
Abstract
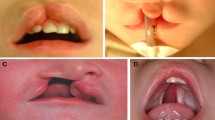
Nonsyndromic orofacial clefts (OFCs) are a heterogeneous group of common craniofacial birth defects with complex etiologies that include genetic and environmental risk factors. OFCs are commonly categorized as cleft lip with or without cleft palate (CL/P) and cleft palate alone (CP), which have historically been analyzed as distinct entities. Genes for both CL/P and CP have been identified via multiple genome-wide linkage and association studies (GWAS); however, altogether, known variants account for a minority of the estimated heritability in risk to these craniofacial birth defects. We performed genome-wide meta-analyses of CL/P, CP, and all OFCs across two large, multiethnic studies. We then performed population-specific meta-analyses in sub-samples of Asian and European ancestry. In addition to observing associations with known variants, we identified a novel genome-wide significant association between SNPs located in an intronic TP63 enhancer and CL/P (p = 1.16 × 10−8). Several novel loci with compelling candidate genes approached genome-wide significance on 4q21.1 (SHROOM3), 12q13.13 (KRT18), and 8p21 (NRG1). In the analysis of all OFCs combined, SNPs near FOXE1 reached genome-wide significance (p = 1.33 × 10−9). Our results support the highly heterogeneous nature of OFCs and illustrate the utility of meta-analysis for discovering new genetic risk factors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nonsyndromic orofacial clefts (OFCs) are among the most common human birth defects, occurring in 1 in 700 live births worldwide (Leslie and Marazita 2013). Nonsyndromic OFCs occur in the absence of other major cognitive or structural abnormalities and have a complex etiology reflecting the combined actions of multiple genetic and environmental risk factors. The focus of much of the OFC genetics research has been on the most common forms: cleft lip with or without cleft palate (CL/P) and cleft palate alone (CP) (Dixon et al. 2011; Leslie and Marazita 2013). Multiple successful genome-wide linkage and association studies have contributed to the substantial progress in identifying potentially causal genes for OFCs over the past 10 years. To date, there have been eight CL/P GWASs (Beaty et al. 2010; Birnbaum et al. 2009; Camargo et al. 2012; Grant et al. 2009; Leslie et al. 2016a; Mangold et al. 2010; Sun et al. 2015; Wolf et al. 2015), a genome-wide meta-analysis of two CL/P GWASs (Ludwig et al. 2012), and two GWASs of CP (Beaty et al. 2011; Leslie et al. 2016b).
Collectively, these studies have demonstrated that OFCs exhibit significant genetic heterogeneity. For CL/P, at least 20 different genetic loci have been identified with compelling statistical and biological support. In contrast, only two GWASs for CP have been published with mixed results. The first study, despite interrogating 400 CP case-parent trios, did not identify any statistically significant SNP main effects (Beaty et al. 2011). The second study identified a single locus associated with CP, but this association signal was limited to European populations because of very low frequencies of the risk allele in other populations (Leslie et al. 2016b; Mangold et al. 2016). For both CL/P and CP, the identified risk loci only account for a modest portion of the genetic variance of OFCs, suggesting that additional genetic risk factors may be involved. CL/P and CP have historically been considered distinct disorders due to the different developmental origins of the lip and palate (Jiang et al. 2006), different prevalence rates among males and females (Mossey and Little 2002), and different proportions of syndromic cases (50% CP vs. 30% for CL/P) (Leslie and Marazita 2013). In the current study, we sought to identify additional genetic risk variants for OFCs, considering the historical groupings of CL/P and CP, but also exploring the possibility of shared etiology. Therefore, we conducted genome-wide meta-analyses for CL/P, CP, and all OFCs, drawing from the two largest CL/P studies published to date and the two published CP studies.
Methods
Contributing GWAS studies
Two consortia contributed to this study (Table 1). The first, hereafter called GENEVA OFC, used a family-based design and included 1604 case-parent trios with CL/P and 475 case-parent trios with CP, respectively, from populations in Europe (Denmark and Norway), the United States, and Asia (Singapore, Taiwan, Philippines, Korea, and China). The specifics of this study were previously described in Beaty et al. (2010, 2011). Briefly, samples were genotyped for 589,945 SNPs on the Illumina Human610-Quadv.1_B BeadChip, genetic data were phased using SHAPEIT, and imputation was performed with IMPUTE2 software to the 1000 Genomes Phase 1 release (June 2011) reference panel. Genotype probabilities were converted to most-likely genotype calls with the GTOOL software (http://www.well.ox.ac.uk/~cfreeman/software/gwas/gtool.html), using a genotype probability threshold of 0.9.
The second consortium included samples contributing to the Pittsburgh Orofacial Cleft (POFC) study, comprising 823 cases and 1319 case-parent trios with CL/P, 78 cases and 165 case-parent trios with CP, plus 1700 unaffected controls. Participants were recruited from 13 countries in North America (United States), Central or South America (Guatemala, Argentina, Colombia, Puerto Rico), Asia (China, Philippines), Europe (Denmark, Turkey, Spain), and Africa (Ethiopia, Nigeria). Additional details on recruitment, genotyping, and quality controls are described in Leslie et al. (2016a, b). Briefly, samples were genotyped for 539,473 SNPs on the Illumina HumanCore + Exome array. Data were phased with SHAPEIT2 and imputed using IMPUTE2 to the 1000 Genomes Phase 3 release (September 2014) reference panel and converted to most-likely genotypes for statistical analysis.
A total of 412 individuals were in both the GENEVA OFC and POFC studies so we excluded these participants from the GENEVA OFC study for this analysis. Informed consent was obtained for all participants and all sites had both local IRB approval and approval at the University of Pittsburgh, the University of Iowa, or Johns Hopkins University.
SNP selection
Quality control procedures were completed in each contributing study and have been described extensively in the original publications (Leslie et al. 2016a, b; Beaty et al. 2010, 2011). In the POFC study, SNPs with minor allele frequencies (MAF) less than 1% or those deviating from Hardy–Weinberg Equilibrium (HWE p < 0.0001) in genetically defined, unrelated European controls were excluded. Similarly, SNPs with MAF <1% or those deviating from HWE were excluded. To account for different marker sets and identifiers between the two imputed datasets, the final analysis included only those overlapping SNPs that were matched on chromosome, nucleotide position, and alleles. A total of 6090,031 SNPs were included in the meta-analysis.
Statistical analysis
We identified three analysis groups from the contributing studies: a case–control subgroup from POFC, an unrelated case-parent trio group from POFC, and an unrelated case-parent trio group from GENEVA OFC. In the case–control subgroup, logistic regression was used to test for association under the additive genetic model while including 18 principal components of ancestry (generated via principal component analysis [PCA] of 67,000 SNPs in low linkage disequilibrium across all ancestry groups) to adjust for population structure (Leslie et al. 2016a). The two case-parent trio subgroups from POFC and GENEVA were analyzed separately using the transmission disequilibrium test (TDT). The resulting effects estimates for the three analysis groups were combined in an inverse variance-weighted fixed-effects meta-analysis. The combined estimate, a weighted log odds ratio, follows a Chi squared distribution with two degrees of freedom under the null hypothesis of no association. Heterogeneity of effects was examined using confidence intervals of the effect estimates. GWAS was performed for all cleft types combined and for the CL/P and CP groups separately.
Subpopulation analyses
Because the contributing studies contained individuals from diverse populations, we also performed stratified analyses of Asian and European ancestry groups defined by PCA (Table 1). We only considered these subpopulations because they were the only ancestry groups represented in both OFC and GENEVA. In these analyses, 5 and 3 principal components of ancestry were included in European and Asian case–control analyses, respectively (Leslie et al. 2016a).
Bioinformatic analysis of top hits
We performed functional annotation enrichment analysis on genes using ToppFun from the ToppGene Suite (Chen et al. 2009) and significance was assessed using Bonferroni adjusted p-values. Enrichment of SNPs in regulatory regions was performed using FORGE v1.2 (Dunham et al. 2014). Individual SNPs were annotated for potential regulatory function using HaploReg v4.1 (Ward and Kellis 2012, 2016).
Results
CL/P
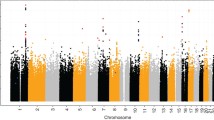
In the CL/P meta-analysis of 823 cases, 1700 controls, and 2811 trios, 1248 SNPs from 13 loci reached genome-wide significance (Table 2; Fig. 1a). Of these, 11 loci have been reported previously, including 8q24, 1q32 (IRF6), and 17p13 (NTN1). The 15q24 (ARID3B) locus, which reached genome-wide significance, was reported as a suggestive signal in the POFC study (Leslie et al. 2016a). We detected a novel association on 3q28 (lead SNP rs76479869, p = 1.16 × 10−8) within the third intron of TP63 (Fig. 2). Six additional loci approached genome-wide significance with p-values less than 5 × 10−7. Three of these loci were suggestive in the contributing studies: 3q21.1 (COL8A1) in the GENEVA OFC study and 5q13.1 (PIK3R1) and 17q21.32 (GOSR2/WNT9B) in the POFC study. The remaining three loci have not been associated with CL/P previously and include 4q21.1 (SHROOM3), 12q13.13 (KRT18), and 8p12 (NRG1) (supplemental Figs. 1–3).

Manhattan plots for genome-wide meta-analyses: a cleft lip with or without cleft palate (CL/P), b cleft palate (CP), c all orofacial clefts (CL/P plus CP). The red line denotes a Bonferroni-corrected genome-wide significant p value (p < 5 × 10−8). Peaks are labeled with the candidate gene or closest gene in the region; colored labels indicate the locus was identified in a previous study, black labels indicate new loci

SNPs in a TP63 enhancer are associated with CL/P. a LocusZoom plot of CL/P meta-analysis results. Points are color-coded based on linkage disequilbrium (r 2) in Europeans. b Annotations for the depicted regions: chromatin state segmentation from ENCODE data in selected cell types, p63 ChIP-Seq and binding motifs from McDade et al. (2012), H3K27Ac and H3K4Meq from ENCODE. c Results from HaploReg analysis of SNPs in high linkage disequilibrium with lead SNP rs76479869. Filled boxes indicate the presence of the annotation
We also stratified our analyses by ancestral group to determine if there were stronger signals in these subgroups. Although we did not detect any new signals in these sub-group analyses, we did identify multiple genome-wide significant signals in each subgroup. In Europeans, we detected five genome-wide significant signals: 1p36 (PAX7), 8q21.3, 8q24, 17q23.2 (TANC2), and 17p13 (NTN1) (supplemental Fig. 4A; supplemental Table 1). In the subset with Asian ancestry, we detected three group-specific signals: 1p22 (ARHGAP29), 1q32 (IRF6), and 17p13 (NTN1) (supplemental Fig. 4B; supplemental Table 2). In addition SNPs on 10q25 (VAX1) and 20q12 (MAFB) approached genome-wide significance. Overall, the stratified CL/P results are in agreement with previous findings (Beaty et al. 2010, 2013; Leslie et al. 2015; Ludwig et al. 2012).
CP
The meta-analysis of CP included a total of 78 cases, 1700 controls, and 616 trios. We observed a single genome-wide significant hit previously identified on 1p36 in GRHL3 (Fig. 1b). The only other hit with a p value less than 1 × 10−5 was on 5p13.2 within UGT3A2 (lead SNP rs604328, p = 5.85 × 10−6; supplemental Fig. 5). In the European subgroup, we identified two suggestive signals (see supplemental Table 1): GRHL3, which we previously identified in Europeans (Leslie et al. 2016b), and a new hit on 11q22.2 (lead SNP rs2260433, p = 8.70 × 10−6; supplemental Fig. 6A). The Asian subgroup was limited to trios from the POFC and GENEVA OFC studies, so we performed a meta-analysis of just these two groups (272 total trios) using a one degree of freedom test. Although there were no genome-wide significant hits, three loci achieved p values <5 × 10−5, and these were driven by the GENEVA OFC trios (supplemental Fig. 6B). Specifically, markers on 8q21.3, 8q24.3 (n.b. this is not the 8q24 peak in Europeans at 8q24.21), and 16p12.1 yielded suggestive evidence (supplemental Table 2; Figs. 7–9).
All OFCs
Historically, CL/P and CP have been analyzed separately. Nevertheless, we hypothesize that there may be genetic risk variants common to both sub-types, and, therefore, analysis of all OFCs (i.e., CL/P plus CP) may yield greater statistical power to identify such shared variants. Therefore, we used the above analytical approach to determine if combined analysis would identify new loci conferring risk to all OFCs. We identified 11 genome-wide significant loci (Fig. 1c). Not surprisingly, all but one of these were genome-wide significant in the CL/P group, which was the largest contributing sample for this analysis. The remaining genome-wide significant signal was on 9q22, immediately downstream of FOXE1 (lead SNP rs12347191, p = 1.33 × 10−9; Fig. 3). This locus was not genome-wide significant in either the CL/P (p = 7.75 × 10−7) or CP analyses (p = 5.42 × 10−4) alone, nor was it significant in either of the contributing studies.

FOXE1 is associated with all OFCs. a LocusZoom plot for OFC meta-analysis results. Points are color coded based on linkage disequilbrium (r 2) in Europeans. Labeled SNPs are denoted by diamonds: lead SNP rs12347191 and rs7850258, a functional SNP from Lidral et al. (2015). b Craniofacial enhancers in the region. In blue, enhancers tested in zebrafish from Lidral et al. (2015) study. In purple, enhances from the VISTA enhancer browser tested in mouse. c Results from HaploReg analysis of SNPs located in craniofacial enhancers. Filled boxes indicate the presence of an annotation
In the all OFCs ancestry-specific subgroups, several previously reported associations were recapitulated. For example, in the European subgroup, the 17p13 (NTN1) and 8q24 loci reached genome-wide significance with 1p36 (PAX7) and 8q21.3 approaching genome-wide significance (supplemental Table 1; Fig. 10A). In contrast, 1p22 (ARHGAP29), 1q32 (IRF6), 10q25 (VAX1), 17p22 (NOG), and 20q12 (MAFB) showed genome-wide significant or suggestive p-values among Asians (supplemental Table 2; Fig. 10B). The strengths of these signals in European and Asian populations are consistent with the results of previous studies. Novel signals with suggestive evidence of association were 6p24.3 (lead SNP rs1333657, p = 8.34 × 10−6; supplemental Fig. 11) in Europeans, and 9q33.3 (lead SNP rs78427461, p = 8.42 × 10−6; supplemental Fig. 12) in Asians.
Bioinformatics analysis of top hits
To further explore the biological relevance of the associated loci, we subjected all genes in proximity to the top SNPs (±100 kb, N = 72 genes) to functional annotation enrichment analysis with ToppFun from the ToppGene Suite (Chen et al. 2009). These loci were enriched for genes expressed in the olfactory pit, olfactory placode, and non-floor plate epithelium identified by RNA-Seq in embryonic mouse tissues (E8.5–E10.5). Of the 17 genes with this expression pattern, several are known OFC risk genes based on recognized Mendelian syndromes in humans and/or mouse models with craniofacial anomalies (e.g., IRF6, TP63, VAX1, and PAX7). However, this analysis can help prioritize candidate genes from associated loci otherwise lacking functional supporting evidence: NRG1, ZFHX4, KRT8, SHTN1, and FILIP1L.
Recognizing that OFC GWAS signals generally occur in non-coding parts of the genome, we also performed a FORGE analysis to look for tissue-specific signals using the Roadmap Epigenomics project data (Dunham et al. 2014). We did not detect any enrichment of signals, which likely reflects the multiple tissue types involved in craniofacial development, and the relative inaccessibility of the key tissue types. However, inspection of individual regions revealed several intriguing findings.
The 3q28 association signal resides within the largest intron of TP63, specifically within an expanse of H3K27Ac and H3K4Me1 histone modifications in NHEK cells (normal human epidermal keratinocytes) (Fig. 2a, b). Recently, Antonini et al. (2015) showed the orthologous region in mouse acts a cis-regulatory element that recapitulates p63 expression and is positively regulated by p63 (Fig. 2b). We annotated all variants in strong linkage disequilibrium with rs76479869 for regulatory functions using HaploReg v4 (Fig. 2c). Among the altered transcription factor motifs were multiple annotations for the Fox family of transcription factors, Pou transcription factors, and CEBPB. Most notable among these is SNP rs55660938 as the risk allele creates a binding site for CEBPB, a protein previously demonstrated to negatively regulate this enhancer (Antonini et al. 2015).
The 9q22 locus near FOXE1 was previously interrogated for craniofacial regulatory elements in zebrafish (Lidral et al. 2015) and mouse (Attanasio et al. 2013) (Fig. 3b). Lidral et al. identified three regulatory elements at −82.4, −67.7, and +22.6 kb from the FOXE1 transcription start site that largely recapitulated endogenous FOXE1 expression in the oral epithelium, heart, and thyroid (Lidral et al. 2015). Independently, three additional elements downstream of FOXE1 showed activity in forebrain and facial mesenchyme of mouse embryos. Because these regulatory elements were already identified, we selected SNPs located within them for bioinformatics analysis (Fig. 3c). Among the annotations, the risk allele at rs925487 destroys a PLAG1 binding site while the risk allele at rs10119853 creates IRF binding sites.
Discussion
We performed meta-analyses of two large GWAS to identify novel loci associated with risk to OFC. We identified new genome-wide significant loci for CL/P (3q28, TP63) and all OFCs (9q22, FOXE1), and recapitulated prior results for multiple loci. Overall, our results are in agreement with previous findings (Beaty et al. 2010; Leslie et al. 2016a, b; Ludwig et al. 2016). Our stratified analyses in Europeans and Asians are consistent with our previous observations that stronger signals are found within the subpopulations with the most statistical power, reflecting the minor allele frequency and information content of SNPs in different populations (Murray et al. 2012).
TP63 is an essential regulator of epidermal morphogenesis. Dominant mutations clustered in TP63 cause six syndromes with overlapping phenotypic features: ectrodactyly-ectodermal dysplasia clefting syndrome (Celli et al. 1999), Hay–Wells syndrome (McGrath et al. 2001), Rapp-Hodgkin syndrome (Kantaputra et al. 2003), split-hand/foot malformation (Ianakiev et al. 2000), limb-mammary syndrome (van Bokhoven and Brunner 2002), and ADULT syndrome (Amiel et al. 2001). Affected individuals are variably affected with ectodermal dysplasia, orofacial clefting, and split-hand/foot malformation, among other features. The phenotypic spectrum may also include nonsyndromic clefts as a de novo mutation was previously reported in an individual with apparently nonsyndromic cleft lip and palate (Leoyklang et al. 2007). Deletion of p63 in mouse results in a similar constellation of defects (Yang et al. 1999).
The complex regulation of TP63 expression occurs in a tissue- and layer-specific manner and depends on two conserved modules within its intronic enhancer (Antonini et al. 2015). These modules positively regulate TP63 when bound by p63 protein but are negatively regulated by Cepba, Cebpb, and Pou3f1 transcription factors. Although there are no polymorphisms in the described p63 binding sites, our bioinformatics analysis showed the risk allele for rs55660938 creates a Cebpb binding site, suggesting misregulation of p63 expression via this enhancer element contributes to development of orofacial clefts.
We identified an association between SNPs on 9q22 and OFCs by combining the CL/P and CP subtypes. This result follows several previous studies on this locus beginning with a genome-wide linkage scan with fine-mapping in CL/P that originally implicated FOXE1 (Marazita et al. 2009), followed by additional fine-mapping in CL/P and CP that narrowed the critical region further to a region near the FOXE1 gene (Moreno et al. 2009). More recently, an independent replication found that two of the top SNPs from previous studies were associated with CL/P, but were more strongly associated when CL/P and CP were analyzed together (Ludwig et al. 2014). Recessive FOXE1 mutations cause Bamforth–Lazarus syndrome, a rare Mendelian disorder characterized by cleft palate and congenital hypothyroidism (Bamforth et al. 1989). Similarly, mice lacking Foxe1 have cleft palate and thyroid dysgenesis (De Felice et al. 1998). Despite the clear connection between OFCs and the FOXE1 gene, this locus has not been identified in any previous GWAS. Ludwig et al. (2014) speculated that this may because the top SNPs from Moreno et al. (2009) are not well-represented on commercial SNP panels and previous candidate studies included only small numbers of SNPs. Therefore, our success may be due to dense genotyping in the region through custom SNP content and imputation of untyped SNPs. In addition, previous studies have not performed GWAS of all OFCs together. There is a growing emphasis on identifying subtype-specific association signals (e.g., CL or CLP), and we and others have contributed to that endeavor (Jia et al. 2015; Ludwig et al. 2012, 2016; Rahimov et al. 2008). However, this study also demonstrates that some signals reflect a shared etiology among the various cleft subtypes that will only be identified when all OFCs are considered together.
There are no common missense polymorphisms in FOXE1 and rare variants identified by sequencing do not account for the association signal, leading to a hypothesis that the functional variants are regulatory. Recently, multiple craniofacial enhancers were identified in a zebrafish screen of multi-species conserved elements or by ChIP-Seq of p300 in mouse craniofacial tissue. In the zebrafish study by Lidral et al. (2015), differential activity was observed for the -67.7 kb element with alleles at rs7850258. The OFC risk allele creates MYC and ARNT binding sites that increase activity of the enhancer (Lidral et al. 2015). In our study, rs7850258 was not among the top SNPs (p = 1.45 × 10−5) and is not in strong linkage disequilibrium with our lead SNP, rs12347191, (r 2 = 0.33, D′ = 0.65 in Europeans, see Fig. 3). We were unable to perform conditional analyses because of the large number of trios contributing to this study, so it remains possible SNP rs7850258 is an independent association signal, which would be consistent with the risk haplotypes described in Moreno et al. (2009). Our bioinformatic analysis of the other craniofacial enhancers identified several motifs altered by OFC risk alleles that are candidates for the molecular validation needed to identify specific functional variants regulating FOXE1. A major contribution of this study is the dense genotyping of this region that could allow comprehensive interrogation of risk alleles for OFCs.
A number of loci approached genome-wide significance in the full CL/P GWAS. Most of these were suggestive in one of the contributing studies, but some were not observed previously, and included biologically relevant genes, SHROOM3 (4q21.1), keratins (12q13.13), and NRG1. SHROOM3 is an actin-binding protein required for neurulation. The 12q13.13 locus contains a cluster of type II keratins, which heteropolymerize to form intermediate filaments in epithelial cells. The GWAS approach has now pointed to several genes involved in the cytoskeleton (Leslie et al. 2012, 2016a). Additional molecular evidence on other OFC-related genes further supports regulation of cytoskeletal dynamics in the pathogenesis of OFCs (Biggs et al. 2014; Caddy et al. 2010; De Groote et al. 2015; Leslie et al. 2012). Similarly SHROOM3 and NRG1 join a list of genes, including GRHL3, IRF6, and TFAP2A, required for neurulation that are also implicated in OFCs (Copp and Greene 2013; Kousa et al. 2013; Wang et al. 2011).
In conclusion, we have performed a multi-ethnic genome-wide meta-analysis of CL/P, CP, and all OFCs combined which revealed two novel, biologically relevant genes, TP63 (for CL/P) and FOXE1 (for all OFCs). Previously reported associations were recapitulated, and several new suggestive loci were implicated. Overall, this study reinforces the notion that OFCs exhibit a high level of genetic heterogeneity and illustrates the utility of combining studies via meta-analysis to yield new discoveries. These findings contribute to our growing understanding of the genetic architecture of OFCs and may one day benefit recurrence prediction and prognosis.
Abbreviations
- OFC:
-
Orofacial cleft
- CL:
-
Cleft lip
- CLP:
-
Cleft lip and palate
- CL/P:
-
Cleft lip with or without cleft palate
- CP:
-
Cleft palate
- TDT:
-
Transmission disequilibrium test
- OR:
-
Odds ratio
- SNP:
-
Single nucleotide polymorphism
- GWAS:
-
Genome-wide association study
- POFC:
-
Pittsburgh orofacial cleft study
- PCA:
-
Principal components analysis
- PC:
-
Principal component
References
Amiel J, Bougeard G, Francannet C, Raclin V, Munnich A, Lyonnet S, Frebourg T (2001) TP63 gene mutation in ADULT syndrome. Eur J Hum Genet 9:642–645. doi:10.1038/sj.ejhg.5200676
Antonini D, Sirico A, Aberdam E, Ambrosio R, Campanile C, Fagoonee S, Altruda F, Aberdam D, Brissette JL, Missero C (2015) A composite enhancer regulates p63 gene expression in epidermal morphogenesis and in keratinocyte differentiation by multiple mechanisms. Nucl Acids Res 43:862–874. doi:10.1093/nar/gku1396
Attanasio C, Nord AS, Zhu Y, Blow MJ, Li Z, Liberton DK, Morrison H, Plajzer-Frick I, Holt A, Hosseini R, Phouanenavong S, Akiyama JA, Shoukry M, Afzal V, Rubin EM, FitzPatrick DR, Ren B, Hallgrimsson B, Pennacchio LA, Visel A (2013) Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342:1241006. doi:10.1126/science.1241006
Bamforth JS, Hughes IA, Lazarus JH, Weaver CM, Harper PS (1989) Congenital hypothyroidism, spiky hair, and cleft palate. J Med Genet 26:49–51
Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin MD, Redett RA, Raymond G, Schwender H, Jin SC, Cooper ME, Dunnwald M, Mansilla MA, Leslie E, Bullard S, Lidral AC, Moreno LM, Menezes R, Vieira AR, Petrin A, Wilcox AJ, Lie RT, Jabs EW, Wu-Chou YH, Chen PK, Wang H, Ye X, Huang S, Yeow V, Chong SS, Jee SH, Shi B, Christensen K, Melbye M, Doheny KF, Pugh EW, Ling H, Castilla EE, Czeizel AE, Ma L, Field LL, Brody L, Pangilinan F, Mills JL, Molloy AM, Kirke PN, Scott JM, Arcos-Burgos M, Scott AF (2010) A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet 42:525–529. doi:10.1038/ng.580
Beaty TH, Ruczinski I, Murray JC, Marazita ML, Munger RG, Hetmanski JB, Murray T, Redett RJ, Fallin MD, Liang KY, Wu T, Patel PJ, Jin SC, Zhang TX, Schwender H, Wu-Chou YH, Chen PK, Chong SS, Cheah F, Yeow V, Ye X, Wang H, Huang S, Jabs EW, Shi B, Wilcox AJ, Lie RT, Jee SH, Christensen K, Doheny KF, Pugh EW, Ling H, Scott AF (2011) Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol 35:469–478. doi:10.1002/gepi.20595
Beaty TH, Taub MA, Scott AF, Murray JC, Marazita ML, Schwender H, Parker MM, Hetmanski JB, Balakrishnan P, Mansilla MA, Mangold E, Ludwig KU, Noethen MM, Rubini M, Elcioglu N, Ruczinski I (2013) Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum Genet 132:771–781. doi:10.1007/s00439-013-1283-6
Biggs LC, Naridze RL, DeMali KA, Lusche DF, Kuhl S, Soll DR, Schutte BC, Dunnwald M (2014) Interferon regulatory factor 6 regulates keratinocyte migration. J Cell Sci 127:2840–2848. doi:10.1242/jcs.139246
Birnbaum S, Ludwig KU, Reutter H, Herms S, Steffens M, Rubini M, Baluardo C, Ferrian M, Almeida de Assis N, Alblas MA, Barth S, Freudenberg J, Lauster C, Schmidt G, Scheer M, Braumann B, Berge SJ, Reich RH, Schiefke F, Hemprich A, Potzsch S, Steegers-Theunissen RP, Potzsch B, Moebus S, Horsthemke B, Kramer FJ, Wienker TF, Mossey PA, Propping P, Cichon S, Hoffmann P, Knapp M, Nothen MM, Mangold E (2009) Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet 41:473–477. doi:10.1038/ng.333
Caddy J, Wilanowski T, Darido C, Dworkin S, Ting SB, Zhao Q, Rank G, Auden A, Srivastava S, Papenfuss TA, Murdoch JN, Humbert PO, Parekh V, Boulos N, Weber T, Zuo J, Cunningham JM, Jane SM (2010) Epidermal wound repair is regulated by the planar cell polarity signaling pathway. Dev Cell 19:138–147. doi:10.1016/j.devcel.2010.06.008
Camargo M, Rivera D, Moreno L, Lidral AC, Harper U, Jones M, Solomon BD, Roessler E, Velez JI, Martinez AF, Chandrasekharappa SC, Arcos-Burgos M (2012) GWAS reveals new recessive loci associated with non-syndromic facial clefting. Eur J Med Genet 55:510–514. doi:10.1016/j.ejmg.2012.06.005
Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, Newbury-Ecob R, Hennekam RC, Van Buggenhout G, van Haeringen A, Woods CG, van Essen AJ, de Waal R, Vriend G, Haber DA, Yang A, McKeon F, Brunner HG, van Bokhoven H (1999) Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99:143–153
Chen J, Bardes EE, Aronow BJ, Jegga AG (2009) ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucl Acids Res 37:W305–W311. doi:10.1093/nar/gkp427
Copp AJ, Greene ND (2013) Neural tube defects—disorders of neurulation and related embryonic processes. Wiley Interdiscip Rev Dev Biol 2:213–227. doi:10.1002/wdev.71
De Felice M, Ovitt C, Biffali E, Rodriguez-Mallon A, Arra C, Anastassiadis K, Macchia PE, Mattei MG, Mariano A, Scholer H, Macchia V, Di Lauro R (1998) A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet 19:395–398. doi:10.1038/1289
De Groote P, Tran HT, Fransen M, Tanghe G, Urwyler C, De Craene B, Leurs K, Gilbert B, Van Imschoot G, De Rycke R, Guerin CJ, Holland P, Berx G, Vandenabeele P, Lippens S, Vleminckx K, Declercq W (2015) A novel RIPK4-IRF6 connection is required to prevent epithelial fusions characteristic for popliteal pterygium syndromes. Cell Death Differ 22:1012–1024. doi:10.1038/cdd.2014.191
Dixon MJ, Marazita ML, Beaty TH, Murray JC (2011) Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet 12:167–178. doi:10.1038/nrg2933
Dunham I, Kulesha E, Iotchkova V, Morganella S, Birney E (2014) FORGE: a tool to discover cell specific enrichments of GWAS associated SNPs in regulatory regions. BioRxiv. doi:10.1101/013045
Grant SF, Wang K, Zhang H, Glaberson W, Annaiah K, Kim CE, Bradfield JP, Glessner JT, Thomas KA, Garris M, Frackelton EC, Otieno FG, Chiavacci RM, Nah HD, Kirschner RE, Hakonarson H (2009) A genome-wide association study identifies a locus for nonsyndromic cleft lip with or without cleft palate on 8q24. J Pediatr 155:909–913. doi:10.1016/j.jpeds.2009.06.020
Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P (2000) Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet 67:59–66. doi:10.1086/302972
Jia Z, Leslie EJ, Cooper ME, Butali A, Standley J, Rigdon J, Suzuki S, Gongorjav A, Shonkhuuz TE, Natsume N, Shi B, Marazita ML, Murray JC (2015) Replication of 13q31.1 association in nonsyndromic cleft lip with cleft palate in Europeans. Am J Med Genet A 167:1054–1060. doi:10.1002/ajmg.a.36912
Jiang R, Bush JO, Lidral AC (2006) Development of the upper lip: morphogenetic and molecular mechanisms. Dev Dyn 235:1152–1166. doi:10.1002/dvdy.20646
Kantaputra PN, Hamada T, Kumchai T, McGrath JA (2003) Heterozygous mutation in the SAM domain of p63 underlies Rapp and Hodgkin ectodermal dysplasia. J Dent Res 82:433–437
Kousa YA, Zhu H, Kinoshita A, Fakhouri W, Dunnwald M, Roushangar RR, Williams TJ, Amendt BA, Chai Y, Finnell RH, Schutte BC (2013) Irf6 homeostasis is required for neurulation through a direct interaction with Tfap2a. In: 63rd Annual Meeting of The American Society of Human Genetics, Boston, MA
Leoyklang P, Suphapeetiporn K, Siriwan P, Desudchit T, Chaowanapanja P, Gahl WA, Shotelersuk V (2007) Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat 28:732–738. doi:10.1002/humu.20515
Leslie EJ, Marazita ML (2013) Genetics of cleft lip and cleft palate. Am J Med Genet C Semin Med Genet 163C:246–258. doi:10.1002/ajmg.c.31381
Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, Dunnwald M, Lidral AC, Marazita ML, Beaty TH, Murray JC (2012) Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res A 94:934–942. doi:10.1002/bdra.23076
Leslie EJ, Taub MA, Liu H, Steinberg KM, Koboldt DC, Zhang Q, Carlson JC, Hetmanski JB, Wang H, Larson DE, Fulton RS, Kousa YA, Fakhouri WD, Naji A, Ruczinski I, Begum F, Parker MM, Busch T, Standley J, Rigdon J, Hecht JT, Scott AF, Wehby GL, Christensen K, Czeizel AE, Deleyiannis FW, Schutte BC, Wilson RK, Cornell RA, Lidral AC, Weinstock GM, Beaty TH, Marazita ML, Murray JC (2015) Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci. Am J Hum Genet. doi:10.1016/j.ajhg.2015.01.004
Leslie EJ, Carlson JC, Shaffer JR, Feingold E, Wehby G, Laurie CA, Jain D, Laurie CC, Doheny KF, McHenry T, Resick J, Sanchez C, Jacobs J, Emanuele B, Vieira AR, Neiswanger K, Lidral AC, Valencia-Ramirez LC, Lopez-Palacio AM, Valencia DR, Arcos-Burgos M, Czeizel AE, Field LL, Padilla CD, Cutiongco-de la Paz EM, Deleyiannis F, Christensen K, Munger RG, Lie RT, Wilcox A, Romitti PA, Castilla EE, Mereb JC, Poletta FA, Orioli IM, Carvalho FM, Hecht JT, Blanton SH, Buxo CJ, Butali A, Mossey PA, Adeyemo WL, James O, Braimah RO, Aregbesola BS, Eshete MA, Abate F, Koruyucu M, Seymen F, Ma L, de Salamanca JE, Weinberg SM, Moreno L, Murray JC, Marazita ML (2016a) A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24.2, 17q23 and 19q13. Hum Mol Genet. doi:10.1093/hmg/ddw104
Leslie EJ, Liu H, Carlson JC, Shaffer JR, Feingold E, Wehby G, Laurie CA, Jain D, Laurie CC, Doheny KF, McHenry T, Resick J, Sanchez C, Jacobs J, Emanuele B, Vieira AR, Neiswanger K, Standley J, Czeizel AE, Deleyiannis F, Christensen K, Munger RG, Lie RT, Wilcox A, Romitti PA, Field LL, Padilla CD, Cutiongco-de la Paz EM, Lidral AC, Valencia-Ramirez LC, Lopez-Palacio AM, Valencia DR, Arcos-Burgos M, Castilla EE, Mereb JC, Poletta FA, Orioli IM, Carvalho FM, Hecht JT, Blanton SH, Buxo CJ, Butali A, Mossey PA, Adeyemo WL, James O, Braimah RO, Aregbesola BS, Eshete MA, Deribew M, Koruyucu M, Seymen F, Ma L, de Salamanca JE, Weinberg SM, Moreno L, Cornell RA, Murray JC, Marazita ML (2016b) A genome-wide association study of nonsyndromic cleft palate identifies an etiologic missense variant in GRHL3. Am J Hum Genet 98:744–754. doi:10.1016/j.ajhg.2016.02.014
Lidral AC, Liu H, Bullard SA, Bonde G, Machida J, Visel A, Uribe LM, Li X, Amendt B, Cornell RA (2015) A single nucleotide polymorphism associated with isolated cleft lip and palate, thyroid cancer and hypothyroidism alters the activity of an oral epithelium and thyroid enhancer near FOXE1. Hum Mol Genet 24:3895–3907. doi:10.1093/hmg/ddv047
Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, AlChawa T, Nasser E, Bohmer AC, Mattheisen M, Alblas MA, Barth S, Kluck N, Lauster C, Braumann B, Reich RH, Hemprich A, Potzsch S, Blaumeiser B, Daratsianos N, Kreusch T, Murray JC, Marazita ML, Ruczinski I, Scott AF, Beaty TH, Kramer FJ, Wienker TF, Steegers-Theunissen RP, Rubini M, Mossey PA, Hoffmann P, Lange C, Cichon S, Propping P, Knapp M, Nothen MM (2012) Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet 44:968–971. doi:10.1038/ng.2360
Ludwig KU, Bohmer AC, Rubini M, Mossey PA, Herms S, Nowak S, Reutter H, Alblas MA, Lippke B, Barth S, Paredes-Zenteno M, Munoz-Jimenez SG, Ortiz-Lopez R, Kreusch T, Hemprich A, Martini M, Braumann B, Jager A, Potzsch B, Molloy A, Peterlin B, Hoffmann P, Nothen MM, Rojas-Martinez A, Knapp M, Steegers-Theunissen RP, Mangold E (2014) Strong association of variants around FOXE1 and orofacial clefting. J Dent Res 93:376–381. doi:10.1177/0022034514523987
Ludwig KU, Ahmed ST, Bohmer AC, Sangani NB, Varghese S, Klamt J, Schuenke H, Gultepe P, Hofmann A, Rubini M, Aldhorae KA, Steegers-Theunissen RP, Rojas-Martinez A, Reiter R, Borck G, Knapp M, Nakatomi M, Graf D, Mangold E, Peters H (2016) Meta-analysis reveals genome-wide significance at 15q13 for nonsyndromic clefting of both the lip and the palate, and functional analyses implicate GREM1 as a plausible causative gene. PLoS Genet 12:e1005914. doi:10.1371/journal.pgen.1005914
Mangold E, Ludwig KU, Birnbaum S, Baluardo C, Ferrian M, Herms S, Reutter H, de Assis NA, Chawa TA, Mattheisen M, Steffens M, Barth S, Kluck N, Paul A, Becker J, Lauster C, Schmidt G, Braumann B, Scheer M, Reich RH, Hemprich A, Potzsch S, Blaumeiser B, Moebus S, Krawczak M, Schreiber S, Meitinger T, Wichmann HE, Steegers-Theunissen RP, Kramer FJ, Cichon S, Propping P, Wienker TF, Knapp M, Rubini M, Mossey PA, Hoffmann P, Nothen MM (2010) Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat Genet 42:24–26. doi:10.1038/ng.506
Mangold E, Bohmer AC, Ishorst N, Hoebel AK, Gultepe P, Schuenke H, Klamt J, Hofmann A, Golz L, Raff R, Tessmann P, Nowak S, Reutter H, Hemprich A, Kreusch T, Kramer FJ, Braumann B, Reich R, Schmidt G, Jager A, Reiter R, Brosch S, Stavusis J, Ishida M, Seselgyte R, Moore GE, Nothen MM, Borck G, Aldhorae KA, Lace B, Stanier P, Knapp M, Ludwig KU (2016) Sequencing the GRHL3 coding region reveals rare truncating mutations and a common susceptibility variant for nonsyndromic cleft palate. Am J Hum Genet 98:755–762. doi:10.1016/j.ajhg.2016.02.013
Marazita ML, Lidral AC, Murray JC, Field LL, Maher BS, McHenry TG, Cooper ME, Govil M, Daack-Hirsch S, Riley B, Jugessur A, Felix T, Morene L, Mansilla MA, Vieira AR, Doheny K, Pugh E, Valencia-Ramirez C, Arcos-Burgos M (2009) Genome scan, fine-mapping, and candidate gene analysis of non-syndromic cleft lip with or without cleft palate reveals phenotype-specific differences in linkage and association results. Hum Hered 68:151–170. doi:10.1159/000224636
McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, Wessagowit V, Kelly A, Atherton DJ, Griffiths WA, Orlow SJ, van Haeringen A, Ausems MG, Yang A, McKeon F, Bamshad MA, Brunner HG, Hamel BC, van Bokhoven H (2001) Hay–Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet 10:221–229
Moreno LM, Mansilla MA, Bullard SA, Cooper ME, Busch TD, Machida J, Johnson MK, Brauer D, Krahn K, Daack-Hirsch S, L’Heureux J, Valencia-Ramirez C, Rivera D, Lopez AM, Moreno MA, Hing A, Lammer EJ, Jones M, Christensen K, Lie RT, Jugessur A, Wilcox AJ, Chines P, Pugh E, Doheny K, Arcos-Burgos M, Marazita ML, Murray JC, Lidral AC (2009) FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum Mol Genet 18:4879–4896. doi:10.1093/hmg/ddp444
Mossey PA, Little J (2002) Epidemiology of oral clefts: an international perspective. In: Wyszynski DF (ed) Cleft lip and palate: from origin to treatment. Oxford University Press, Oxford, pp 127–158
Murray T, Taub MA, Ruczinski I, Scott AF, Hetmanski JB, Schwender H, Patel P, Zhang TX, Munger RG, Wilcox AJ, Ye X, Wang H, Wu T, Wu-Chou YH, Shi B, Jee SH, Chong S, Yeow V, Murray JC, Marazita ML, Beaty TH (2012) Examining markers in 8q24 to explain differences in evidence for association with cleft lip with/without cleft palate between Asians and Europeans. Genet Epidemiol 36:392–399. doi:10.1002/gepi.21633
Rahimov F, Marazita ML, Visel A, Cooper ME, Hitchler MJ, Rubini M, Domann FE, Govil M, Christensen K, Bille C, Melbye M, Jugessur A, Lie RT, Wilcox AJ, Fitzpatrick DR, Green ED, Mossey PA, Little J, Steegers-Theunissen RP, Pennacchio LA, Schutte BC, Murray JC (2008) Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet 40:1341–1347. doi:10.1038/ng.242
Sun Y, Huang Y, Yin A, Pan Y, Wang Y, Wang C, Du Y, Wang M, Lan F, Hu Z, Wang G, Jiang M, Ma J, Zhang X, Ma H, Ma J, Zhang W, Huang Q, Zhou Z, Ma L, Li Y, Jiang H, Xie L, Jiang Y, Shi B, Cheng J, Shen H, Wang L, Yang Y (2015) Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat Commun 6:6414. doi:10.1038/ncomms7414
van Bokhoven H, Brunner HG (2002) Splitting p63. Am J Hum Genet 71:1–13. doi:10.1086/341450
Wang WD, Melville DB, Montero-Balaguer M, Hatzopoulos AK, Knapik EW (2011) Tfap2a and Foxd3 regulate early steps in the development of the neural crest progenitor population. Dev Biol 360:173–185. doi:10.1016/j.ydbio.2011.09.019
Ward LD, Kellis M (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucl Acids Res 40:D930–D934. doi:10.1093/nar/gkr917
Ward LD, Kellis M (2016) HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucl Acids Res 44:D877–D881. doi:10.1093/nar/gkv1340
Wolf ZT, Brand HA, Shaffer JR, Leslie EJ, Arzi B, Willet CE, Cox TC, McHenry T, Narayan N, Feingold E, Wang X, Sliskovic S, Karmi N, Safra N, Sanchez C, Deleyiannis FW, Murray JC, Wade CM, Marazita ML, Bannasch DL (2015) Genome-wide association studies in dogs and humans identify ADAMTS20 as a risk variant for cleft lip and palate. PLoS Genet 11:e1005059. doi:10.1371/journal.pgen.1005059
Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398:714–718. doi:10.1038/19539
Acknowledgements
Many thanks to the participating families world-wide who made this project possible. The tireless efforts by the dedicated field staff and collaborators were similarly essential to the success of this study. This work was supported by grants from the National Institutes of Health (NIH) including: K99-DE025060 [EJL], X01-HG007485 [MLM, EF], R01-DE016148 [MLM, SMW], U01-DE024425 [MLM], R37-DE008559 [JCM, MLM], R01-DE009886 [MLM], R21-DE016930 [MLM], R01-DE014667 [LMM], R01-DE012472 [MLM], R01-DE011931 [JTH], R01-DE011948 [KC], U01-DD000295 [GLW], K99 -DE024571 [CJB], R25-MD007607 [CJB], R01-DE014581 [TB], U01-DE018993 [TB]. Genotyping and data cleaning were provided via an NIH contract to the Johns Hopkins Center for Inherited Disease Research: HHSN268201200008I. Additional support provided by the Robert Wood Johnson Foundation, AMFDP Grant 72429 [AB]; an intramural grant from the Research Institute of the Children’s Hospital of Colorado [FWD]; operating costs support in the Philippines was provided by the Institute of Human Genetics, National Institutes of Health, University of the Philippines, Manila [CP]; grants through FAPERJ, Brazil [IMO]: grant numbers: E-26/102.797/2012, E-26/110.140/2013; grants through CNPq, Brazil [IMO]: grant numbers: 481069/2012-7, 306396/2013-0, 400427/2013-3.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Leslie, E.J., Carlson, J.C., Shaffer, J.R. et al. Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum Genet 136, 275–286 (2017). https://doi.org/10.1007/s00439-016-1754-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1754-7