text{OR}}\left( {{\text{CL}}/{\text{P}}|{\text{G no E}}} \right) = { \exp }(\hat{\beta }_{\text{G}} ) \)); and the corresponding \( {\text{OR}}\left( {{\text{CL}}/{\text{P}}|{\text{G and E}}} \right) = { \exp }(\hat{\beta }_{\text{G}} + \hat{\beta }_{\text{GxE}} ) \). We used the computationally efficient method of Schwender et al. (2012) implemented in the R package TRIO (version 1.7.0), available at http://cran.r-project.org to estimate these odds ratios with and without exposure.
Schwender et al. (2012) also show this conditional logistic regression approach can also be extended to consider gene–gene (G×G) interaction. Following the approach of Cordell (2002), the conditional logistic regression model was thus extended to compare the observed 2-locus genotype in the case to the remaining 15 “pseudo-controls” created when considering all 16 possible genotypes from any parental mating as a single matched set. A 4 df likelihood ratio test (LRT) for two-way interaction was calculated (Cordell 2009b). To minimize false-positive signals, we only considered markers in unlinked genes because even long-distance linkage disequilibrium (LD) between two SNPs can lead to spurious significance in a test for G×G interaction. To minimize the total number of tests, we limited tests for G×G interaction to genes showing some evidence of a marginal SNP effect.
Because this replication study used an independent sample of case–parent trios and only markers in regions identified from a previous GWAS or some other study were examined, significance levels were adjusted for the number of SNPs used in each individual candidate gene. Furthermore, because the original GWAS sample from an international consortium was composed of two distinct racial groups (European and Asian ancestry groups), we conducted additional stratified analyses on trios of European and Southeast Asian ancestry groups separately to check for racial difference in the statistical evidence from this replication study.
To assess the consistency of evidence in terms of both significance and direction of effect between the original GWAS sample and this replication study, we also conducted a meta-analysis in regions of interest imputing genotypes in the GWAS case–parent trios. Imputed SNPs were generated by the GENEVA Coordinating Center using the IMPUTE2 program (Howie et al. 2009) with ‘pre-phasing’ of haplotypes to improve accuracy. Prior to imputation, the original GWAS data were quality filtered both at the marker level and at the sample level (Laurie et al. 2010). Any GWAS sample where DNA had been generated by whole-genome amplification was excluded. The imputation target was based on the June 2011 release of 1,094 samples from the 1,000 Genomes Project (Durbin 2010). For our analysis, imputed genotypes were assigned to all members of the GWAS trios if the most likely genotype had a probability >90 %, and these were used in the gTDT separately on GWAS trios of European and Asian ancestry (which included some Filipino trios along with trios of East Asian ancestry). The resulting p values were generated via a meta-analysis over the GWAS discovery trios and these replication trios using the METAL program (Willer et al. 2010).
Results
The four genes/regions identified by Beaty et al. (2010) as reaching genome-wide significance included two with peak signals in recognized genes/regions (IRF6 and 8q24) and two genes that were novel at that time (ABC4A and MAFB). All four of these genes/regions yielded significant evidence of linkage and association in this independent sample of 1,108 case–parent trios, although SNPs in MAFB gave less dramatic significance in this replication sample (see Fig. 1). Stratified analysis of the European and Filipino trios separately showed much greater significance in the evidence of linkage and association among European trios (see Supplemental Figure 1). European trios provided highly significant results for many 8q24 markers, while the Filipino trios yielded only marginal significance for these same markers. Of the 53 SNPs in the 8q24 region, the mean gene diversity over all markers (equivalent to heterozygosity for biallelic SNPs) among European parents was higher than seen among Filipino parents (0.43 vs. 0.35).

Evidence for linkage and association from genotypic TDT in 1,108 CL/P case–parent trios for four genes/regions identified as genome-wide significant by Beaty et al. (2010). (note differences in scale of Y-axis for chr. 8q24)
Ludwig et al. (2012) showed several genes approaching but not attaining genome-wide significance in the GWAS (typically called ‘second tier hits’) yielded genome-wide significance when combined in a meta-analysis of the German case–control GWAS (Birnbaum et al. 2009; Mangold et al. 2010) and the case–parent trio GWAS of Beaty et al. (2010). The most significant SNP in PAX7 (rs742071) gave p = 7 × 10−9 with an estimated OR(CL/P) = 1.32 (95 %CI = 1.13–1.54) in the meta-analysis of Ludwig et al. (2012). Several SNPs in PAX7 on chr. 1p36.13 showed significant linkage and association in both European and Filipino trios from the current study (see Fig. 2). This same SNP yielded an estimated odds ratio of 1.43 (95 %CI = 1.24–1.66; p = 1.59 × 10−7) when comparing heterozygotes to the wild-type homozygote under an additive model in the total replication sample (see Table 2). When stratified into European and Filipino groups, these estimated odds ratio were 1.55 (95 %CI = 0.96–2.53; p = 0.07; MAF = 0.04) among Filipino trios and 1.44 (95 %CI = 1.23–1.68; p = 4 × 10−6; MAF = 0.45) among European trios.

Significance of meta-analyses of p values from genotypic TDT on CL/P case–parent trios from the replication study and the original GWAS for three genes identified as second tier hits: NTN1, PAX7 and COL8A1/FILIP1L1. Each of these genes achieved genome-wide significance in this meta-analysis based on called genotypes from imputed genotypic probabilities in the GWAS trios and the observed genotypes in the replication trios (n.b. the scale of the Y-axis varies)
Markers in THADA on chr. 2p21 were also identified as reaching genome-wide significance in the meta-analysis of Ludwig et al. (2012), and SNP rs4372955 was nominally significant in this study (although Bonferroni correction for all unflagged SNPs left only marginal significance at p = 0.06). While the most significant SNP (rs7590268) in THADA from Ludwig et al. (2012) was not significant among either Europeans or Filipinos in this replication sample, several nearby SNPs did achieve significance (including rs4372955, located 26 kb away; MAF = 0.13 among European parents and MAF = 0.01 among Filipino parents). Similarly, the region containing DCAF4L2 on chr. 8q21.3 included one SNP achieving genome-wide significance in Ludwig et al. (2012), and our analysis of 12 SNPs spanning this region also showed evidence of linkage and association, although other SNPs yielded stronger significance. Supplemental information from Ludwig et al. (2012) showed rs1880646 in NTN1 on chr. 17p13 approached genome-wide significance in their meta-analysis, and this same SNP was again significant in our data (p = 1.20 × 10−3 after Bonferroni correction for 54 unflagged SNPs spanning this gene; this SNP had MAF = 0.25 among European parents and MAF = 0.43 among Filipino parents). Table 2 lists eight ‘second tier hits’ from the GWAS showing significance to varying degrees in this independent replication data set, of which four were also identified by Ludwig et al. (2012).
We also conducted a stratified analysis of CL and CLP trios to test for the possibility of heterogeneity between these two anatomically distinct types of oral clefts. Comparing the estimated regression coefficients from these separate models showed no evidence of heterogeneity in SNP effects between CL and CLP trios. The 33 genes examined here, however, did not include the genomic region near SPRY2 on 13q31.1, which yielded evidence of significance only for CLP (not CL) in Ludwig et al. (2012).
Meta-analysis with GWAS trios
We conducted a meta-analysis between European and Filipino case–parent trios using the observed genotypes from this replication study and called genotypes based on imputed probabilities for SNPs in case–parent trios of European and Asian ancestry from the original GWAS of Beaty et al. (2010). Any individual from the original GWAS whose most likely genotype call probability was <90 % was set to missing for this meta-analysis. Figure 2 shows the results for the combined data set for three genes which achieved genome-wide levels of significance: PAX7 on chr. 1p36.13, NTN1 on chr. 17p13 and COL8A1/FILIP1L on chr. 3q12.3). Supplemental Figure 2 shows corresponding results of stratified analysis. SNPs yielding the greatest significance fell in specific regions in and around these three genes. For PAX7, the most significant SNPs were in intron 3; for COL8A1/FILIP1L, the region of signal spanned both genes although almost all SNPs were intronic; for NTN1, intronic SNPs again gave the lowest p values.
G×G interaction
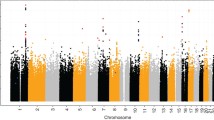
We tested for possible G×G interaction using Cordell’s test implemented in the R package TRIO. All markers in or near 8q24, IRF6, ABCA4, MAFB, PAX7, THADA, COL8A1, DCAF4L2 were used, but no pairs of SNPs within a gene were considered in this test for two-way interaction.
Cordell’s 4 df test was computed for all pairwise combinations of SNPs across genes. Overall, the resulting QQ plot revealed little evidence of G×G interaction (see Fig. 3). Given the large number of tests (173,644), none of these observed test statistics would retain their significance after strict Bonferroni correction (which would require p < 3 × 10−7). While this analysis does not argue for substantial interaction between markers across these genes, Supplemental Table 2 lists the ten most significant SNP pairs showing nominal significance. Interestingly, one SNP in NTN1 (rs11650357) showed evidence of possible G×G interaction with three different SNPs in IRF6, including the most significant individual SNP in this gene (rs6685182).

QQ plots for tests of G×G interaction using 647 SNPs in 12 genes showing some marginal effect of genotype
G×E interaction
Maternal exposure data were available on maternal smoking during pregnancy, and this was used to test for G×E interaction using the Trio package. Because the rates of exposure to maternal smoking differed substantially between European and Filipino trios (18 vs. 7 %, respectively, see Table 3), we conducted a stratified analysis. Two genes showed some evidence of G×Smoking interaction: GRID2 on chr. 4 and ELAVL2 on chr. 9. As expected the stronger evidence came from European trios where exposure rates were higher, and Fig. 4 illustrates the strength of G×Smoking interaction as the −log10(p) for all SNPs in these two genes among 645 European trios (Supplemental Figure 3 shows the corresponding plots for 439 Filipino trios). As apparent in the lower part of this figure, there was little evidence of strong LD across markers in these two genes (where gray indicates effectively no pairwise LD). Evidence for G×Smoking interaction were limited to specific regions of these genes, and only the interaction term itself was significant (i.e., the regression coefficient β G for SNP effects was not different from zero—see Supplemental Tables 3 and 4 for estimated OR, test statistics, p values and MAF for SNPs showing evidence of G×Smoking interaction, marked by the box in Fig. 4). Of course, stratification into European and Filipino groups resulted in some loss of statistical power to detect interaction. Although the median difference in MAF between European and Filipino parents was only 0.09 for the 25 SNPs in GRID2 and 0.13 for the 26 SNPs in ELAVL2, some SNPs showed larger differences. For example, SNP rs4380540 in GRID2 gave the strongest evidence for G×Smoking interaction among Europeans, but the MAF for this SNP among Filipino parents was too low to even permit estimation of β G×E (MAF = 0.144 among European parents vs. 0.003 among Filipino parents). Figure 5 shows the estimated odd ratios (with 95 %CI) for European children carrying the risk allele at SNPs in these two genes in the presence and absence of maternal smoking. Neither GRID2 nor ELAVL2 had shown any significant evidence of SNP effects alone when G×Smoking interaction was ignored.

Significance of genotypic TDT model including a term for G×Smoking interaction in two genes among 654 European CL/P case–parent trios (where the exposure rate for maternal smoking was 18 %). Blocks of SNPs showing evidence of G×Smoking interaction are highlighted in black

Predicted OR(CL/P|unexposed carrier) and OR(CL/P|exposed carrier) in two genes showing evidence of G×Smoking interaction among 645 European CL/P case–parent trios. Open circles represent exposed infants with one risk allele (i.e., those whose mothers smoked) and black dots represent unexposed carrier infants. P values from 1 df test for G×Smoking interaction are shown along the x-axis. a 25 SNPs in GRID2, b 26 SNPs in ELAVL2
Discussion
Although numerous candidate gene and linkage studies have identified a number of genes as likely to play some role in the etiology of CL/P, it has been difficult to achieve consistency across studies. The GWAS approach has rapidly identified and confirmed several additional regions or genes (Mangold et al. 2011). The current replication study confirmed the influence of the four genes/regions (IRF6, ABCA4 and MAFB, plus the 8q24 region) achieving genome-wide significance in our previous case–parent trio GWAS of CL/P (Beaty et al. 2010). Because this replication study included both trios of European and Asian ancestry (here Filipino), we were also able to document differences in the strength of statistical evidence for linkage and association across these two racial groups, where exceptionally strong significance for markers in chr. 8q24 was only seen among Europeans. As discussed by Murray et al. (2012), the marker and haplotype diversity in 8q24 were greater among European parents compared to Asian parents in our original GWAS data which leads to less power among Asian trios, but there was no evidence of any distinct biological mechanism(s). In this replication sample, the level of gene diversity on 8q24 among Filipino parents (Southeast Asian) was again slightly lower than among European parents, but in general the direction of SNP effects on risk was similar (although their statistical significance was much diminished among Filipino trios; see Supplemental Figure 1). This difference in marker diversity could account for some of the inconsistencies across studies from different populations.
Several mutations in IRF6 are causal for Van der Woude syndrome (Leslie et al. 2012a), the most common autosomal-dominant malformation syndrome regularly including CL/P, but multiple studies have shown strong evidence of association between polymorphic markers in IRF6 and isolated, nonsyndromic CL/P cases (Rutledge et al. 2010; Mangold et al. 2011). Trios of European and Southeast Asian ancestry gave evidence that polymorphic markers in IRF6 are linked to and associated with an unobserved causal element for CL/P.
Beaty et al. (2010) was the first study to find evidence of association between markers in ABCA4 and CL/P, a gene known to be causal for inherited retinopathies, although this association has been since confirmed among Brazilians in a case–control study (Fontoura et al. 2012). The CL/P trios used here yielded strong evidence of linkage and association between markers in and between ABCA4 and the neighboring ARHGAP29 gene. ARHGAP29 is expressed in the developing craniofacial region (unlike ABCA4 itself), and Leslie et al. (2012b) identified several rare mutations and variants in ARHGAP29 suggesting this gene may play an etiologic role. The peak signal occurred in an intron of ABCA4 which contains a transcriptional enhancer in the homologous mouse region active in craniofacial tissues during embryonic development (A. Visel, personal communication). Conceivably, the human region may also contain a craniofacial enhancer of the neighboring ARHGAP29 gene, if not ABCA4 itself.
SNPs near MAFB on chr. 20 also showed strong evidence in Beaty et al. (2010), especially among Asian case–parent trios. The exact SNPs reported by Beaty et al. (2010) failed QC in this custom marker panel; however, SNP rs6029326 (at position 38845294) yielded p = 0.0003 in the gTDT, and remained significant after Bonferroni correction for the 93 unflagged SNPs typed around this small gene.
Recently, Ludwig et al. (2012) conducted a meta-analysis using the entire genome-wide marker panel from their cases and unrelated controls, and the corresponding genome-wide markers used in the case–parent trio GWAS of Beaty et al. (2010). This meta-analysis identified a total of 12 genes achieving genome-wide significance in tests of association with CL/P across these GWAS (the four mentioned above plus VAX1, NOG, PAX7, THADA, EPHA3, DCAF4L2, SPRY2 and TPM1). While 204 of the affected cases from German trios used here were also in the case–control GWAS of Mangold et al. (2010) (precluding a meta-analysis over both GWAS groups and these replication trios), these additional independent European and Southeast Asian case–parent trios highlighted the potential importance of 4 additional ‘second tier hits’ in influencing risk to CL/P (COL8A1/FILIP1L on chr. 3p12.3; GADD45G on 9q22.1; RBFOX3 on 17q25.3; and to a lesser extent FOXE1 on 9q22.33). Linkage studies using multiplex CL/P families provided strong evidence for a causal gene near FOXE1 (Marazita et al. 2009; Moreno et al. 2009), although GWAS studies have yielded only limited support for this gene to date and the corrected p value seen here was marginal (see Table 2).
Our formal tests for G×G interaction failed to show compelling evidence for 2-way interaction among SNPs in genes yielding evidence of marginal gene effects, but the number of tests involved is very large so even this replication study of over a thousand case–parent trios may not have sufficient statistical power to detect moderate levels of G×G interaction. This negative finding cannot exclude the possibility of biological interaction between one or more genes in determining the risk of CL/P, and further exploration is warranted.
We did identify two genes (GRID2 and ELAVL2) showing evidence of G×E interaction in the absence of any marginal gene effects among European case–parent trios (where the exposure rate was higher). This finding underscores the need to explore further the effects of genes, both recognized and novel, and their interactions with environmental exposures to the mother to expand our understanding of the genetic etiology of CL/P, which is highly complex and heterogeneous. To summarize, the number of genes contributing to the etiology of CL/P continues to expand (see Table 4).
References
Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin MD, Redett RA, Raymond G, Schwender H, Jin SC, Rose M, Cooper ME, Dunnwald M, Mansilla MA, Leslie E, Bullard S, Lidral A, Moreno LM, Menezes R, Vieira AR, Petrin A, Wilcox A, Lie RT, Jabs EW, Wu-Chou YH, Wang H, Ye X, Huang S, Yeow V, Chong SS, Jee SH, Shi B, Christensen K, Doheny K, Pugh EW, Ling H, Castilla EE, Czeizel AE, Ma L, Field LL, Brody LC, Pangilinan F, Mills JL, Molloy AM, Kirke PN, Scott JM, Arcos-Burgos M, Scott AF (2010) A genome wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet 42:525–529. doi:10.38/ng.580
Birnbaum S, Ludwig KU, Reutter H, Herms S, Steffens M, Rubini M, Baluardo C, Ferrian M, Almeida de Assis N, Alblas MA, Barth S, Freudenberg J, Lauster C, Schmidt G, Scheer M, Braumann B, Bergé SJ, Reich RH, Schiefke F, Hemprich A, Pötzsch S, Steegers-Theunissen RP, Pötzsch B, Moebus S, Horsthemke B, Kramer FJ, Wienker TF, Mossey PA, Propping P, Cichon S, Hoffmann P, Knapp M, Nöthen MM, Mangold E (2009) Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet 41:473–477. doi:10.1038/ng.333
Cordell HJ (2002) Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet 11:2463–2468. doi:10.1093/hmg/11.20.2463
Cordell HJ (2009a) Estimation and testing of gene-environment interactions in family-based association studies. Genomics 93:5–9. doi:org/10.1016/j.ygeno.2008.05.002
Cordell HJ (2009b) Detecting gene–gene interactions that underlie human diseases. Nat Rev 10:392–404. doi:10.1038/nrg2579
Dixon MJ, Marazita ML, Beaty TH, Murray JC (2011) Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet 12:167–178. doi:10.1038/nrg2933
Durbin RM, The 1000 Genomes Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. doi:10.1038/nature09534
Fallin MD, Beaty TH, Liang KY, Chen W (2002) Power comparisons for genotypic vs. allelic TDT methods with > 2 alleles. Genet Epidemiol 23:458–461. doi:10.1002/gepi10192
Fontoura C, Silva RM, Granjeiro JM, Letra A (2012) Further evidence of association of the ABCA4 gene with cleft lip/palate. Eur J Oral Sci 120:553–557. doi:10.1111/eos.12001
Grant SF, Wang K, Zhang H, Glaberson W, Annaiah K, Kim CE, Bradfield JP, Glessner JT, Thomas KA, Garris M, Frackelton EC, Otieno FG, Chiavacci RM, Nah HD, Kirschner RE, Hakonarson H (2009) A genome-wide association study identifies a locus for nonsyndromic cleft lip with or without cleft palate on 8q24. J Pediatr 155:909–913. doi:10.1016/j.jpeds.2009.06.020
Howie B, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529. doi:10.1371/journal.pgen.1000529
Laurie CC, Doheny KF, Mirel DB, Pugh EW, Bierut LJ, Bhangale T, Boehm F, Caporaso NE, Cornelis MF, Edenberg HL, Gabriel SB, Harris EL, Hu FB, Jacobs KB, Kraft P, Landi MT, Lumley T, Manolio TA, McHugh C, Painter I, Paschall J, Rice JP, Rice KM, Zheng X, Weir BS for the GENEVA Investigators (2010) Quality control and quality assurance in genotypic data for genome-wide association studies. Genetic Epidemiol 34:591–602 doi:10.1002/gepi.20516
Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, Dunnwald M, Lidral AC, Marazita ML, Beaty TH, Murray JC (2012a) Mutation screen and expression analysis implicate ARHGAP29 as the etiologic gene for nonsyndromic cleft lip/palate first localized by GWAS. Birth Defects Res A 94:934–942. doi:10.1002/bdra.23076
Leslie EJ, Standley J, Compton J, Bale S, Schutte BC, Murray JC (2012b) Comparative analysis of IRF6 variants in families with Van der Woude syndrome and popliteal pterygium syndrome using public whole-exome databases. Genet Med (epub). doi:10.1038/gim.2012.141
Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, AlChawa T, Nasser E, Boehmer A, Mattheisen M, Alblas MA, Barth S, Kluck N, Lauster C, Braumann B, Reich RH, Hemprich A, Poetzsch S, Blaumeiser B, Daratsianos N, Kreusch T, Murray JC, Marazita ML, Scott AF, Beaty TH, Ruczinski I, Kramer FJ, Wienker TF, Steegers-Theunissen RP, Rubini M, Mossey PA, Hoffmann P, Lange C, Cichon S, Propping P, Knapp M, Noethen MM (2012) First genome wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci including one subtype specific locus. Nat Genet 44:968–971. doi:10.1038/ng.2360
Mangold E, Ludwig KU, Birnbaum S, Baluardo C, Ferrian M, Herms S, Reutter H, de Assis NA, Chawa TA, Mattheisen M, Steffens M, Barth S, Kluck N, Paul A, Becker J, Lauster C, Schmidt G, Braumann B, Scheer M, Reich RH, Hemprich A, Pötzsch S, Blaumeiser B, Moebus S, Krawczak M, Schreiber S, Meitinger T, Wichmann HE, Steegers-Theunissen RP, Kramer FJ, Cichon S, Propping P, Wienker TF, Knapp M, Rubini M, Mossey PA, Hoffmann P, Nöthen MM (2010) Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nature Genet 42:24–26. doi:10.1038/ng.506
Mangold E, Ludwig K, Noethen MM (2011) Breakthroughs in the genetics of orofacial clefting. Trends Molec Med 17:725–733. doi:org/10.1016/j.molmed.2011.07.007
Marazita ML (2012) The evolution of human genetic studies of Cleft Lip and Cleft Palate. Annu Rev Genomics Hum Genet 13:263–283. doi:10.1146/annurev-genom-090711-163729
Marazita ML, Lidral AC, Murray JC, Field LL, Maher BS, McHenry TG, Cooper ME, Govil M, Daack-Hirsch S, Riley B, Jugessur A, Felix T, Moreno L, Mansilla MA, Vieira AR, Doheny K, Pugh E, Valencia-Ramirez C, Arcos-Burgos M (2009) Genome scan, fine-mapping, and candidate gene analysis of non-syndromic cleft lip with or without cleft palate reveals phenotype specific differences in linkage and association results. Hum Hered 68:151–170. doi:10.1159/000224636
Moreno LM, Mansilla MA, Bullard SA, Cooper ME, Busch TD, Machida J, Johnson MK, Brauer D, Krahn K, Daack-Hirsch S, L’heureux J, Valencia-Ramirez C, Rivera D, Lopez AM, Moreno MA, Hing A, Lammer EJ, Jones M, Christensen K, Lie RT, Jugessur A, Wilcox AJ, Chines P, Pugh E, Doheny K, Arcos-Burgos M, Marazita ML, Murray JC, Lidral AC (2009) FOXE1 association with both isolated cleft lip with or without cleft palate and isolated cleft palate. Hum Molec Genet 18:4879–4896. doi:10.1093/hmg/ddp444
Murray T, Taub MA, Ruczinski I, Scott AF, Hetmanski JB, Schwender H, Patel P, Zhang TX, Munger RG, Wilcox AJ, Ye X, Wang H, Wu-Chou YH, Shi B, Jee SH, Chong SS, Yeow V, Murray JC, Marazita ML, Beaty TH (2012) Examining 8q24 to explain differences in evidence of association with cleft lip/palate between Asians and Europeans. Genetic Epidemiol 6:392–399. doi:10.1002/gepi.21633
Rutledge KD, Barger C, Grant JH, Robin NH (2010) IRF6 mutations in mixed isolated familial clefting. Am J Med Genet A 152:2107–2019. doi:10.1002/ajmg.a.33053
Schaid DJ (1996) General score tests for associations of genetic markers with disease using cases and their parents. Genetic Epidemiol 13:423–449
Schaid DJ (1999) Likelihoods and TDT for the case–parents design. Genetic Epidemiol 16:250–260. doi:10.1002/(SICI)1098-2272
Schwender H, Taub MA, Beaty TH, Marazita ML, Ruczinski I (2012) Rapid testing of SNPs and gene-environment interactions in case–parent trio data based on exact analytical parameter estimation. Biometrics 68:766–773. doi:10.1111/j.1541-0420.2011.01713.x
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191. doi:10.1093/bioinformatics/btq340
Acknowledgments
We sincerely thank all families at each recruitment site for participating in this international replication study, and we gratefully acknowledge the invaluable assistance of clinical, field and laboratory staff who made this work possible. Funding to support data collection, genotyping and analysis came from several sources, some to individual investigators (R01-DE-014581 T.H.B.; R01-DE-016148 M.L.M.; P50-DE016215 J.C.M.) and some to the International Cleft Consortium itself (supported by a supplement to U01-DE-018993; T.H. Beaty PI) which was part of the Gene, Environment Association Studies (GENEVA) Consortium. Recruitment of German trios was supported by the Deutsche Forschungsgemeinschaft (FOR 423 and individual grants MA 2546/3-1, KR 1912/7-1, NO 246/6-1, WI 1555/5-1). Genotyping services were provided by the Center for Inherited Disease Research (CIDR) which is funded through a federal contract from the NIH to Johns Hopkins University (contract number HHSN268200782096C).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Beaty, T.H., Taub, M.A., Scott, A.F. et al. Confirming genes influencing risk to cleft lip with/without cleft palate in a case–parent trio study. Hum Genet 132, 771–781 (2013). https://doi.org/10.1007/s00439-013-1283-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-013-1283-6