
Abstract
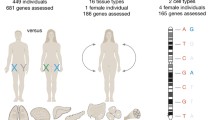
To achieve a balanced gene expression dosage between males (XY) and females (XX), mammals have evolved a compensatory mechanism to randomly inactivate one of the female X chromosomes. Despite this chromosome-wide silencing, a number of genes escape X inactivation: in women about 15% of X-linked genes are bi-allelically expressed and in mice, about 3%. Expression from the inactive X allele varies from a few percent of that from the active allele to near equal expression. While most genes have a stable inactivation pattern, a subset of genes exhibit tissue-specific differences in escape from X inactivation. Escape genes appear to be protected from the repressive chromatin modifications associated with X inactivation. Differences in the identity and distribution of escape genes between species and tissues suggest a role for these genes in the evolution of sex differences in specific phenotypes. The higher expression of escape genes in females than in males implies that they may have female-specific roles and may be responsible for some of the phenotypes observed in X aneuploidy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dosage compensation mechanisms have evolved in mammals because of the divergence of the sex chromosome complement between males (XY) and females (XX). Chromosome-wide transcriptional silencing of one X in females evens gene expression dosage differences between the sexes (Lyon 1961). X inactivation is a chromosome-wide phenomenon and thus the majority of genes on the inactive X are silenced. However, some genes escape X inactivation (escape genes), i.e., remain expressed from both the active and inactive X alleles (Prothero et al. 2009). In this review, we will focus on this particular subset of X-linked genes, in terms of differences between species, molecular mechanisms of escape, sex differences caused by differential expression and roles in disease.
We will first briefly summarize the main features of X inactivation, as this will be extensively discussed in this issue. In mouse, X inactivation is initiated in female embryos by transcription of the long noncoding RNA (ncRNA) Xist from the X destined to become the inactive X (Xi), while Xist is repressed on the active X (Xa) by the antisense Tsix (Brown et al. 1991; Lee et al. 1999). Expression of Xist is regulated by factors that control pluripotency during mouse development and keep Xist repressed prior to X inactivation (Navarro et al. 2008). Additional factors that have been implicated in the initiation of mouse X inactivation comprise the ncRNA Jpx and the E3 ubiquitin ligase RNF12 (Barakat et al. 2011; Tian et al. 2010). X inactivation is initially imprinted in early mouse embryos with the paternal X (Xp) always being silenced (Takagi and Sasaki 1975; West et al. 1977). However, in the blastocyst, the Xp reactivates and random X inactivation takes place. Recent evidence suggests that a bias toward inactivation of the Xp persists in mouse neonatal brain, perhaps due to incomplete erasure of the Xp imprint and/or preferential growth of cells with the active maternal X (Xm) (Wang et al. 2010). In humans and in rabbits, X inactivation also involves increased XIST expression but it occurs later in development, is random and not imprinted (Okamoto et al. 2011). As a result of random X inactivation, somatic tissues in females are mosaic with cells that differ in the parental origin of the X that is inactivated (Migeon 2007) (Fig. 1). Following the onset of X inactivation, a series of epigenetic modifications that implement chromosome silencing and heterochromatin formation takes place (Heard and Disteche 2006). Xist cis-spreading along the X is still not well understood, but specific motifs may be involved. Notably, repeat elements such as interspersed repetitive elements (LINE-1) are critical for the formation of a silenced compartment and AT-rich motifs may also be involved (Agrelo et al. 2009; Chow et al. 2010; Nguyen et al. 2011). Xist recruits the polycomb complex PRC2, which mediates the initial histone changes (e.g., methylation of lysine 27 of histone H3) on the inactive X (Plath et al. 2003). Additional changes include methylation at CpG islands and late replication.

X inactivation and escape patterns. Before differentiation, the paternal X (Xp) and the maternal X (Xm) chromosomes are active. As cells differentiate, random X inactivation is initiated by coating one X chromosome with Xist RNA (pink cloud). This will become the inactive X, while the other X remains active (green chromosome). Since the process is random, either the Xp or the Xm is inactivated in a given cell, resulting in mosaicism in females. Some genes escape X inactivation, i.e., are expressed from both the Xi and the Xa (yellow bars) in all cells. Other genes escape from X inactivation in a subset of cells in a given tissue resulting in mosaicism of escape patterns. An additional layer of variability in escape patterns results from differences between individuals (Carrel and Willard 2005; Yang et al. 2010)
Genes that escape X inactivation are located throughout the X chromosome, but they predominate in the small regions of homology and pairing that persist on the sex chromosomes called the pseudoautosomal regions (PAR). As expected, genes within the PAR are usually not subject to X inactivation since functional, equivalent alleles are present on the X and Y chromosomes in males and on both X alleles in females. Not surprisingly, non-pseudoautosomal genes that retain a Y-linked copy also often escape X inactivation and thus have two expressed alleles in both male and female somatic tissues (Disteche et al. 2002). Nonetheless, a number of escape genes have lost or differentiated their Y-linked copy and would be predicted to have higher expression in females, which may cause phenotypic sex differences (see below). This suggests either that establishment of X inactivation lags behind Y degeneration, or that specific mechanisms exist to maintain expression of a subset of genes from the Xi due to selective advantages (Park et al. 2010).
Escape from X inactivation not only occurs in female soma, but has also been observed in male meiotic cells where meiotic sex chromosome inactivation (MSCI) takes place. In this cell type, X-linked microRNAs (miRNAs) escape silencing by MSCI, but the effects are unknown (Song et al. 2009). In the present review, we will limit our discussion of escape from X inactivation to female somatic cells. We will summarize the progress made in comparing the number and distribution of human and mouse escape genes, examine possible molecular mechanisms involved in facilitating escape from X inactivation and discuss the role of escape genes in sex differences and disease.
Escape from X inactivation in human and mouse
A systematic survey of human genes has shown that about 15% of X-linked genes consistently escape, based on expression analyses in rodent x human hybrid cell lines that retain a human Xi (Carrel and Willard 2005). In contrast to the situation in human, only 3% of mouse genes escape X inactivation as shown by our next-generation RNA-sequencing study to survey allele-specific expression based on SNPs in a somatic cell line derived from a cross between two mouse species (Yang et al. 2010). Thus, X inactivation in mice is more complete than in humans, although additional analyses will be necessary to extend these findings to other cell types and tissues. Measurements of the relative expression of each allele identified by SNPs in human and mouse cells demonstrate that expression from the Xi is significantly lower than from the Xa (Carrel and Willard 2005; Yang et al. 2010). Expression from the Xi ranges from a few percent to equivalent expression to that from the Xa, likely because of partial spreading of silencing in some of the escape domains. While these findings demonstrate a continuum between complete silencing and escape, genes with a minimum expression level from the Xi that represents 10% of that from the Xa are usually considered escape genes.
The distribution of escape genes along the X is not random and differs between human and mouse. In humans, escape genes are clustered (as many as 13 adjacent genes in large domains ranging in size between approximately 100 kb and 7 Mb), whereas in mouse, single escape genes are embedded in regions of silenced chromatin. This suggests that escape from X inactivation in mouse may be controlled at the level of individual genes rather than chromatin domains (Carrel and Willard 2005; Tsuchiya et al. 2004; Yang et al. 2010). Most escape genes are located on the short arm of the human X, likely because this region has most recently diverged from the Y (Carrel and Willard 2005; Lahn and Page 1999; Ross et al. 2005). Another factor may be the centromeric heterochromatin of the human X, which could prevent proper cis-spreading of XIST RNA from its location on the long arm (Disteche 1999; Duthie et al. 1999). In mouse where the centromere is located at one end of the X, spreading of Xist RNA may be facilitated, contributing to fewer escape genes (Yang et al. 2010).
Little is known about the number and distribution of escape genes in other mammalian species. However, some information exists in marsupials, where key features of eutherian X inactivation are absent. Notably, Xist is absent in marsupials (Duret et al. 2006) and the paternal X is always silenced in somatic tissues (Graves 1996). Variable escape from X inactivation has been reported for a number of marsupial genes in a tissue-dependent manner that is incomplete and stochastic, suggesting that X inactivation is more heterogeneous (Al Nadaf et al. 2010; Deakin et al. 2009).
Variable escape
In eutherian mammals, escape from X inactivation can vary between tissues and/or individuals, but this variability appears to affect a minority of genes (Fig. 1). In human, about 10% of X-linked genes show variable escape, as shown by expression in some but not all hybrid cell lines retaining a human Xi (Carrel and Willard 2005). This variability has also been observed at the level of individuals (differences between women), and tissues or individual cells within a tissue (differences within an individual) (Anderson and Brown 1999; Carrel and Willard 1999, 2005). TIMP1 provides an example of a gene with variable escape between women and in different tissues (Anderson and Brown 1999). When analyzed in primary diploid human female cell lines, REP1 is expressed solely from the Xa in some cell lines, but is bi-allelic in others (Carrel and Willard 1999). Furthermore, expression from the Xi may change over time during development or adulthood: indeed, escape genes may be initially silenced, followed by reactivation during development or with aging (Schoeftner et al. 2009). For example, Kdm5c, a gene that consistently escapes X inactivation in adult mouse cells and tissues, is silenced in a number of embryonic cells, the number of which progressively diminishes during development (Lingenfelter et al. 1998). Changes in expression from the Xi can also be observed during normal aging. Reactivation of Otc has been detected in individual cells within tissues of aged mice (Wareham et al. 1987). Analyses of the chromatin structure of mouse escape genes in embryos and adult tissues also suggest changes in escape from X inactivation during development (Yang et al. 2010) (see below). Thus, while a subset of X-linked genes are stably inactivated, or stably escape X inactivation, another category of genes displays plasticity in terms of allelic expression. It has been suggested that this may contribute to phenotypic differences between women (Fig. 1) (Carrel and Willard 2005; Migeon 2007).
Molecular mechanisms of escape
While many features of the initiation of X inactivation are now understood, the process of heterochromatization of the entire X is still not fully clarified. How does Xist RNA coat the Xi, and how is the silent compartment formed? The study of escape genes can significantly help in figuring out the process of spreading and heterochromatin formation. Indeed, one would expect that escape genes are depleted of epigenetic changes characteristic of genes silenced by X inactivation. Importantly, escape genes, for example Kdm5c and Kdm6a, appear to be devoid of Xist RNA coating (Murakami et al. 2009) (Fig. 2). The distribution of repeat elements may also be important in establishment of domains of escape and inactivation. Overrepresentation of LINE-1 repeats on the X has been proposed as an important factor that may serve to anchor Xist RNA, thus aiding its spreading (Lyon 1998) (Fig. 2). Furthermore, a recent study in mouse shows that LINE-1 repeats themselves recruit Xist and are essential for the formation of a condensed heterochromatic core (Chow et al. 2010). Studies of escape genes have lent support to the LINE-1 hypothesis by demonstrating that such genes have fewer LINE-1 repeats (Bailey et al. 2000; Carrel et al. 2006; Ross et al. 2005).

Molecular characteristics of escape and inactivated X-linked genes. This model shows that silenced regions on the inactive X are coated with the non-coding Xist RNA (pink cloud). Nucleosomes in the inactive X territory contain histones decorated with modifications associated with transcriptional repression and chromatin condensation, for example, H3K27me3 and H3K9me3, as well as the variant histone macroH2A (green circles). In addition, the CpG islands at the 5′ end of inactivated genes are methylated (solid black circles). Silenced chromatin also harbors specific DNA motifs (e.g., LINE-1 elements and AT-rich motifs) (yellow and blue boxes, respectively) to facilitate Xist RNA binding as well as binding of specific proteins such as SATB1. In contrast, genes that escape X inactivation are not coated with Xist, contain nucleosomes with modifications associated with active transcription, for example, H3 and H4 acetylation and H3K4me3, and their CpG islands are unmethylated (open circles). Chromatin between inactivated and escape genes may be bound by the chromatin insulator CTCF preventing the spread of heterochromatin into the escape regions or, vice versa, the spread of euchromatin into the silenced regions (Filippova et al. 2005)
In addition to LINE-1 elements, other repeats and sequence motifs may also play a role in escape. In mouse, long terminal repeats (LTRs) are depleted at escape genes (Tsuchiya et al. 2004). There is also evidence that AT-rich motifs are depleted at escape genes (Fig. 2). Wang et al. (2006) reported that the majority of 3-mers and 5-mers enriched around human genes subject to X inactivation, but not around escape genes, are AT-rich (Wang et al. 2006). This is consistent with our recent observations that mouse escape genes contain fewer AT-rich motifs than genes subject to X inactivation (Nguyen et al. 2011). The role of AT-rich motifs in the process of silencing by X inactivation is supported by findings that SATB1, a protein that specifically binds AT-rich DNA, both binds to the Xi and causes disruption in the silencing function of Xist if mutated in mouse cells (Agrelo et al. 2009). Taken together, these observations suggest that the distribution and content of DNA sequence motifs on the X influence silencing and escape, via controlling Xist RNA recruitment and/or binding of specific proteins that silence genes on the Xi. In addition to Xist, other ncRNAs could be involved in establishing escape and inactivation domains as suggested in a study in which genes that transcribe ncRNAs with a female-biased expression were found adjacent to known protein-coding escape genes (Reinius et al. 2010).
Among the epigenetic marks associated with X silencing, tri-methylation at lysine 27 of histone H3 (H3K27me3) is a repressive chromatin modification catalyzed by the PRC2 complex (Plath et al. 2003). Other repressive marks enriched on the Xi include tri-methylation at lysine 9 of histone H3 (H3K9me3) and incorporation of the histone variant macroH2A (Changolkar and Pehrson 2006; Heard et al. 2001; Peters et al. 2002) (Fig. 2). Such repressive marks are depleted at escape genes (Boggs et al. 2002; Yang et al. 2010; Goto and Kimura 2009; Changolkar et al. 2010). In contrast, active marks such as acetylation of H3 and H4 and tri-methylation at lysine 4 of histone H3 (H3K4me3) are lost from silenced chromatin, but are retained within euchromatic regions of escape (Goto and Kimura 2009; Jeppesen and Turner 1993; Marks et al. 2009; Khalil and Driscoll 2007) (Fig. 2).
How do escape and silenced regions on the Xi co-exist adjacent to each other? There is evidence of boundary elements, such as chromatin insulator proteins that may block either the spreading of heterochromatin into escape regions, or vice versa the spreading of euchromatin within the silenced regions. The chromatin insulator protein CTCF binds to the transition region between the escape gene Kdm5c and the inactivated gene Iqsec2 in mouse (Filippova et al. 2005). In contrast, CTCF does not bind to the corresponding transition region in human where both genes escape X inactivation. Another region of transition between the escape gene Eif2s3x and the silenced gene Klhl15 also binds CTCF. We have also determined that the CpG island at the 5′ end of Kdm5c remains hypomethylated throughout mouse development, possibly because it is bound by CTCF (Filippova et al. 2005) (Fig. 2). Kdm5c retains its escape status when it and its flanking regions are inserted into inactivated loci on the Xi, indicating that the gene itself together with limited flanking sequences is sufficient to elicit escape (Li and Carrel 2008). However, there is evidence that CTCF-binding alone is not sufficient to protect from silencing. Indeed, insertion of CTCF-binding sites from the HS4 insulator site (from the chicken β-globin gene cluster) at each end of a short reporter gene does not prevent its silencing when inserted within an inactivated gene on the mouse Xi (Ciavatta et al. 2006). Further studies are needed to sort out the role of CTCF in escape from X inactivation. Spatial orientation of chromatin is probably critical in maintaining escape from X inactivation. One attractive hypothesis is that escape results from looping of specific domains out of the condensed Xi territory (Heard and Bickmore 2007; Filippova et al. 2005) (Fig. 2). This hypothesis is further supported by evidence that LINE-1 elements are important for the formation of a condensed core of heterochromatin, which would nucleate X-linked repeats, around which silenced genes would be arranged within the Xist cloud, while escape genes would occupy the most outer layer (Chow and Heard 2009). CTCF could anchor chromatin loops that contain the escape genes, thus insuring that flanking regions remain within the condensed heterochromatin; in this case, the role of CTCF would be to protect silenced domains from unraveling and from reactivation.
As discussed above, a subset of escape genes appears to have variable patterns of escape (Fig. 1). That such genes would also have tissue-specific chromatin modifications is supported by our chromatin profiles of the mouse X, which reveal changes in H3K27me3 in a developmental stage and tissue-specific manner (Yang et al. 2010). For example, Mid1 is enriched in H3K27me3 in embryos, but not in liver. Specific enzymes such as the histone demethylases KDM6A and KDM6B may play an important role in removing H3K27me3 at escape genes in specific cell types (Agger et al. 2007; Hong et al. 2007; Lan et al. 2007).
Escape genes and sex differences
Dosage compensation has evolved to insure equal expression from the X chromosome and autosomes between the sexes. However, evidence exists that sexually dimorphic genes are predominant on the X (Arnold and Burgoyne 2004; Saifi and Chandra 1999; Vawter et al. 2004; Yang et al. 2006). Sexual dimorphisms are largely attributed to the action of sex-specific steroid hormones. Nevertheless, prior to the presence of serum testosterone in rodent embryos, sex differences are already detectable in transcription of specific genes, arguing the involvement of additional factors in sexual differentiation (Burgoyne et al. 1995; Carruth et al. 2002; Dewing et al. 2003; Reisert and Pilgrim 1991; Sanchez and Vilain 2010). Development of the four-core genotype (FCG) mouse model has made it possible to study the phenotypic effects of sex chromosome complement independent of sex-specific hormones. Briefly, when gonadal males with the Sry gene deleted from the Y chromosome and inserted on an autosome (XY− Sry) are mated with XX females, four-core genotypes are produced: XX females, XY− females, XXSry males and XY− Sry males. The FCG system has clearly shown that the sex chromosome complement as well as the gender can cause sex differences (reviewed in Arnold and Chen 2009). In addition to Y-linked genes, clearly unique to males, X-linked genes, especially escape genes, would also contribute to sex differences. Differential expression between the sexes makes escape genes plausible candidates for causing gender/sex differences in development and physiology (Arnold 2009; Jazin and Cahill 2010; Xu and Disteche 2006; Bermejo-Alvarez et al. 2011). Furthermore, the sex chromosome complement also influences expression of autosomal genes, suggesting a global effect (Wijchers et al. 2010).
For genes outside the PAR, escape from X inactivation results in higher expression in females because of bi-allelic expression compared to mono-allelic expression in males. Expression from the Xi being significantly less than from the Xa (see above), the total expression level in females is usually less than twice that in males. The contribution of escape genes to sex differences has been studied by comparing expression levels in male and female cell lines and tissues in human and mouse (Talebizadeh et al. 2006; Yang et al. 2006; Johnston et al. 2008). In human lymphoblastoid cell lines, the contribution of escape genes to sex differences in gene expression has been found to be consistent between multiple individuals, but differences between the sexes are relatively modest (Johnston et al. 2008).
Escape genes may be especially important in brain function and this could lead to sex differences (Xu and Disteche 2006). About 10% of X-linked genes but only 3% of autosomal genes cause intellectual disability when mutated (Ropers 2008; Ropers and Hamel 2005; Zechner et al. 2001). This makes the X chromosome an attractive target for study with regard to brain development and function. This pro-brain characteristic of X-linked genes seems to be especially pronounced among X escape genes. Despite fewer escape genes in mouse than human (Berletch et al. 2010; Lopes et al. 2010; Yang et al. 2010), the majority of the mouse escape genes identified thus far play a role in brain development and are conserved in human where they have been implicated in intellectual disability syndromes (Ropers 2010). For example, Kdm5c is a key player in mouse brain development, which regulates neuronal differentiation, neuronal cell survival and dendritic outgrowth (Iwase et al. 2007; Tahiliani et al. 2007). In humans, KDM5C mutations result in intellectual disability, epilepsy, aggression and autism (Abidi et al. 2009; Adegbola et al. 2008; Jensen et al. 2005). Other examples are Ube1x, a ubiquitin activating enzyme that protects the functional integrity of synapses in response to chemical imbalance, and Ddx3x, an RNA helicase that helps transport mRNAs from the nucleus to active synapses for local protein translation (Kanai et al. 2004; Wishart et al. 2007). These escape genes are transcribed more highly in females than males, and the difference is not compensated by their Y-linked paralogues, which are either not transcribed (e.g., Ube1y and Usp9y), transcribed at significantly lower levels than the X paralogues (e.g., Kdm5d) or not translated (e.g., DDX3Y) in the brain (Ditton et al. 2004; Koopman et al. 1989; Xu et al. 2002, 2008a, 2008b). A number of ncRNAs also escape X inactivation in particular regions of the mouse brain, suggesting that they may have a role in mental processes specific to these regions (Reinius et al. 2010).
X inactivation and escape could not only enhance phenotypic differences between males and females, but could also enhance variability within the female sex due to mosaicism for cells with the Xm or Xp inactivated and to variable escape from X inactivation (Fig. 1). For example, some females can perceive significantly more colors in comparison to males, due to mosaicism for expression of the two X-linked retinal photopigment alleles (Jameson et al. 2001; Migeon 2007). In addition, monozygotic female twins are less similar than monozygotic male twins in traits such as social interaction and verbal skill (Loat et al. 2004).
Escape genes and disease
Escape genes, including PAR genes, play important roles in human diseases as women with a single X (X chromosome monosomy; 45,X) have Turner syndrome with severe phenotypes including ovarian dysgenesis, short stature, webbed neck and other physical abnormalities. In addition, as many as 99% of 45,X embryos die in utero (Hook and Warburton 1983). Deficiency in escape genes is thought to play a major role in phenotypes observed in Turner individuals (Zinn and Ross 2001). Since the Y chromosome protects men from these deficiencies, the most likely Turner syndrome candidate genes would have a Y copy, except for genes that control female-specific phenotypes such as ovarian function or processes such as X inactivation itself. Few specific genes have been directly identified as causing Turner syndrome. Thus far, the PAR gene SHOX that encodes a homeodomain transcription factor important for limb development in human has been implicated in the short stature phenotype (Clement-Jones et al. 2000). Interestingly, early lethality of 45,X embryos may be due to a defect in placenta development, which is supported by the finding that many placental genes have much higher expression in 46,XX versus 45,X cells (Urbach and Benvenisty 2009). Urbach and Benvenisty (2009) posit that placental defects in 45,X fetuses could result from the significantly lower (ninefold) expression of the PAR gene CSF2RA that encodes a receptor for a hematopoietic differentiation factor.
The fact that few escape genes exist in mouse is consistent with the significant differences in the impact of X monosomy in female mice and in women (Yang et al. 2010). Candidate genes for the severe phenotypes in Turner syndrome would likely be either genes located within the large human PAR (e.g., SHOX) but located on autosomes in mouse where the PAR contains a single gene (Sts), or non-pseudoautosomal genes that escape from X inactivation in human but are subject to X inactivation in mouse. Interestingly, deletion mapping has identified an 8.3-Mb interval at Xp22.3 involved in the neurocognitive symptoms of Turner syndrome, which contains two human escape genes, STS and NLGN4X, both involved in synaptic/dendritic development (Jamain et al. 2003; Laumonnier et al. 2004; Reed et al. 2005; Zinn et al. 2007).
Escape from X inactivation can also cause phenotypes in individuals with additional copies of the X chromosome. While these supernumerary X copies are inactivated, genes that escape would be present in triple rather than double dose. For example, phenotypes associated with trisomy X (47,XXX) could be a result of over-expression of genes escaping X inactivation (Tartaglia et al. 2010b). Neurological studies of individuals with sex chromosome aneuploidy suggest that X inactivation and escape make important contributions to brain development and function. Indeed, X aneuploidy causes disruptions in cognitive, emotional, and/or behavioral development, for example in 47,XXY Klinefelter’s syndrome (Geschwind et al. 2000; Tartaglia et al. 2010a).
Another potential role for escape from X inactivation is in aging. A recent study has found epigenetic alterations including X reactivation in a mouse model of accelerated aging due to telomere shortening (Schoeftner et al. 2009). As mentioned above, inappropriate reactivation of Otc has been reported in mouse tissues (Wareham et al. 1987). So far, no such reactivation of X-linked genes has been observed in human (Migeon 2007). It will be important to determine whether environmental factors could cause inappropriate escape from X inactivation due to changes in epigenetic marks. This could result in disease phenotypes due to increase expression of genes that are normally repressed or in expression of X-linked mutations in women carriers.
References
Abidi F, Holloway L, Moore CA, Weaver DD, Simensen RJ, Stevenson RE, Rogers RC et al (2009) Novel human pathological mutations. Gene symbol: JARID1C. Disease: mental retardation, X-linked. Hum Genet 125(3):345
Adegbola A, Gao H, Sommer S, Browning M (2008) A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD). Am J Med Genet A 146A(4):505–511. doi:10.1002/ajmg.a.32142
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I et al (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449(7163):731–734. doi:10.1038/nature06145
Agrelo R, Souabni A, Novatchkova M, Haslinger C, Leeb M, Komnenovic V, Kishimoto H et al (2009) SATB1 defines the developmental context for gene silencing by Xist in lymphoma and embryonic cells. Dev Cell 16(4):507–516. doi:10.1016/j.devcel.2009.03.006
Al Nadaf S, Waters PD, Koina E, Deakin JE, Jordan KS, Graves JA (2010) Activity map of the tammar X chromosome shows that marsupial X inactivation is incomplete and escape is stochastic. Genome Biol 11 (12):R122. doi:10.1186/gb-2010-11-12-r122
Anderson CL, Brown CJ (1999) Polymorphic X-chromosome inactivation of the human TIMP1 gene. Am J Hum Genet 65(3):699–708. doi:10.1086/302556
Arnold AP (2009) Mouse models for evaluating sex chromosome effects that cause sex differences in non-gonadal tissues. J Neuroendocrinol 21(4):377–386. doi:10.1111/j.1365-2826.2009.01831.x
Arnold AP, Burgoyne PS (2004) Are XX and XY brain cells intrinsically different? Trends Endocrinol Metab 15(1):6–11. doi:S104327600300242X[pii]
Arnold AP, Chen X (2009) What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol 30(1):1–9. doi:10.1016/j.yfrne.2008.11.001
Bailey JA, Carrel L, Chakravarti A, Eichler EE (2000) Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci USA 97(12):6634–6639. doi:97/12/6634[pii]
Barakat TS, Gunhanlar N, Pardo CG, Achame EM, Ghazvini M, Boers R, Kenter A et al (2011) RNF12 activates Xist and is essential for X chromosome inactivation. PLoS Genet 7 (1):e1002001. doi:10.1371/journal.pgen.1002001
Berletch JB, Yang F, Disteche CM (2010) Escape from X inactivation in mice and humans. Genome Biol 11 (6):213. doi:10.1186/gb-2010-11-6-213
Bermejo-Alvarez P, Rizos D, Lonergan P, Gutierrez-Adan A (2011) Transcriptional sexual dimorphism in elongating bovine embryos: implications for XCI and sex determination genes. Reproduction. doi:10.1530/REP-11-0006
Boggs BA, Cheung P, Heard E, Spector DL, Chinault AC, Allis CD (2002) Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosomes. Nat Genet 30(1):73–76. doi:10.1038/ng787
Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF (1991) A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349(6304):38–44. doi:10.1038/349038a0
Burgoyne PS, Thornhill AR, Boudrean SK, Darling SM, Bishop CE, Evans EP (1995) The genetic basis of XX–XY differences present before gonadal sex differentiation in the mouse. Philos Trans R Soc Lond B Biol Sci 350 (1333):253–260 (discussion 260–251). doi:10.1098/rstb.1995.0159
Carrel L, Willard HF (1999) Heterogeneous gene expression from the inactive X chromosome: an X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others. Proc Natl Acad Sci USA 96(13):7364–7369
Carrel L, Willard HF (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434(7031):400–404
Carrel L, Park C, Tyekucheva S, Dunn J, Chiaromonte F, Makova KD (2006) Genomic environment predicts expression patterns on the human inactive X chromosome. PLoS Genet 2 (9):e151. doi:10.1371/journal.pgen.0020151
Carruth LL, Reisert I, Arnold AP (2002) Sex chromosome genes directly affect brain sexual differentiation. Nat Neurosci 5(10):933–934. doi:10.1038/nn922
Changolkar LN, Pehrson JR (2006) macroH2A1 histone variants are depleted on active genes but concentrated on the inactive X chromosome. Mol Cell Biol 26(12):4410–4420. doi:10.1128/MCB.02258-05
Changolkar LN, Singh G, Cui K, Berletch JB, Zhao K, Disteche CM, Pehrson JR (2010) Genome-wide distribution of macroH2A1 histone variants in mouse liver chromatin. Mol Cell Biol 30(23):5473–5483. doi:10.1128/MCB.00518-10
Chow J, Heard E (2009) X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol 21(3):359–366. doi:10.1016/j.ceb.2009.04.012
Chow JC, Ciaudo C, Fazzari MJ, Mise N, Servant N, Glass JL, Attreed M et al (2010) LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141(6):956–969. doi:10.1016/j.cell.2010.04.042
Ciavatta D, Kalantry S, Magnuson T, Smithies O (2006) A DNA insulator prevents repression of a targeted X-linked transgene but not its random or imprinted X inactivation. Proc Natl Acad Sci USA 103(26):9958–9963. doi:10.1073/pnas.0603754103
Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, Robson SC et al (2000) The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet 9(5):695–702
Deakin JE, Chaumeil J, Hore TA, Marshall Graves JA (2009) Unravelling the evolutionary origins of X chromosome inactivation in mammals: insights from marsupials and monotremes. Chromosome Res 17(5):671–685. doi:10.1007/s10577-009-9058-6
Dewing P, Shi T, Horvath S, Vilain E (2003) Sexually dimorphic gene expression in mouse brain precedes gonadal differentiation. Brain Res Mol Brain Res 118(1–2):82–90. doi:S0169328X03003395[pii]
Disteche CM (1999) Escapees on the X chromosome. Proc Natl Acad Sci USA 96(25):14180–14182
Disteche CM, Filippova GN, Tsuchiya KD (2002) Escape from X inactivation. Cytogenet Genome Res 99(1–4):36–43. doi:10.1159/000071572
Ditton HJ, Zimmer J, Kamp C, Rajpert-De Meyts E, Vogt PH (2004) The AZFa gene DBY (DDX3Y) is widely transcribed but the protein is limited to the male germ cells by translation control. Hum Mol Genet 13(19):2333–2341. doi:10.1093/hmg/ddh240
Duret L, Chureau C, Samain S, Weissenbach J, Avner P (2006) The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312(5780):1653–1655. doi:10.1126/science.1126316
Duthie SM, Nesterova TB, Formstone EJ, Keohane AM, Turner BM, Zakian SM, Brockdorff N (1999) Xist RNA exhibits a banded localization on the inactive X chromosome and is excluded from autosomal material in cis. Hum Mol Genet 8(2):195–204. doi:ddc032[pii]
Filippova GN, Cheng MK, Moore JM, Truong JP, Hu YJ, Nguyen DK, Tsuchiya KD et al (2005) Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev Cell 8(1):31–42
Geschwind DH, Boone KB, Miller BL, Swerdloff RS (2000) Neurobehavioral phenotype of Klinefelter syndrome. Ment Retard Dev Disabil Res Rev 6(2):107–116. doi:10.1002/1098-2779(2000)6:2<107:AID-MRDD4>3.0.CO;2-2
Goto Y, Kimura H (2009) Inactive X chromosome-specific histone H3 modifications and CpG hypomethylation flank a chromatin boundary between an X-inactivated and an escape gene. Nucleic Acids Res 37(22):7416–7428. doi:10.1093/nar/gkp860
Graves JA (1996) Mammals that break the rules: genetics of marsupials and monotremes. Annu Rev Genet 30:233–260. doi:10.1146/annurev.genet.30.1.233
Heard E, Bickmore W (2007) The ins and outs of gene regulation and chromosome territory organisation. Curr Opin Cell Biol 19(3):311–316. doi:10.1016/j.ceb.2007.04.016
Heard E, Disteche CM (2006) Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev 20(14):1848–1867
Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL (2001) Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 107(6):727–738. doi:S0092-8674(01)00598-0[pii]
Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K (2007) Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA 104(47):18439–18444. doi:10.1073/pnas.0707292104
Hook EB, Warburton D (1983) The distribution of chromosomal genotypes associated with Turner’s syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum Genet 64(1):24–27
Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR et al (2007) The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128(6):1077–1088. doi:10.1016/j.cell.2007.02.017
Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H et al (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34(1):27–29. doi:10.1038/ng1136
Jameson KA, Highnote SM, Wasserman LM (2001) Richer color experience in observers with multiple photopigment opsin genes. Psychon Bull Rev 8(2):244–261
Jazin E, Cahill L (2010) Sex differences in molecular neuroscience: from fruit flies to humans. Nat Rev Neurosci 11(1):9–17. doi:10.1038/nrn2754
Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR et al (2005) Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet 76(2):227–236. doi:10.1086/427563
Jeppesen P, Turner BM (1993) The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell 74(2):281–289. doi:0092-8674(93)90419-Q[pii]
Johnston CM, Lovell FL, Leongamornlert DA, Stranger BE, Dermitzakis ET, Ross MT (2008) Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS Genet 4 (1):e9. doi:10.1371/journal.pgen.0040009
Kanai Y, Dohmae N, Hirokawa N (2004) Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43(4):513–525. doi:10.1016/j.neuron.2004.07.022
Khalil AM, Driscoll DJ (2007) Trimethylation of histone H3 lysine 4 is an epigenetic mark at regions escaping mammalian X inactivation. Epigenetics 2(2):114–118. doi:4612[pii]
Koopman P, Gubbay J, Collignon J, Lovell-Badge R (1989) Zfy gene expression patterns are not compatible with a primary role in mouse sex determination. Nature 342(6252):940–942. doi:10.1038/342940a0
Lahn BT, Page DC (1999) Four evolutionary strata on the human X chromosome. Science 286(5441):964–967
Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S et al (2007) A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449(7163):689–694. doi:10.1038/nature06192
Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M et al (2004) X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet 74(3):552–557. doi:10.1086/382137
Lee JT, Davidow LS, Warshawsky D (1999) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21(4):400–404. doi:10.1038/7734
Li N, Carrel L (2008) Escape from X chromosome inactivation is an intrinsic property of the Jarid1c locus. Proc Natl Acad Sci USA 105(44):17055–17060. doi:10.1073/pnas.0807765105
Lingenfelter PA, Adler DA, Poslinski D, Thomas S, Elliott RW, Chapman VM, Disteche CM (1998) Escape from X inactivation of Smcx is preceded by silencing during mouse development. Nat Genet 18(3):212–213
Loat CS, Asbury K, Galsworthy MJ, Plomin R, Craig IW (2004) X inactivation as a source of behavioural differences in monozygotic female twins. Twin Res 7(1):54–61. doi:10.1375/13690520460741444
Lopes AM, Burgoyne PS, Ojarikre A, Bauer J, Sargent CA, Amorim A, Affara NA (2010) Transcriptional changes in response to X chromosome dosage in the mouse: implications for X inactivation and the molecular basis of Turner Syndrome. BMC Genomics 11:82. doi:10.1186/1471-2164-11-82
Lyon MF (1961) Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190:372–373
Lyon MF (1998) X-chromosome inactivation: a repeat hypothesis. Cytogenet Cell Genet 80(1–4):133–137. doi:ccg80133[pii]
Marks H, Chow JC, Denissov S, Francoijs KJ, Brockdorff N, Heard E, Stunnenberg HG (2009) High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res 19(8):1361–1373. doi:10.1101/gr.092643.109
Migeon BR (2007) Females are mosaics: X inactivation and sex differences in disease. Oxford University Press, Oxford
Murakami K, Ohhira T, Oshiro E, Qi D, Oshimura M, Kugoh H (2009) Identification of the chromatin regions coated by non-coding Xist RNA. Cytogenet Genome Res 125(1):19–25. doi:10.1159/000207514
Navarro P, Chambers I, Karwacki-Neisius V, Chureau C, Morey C, Rougeulle C, Avner P (2008) Molecular coupling of Xist regulation and pluripotency. Science 321(5896):1693–1695. doi:10.1126/science.1160952
Nguyen DK, Yang F, Kaul R, Alkan C, Antonellis A, Friery KF, Zhu B et al (2011) Clcn4–2 genomic structure differs between the X locus in Mus spretus and the autosomal locus in Mus musculus: AT motif enrichment on the X. Genome Res 21(3):402–409. doi:10.1101/gr.108563.110
Okamoto I, Patrat C, Thepot D, Peynot N, Fauque P, Daniel N, Diabangouaya P et al (2011) Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature. doi:10.1038/nature09872
Park C, Carrel L, Makova KD (2010) Strong purifying selection at genes escaping X chromosome inactivation. Mol Biol Evol 27(11):2446–2450. doi:10.1093/molbev/msq143
Peters AH, Mermoud JE, O’Carroll D, Pagani M, Schweizer D, Brockdorff N, Jenuwein T (2002) Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet 30(1):77–80. doi:10.1038/ng789
Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC et al (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300(5616):131–135. doi:10.1126/science.1084274
Prothero KE, Stahl JM, Carrel L (2009) Dosage compensation and gene expression on the mammalian X chromosome: one plus one does not always equal two. Chromosome Res 17(5):637–648
Reed MJ, Purohit A, Woo LW, Newman SP, Potter BV (2005) Steroid sulfatase: molecular biology, regulation, and inhibition. Endocr Rev 26(2):171–202. doi:10.1210/er.2004-0003
Reinius B, Shi C, Hengshuo L, Sandhu KS, Radomska KJ, Rosen GD, Lu L et al (2010) Female-biased expression of long non-coding RNAs in domains that escape X-inactivation in mouse. BMC Genomics 11:614. doi:10.1186/1471-2164-11-614
Reisert I, Pilgrim C (1991) Sexual differentiation of monoaminergic neurons–genetic or epigenetic? Trends Neurosci 14(10):468–473
Ropers HH (2008) Genetics of intellectual disability. Curr Opin Genet Dev 18(3):241–250. doi:10.1016/j.gde.2008.07.008
Ropers HH (2010) Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 11:161–187. doi:10.1146/annurev-genom-082509-141640
Ropers HH, Hamel BC (2005) X-linked mental retardation. Nat Rev Genet 6(1):46–57. doi:10.1038/nrg1501
Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M et al (2005) The DNA sequence of the human X chromosome. Nature 434(7031):325–337. doi:10.1038/nature03440
Saifi GM, Chandra HS (1999) An apparent excess of sex- and reproduction-related genes on the human X chromosome. Proc Biol Sci 266(1415):203–209. doi:10.1098/rspb.1999.0623
Sanchez FJ, Vilain E (2010) Genes and brain sex differences. Prog Brain Res 186:65–76. doi:10.1016/B978-0-444-53630-3.00005-1
Schoeftner S, Blanco R, de Silanes IL, Munoz P, Gomez-Lopez G, Flores JM, Blasco MA (2009) Telomere shortening relaxes X chromosome inactivation and forces global transcriptome alterations. Proc Natl Acad Sci USA 106(46):19393–19398
Song R, Ro S, Michaels JD, Park C, McCarrey JR, Yan W (2009) Many X-linked microRNAs escape meiotic sex chromosome inactivation. Nat Genet 41(4):488–493
Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J et al (2007) The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 447(7144):601–605. doi:10.1038/nature05823
Takagi N, Sasaki M (1975) Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 256(5519):640–642
Talebizadeh Z, Simon SD, Butler MG (2006) X chromosome gene expression in human tissues: male and female comparisons. Genomics 88(6):675–681. doi:10.1016/j.ygeno.2006.07.016
Tartaglia N, Cordeiro L, Howell S, Wilson R, Janusz J (2010a) The spectrum of the behavioral phenotype in boys and adolescents 47, XXY (Klinefelter syndrome). Pediatr Endocrinol Rev 8(Suppl 1):151–159
Tartaglia NR, Howell S, Sutherland A, Wilson R, Wilson L (2010b) A review of trisomy X (47, XXX). Orphanet J Rare Dis 5:8. doi:10.1186/1750-1172-5-8
Tian D, Sun S, Lee JT (2010) The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 143(3):390–403. doi:10.1016/j.cell.2010.09.049
Tsuchiya KD, Greally JM, Yi Y, Noel KP, Truong JP, Disteche CM (2004) Comparative sequence and x-inactivation analyses of a domain of escape in human xp11.2 and the conserved segment in mouse. Genome Res 14 (7):1275–1284. doi:10.1101/gr.2575904
Urbach A, Benvenisty N (2009) Studying early lethality of 45, XO (Turner’s syndrome) embryos using human embryonic stem cells. PLoS One 4(1):e4175
Vawter MP, Evans S, Choudary P, Tomita H, Meador-Woodruff J, Molnar M, Li J et al (2004) Gender-specific gene expression in post-mortem human brain: localization to sex chromosomes. Neuropsychopharmacology 29(2):373–384. doi:10.1038/sj.npp.1300337
Wang Z, Willard HF, Mukherjee S, Furey TS (2006) Evidence of influence of genomic DNA sequence on human X chromosome inactivation. PLoS Comput Biol 2 (9):e113. doi:10.1371/journal.pcbi.0020113
Wang X, Soloway PD, Clark AG (2010) Paternally biased X inactivation in mouse neonatal brain. Genome Biol 11 (7):R79. doi:10.1186/gb-2010-11-7-r79
Wareham KA, Lyon MF, Glenister PH, Williams ED (1987) Age related reactivation of an X-linked gene. Nature 327(6124):725–727
West JD, Frels WI, Chapman VM, Papaioannou VE (1977) Preferential expression of the maternally derived X chromosome in the mouse yolk sac. Cell 12(4):873–882. doi:0092-8674(77)90151-9[pii]
Wijchers PJ, Yandim C, Panousopoulou E, Ahmad M, Harker N, Saveliev A, Burgoyne PS et al (2010) Sexual dimorphism in mammalian autosomal gene regulation is determined not only by Sry but by sex chromosome complement as well. Dev Cell 19(3):477–484. doi:10.1016/j.devcel.2010.08.005
Wishart TM, Paterson JM, Short DM, Meredith S, Robertson KA, Sutherland C, Cousin MA et al (2007) Differential proteomics analysis of synaptic proteins identifies potential cellular targets and protein mediators of synaptic neuroprotection conferred by the slow Wallerian degeneration (Wlds) gene. Mol Cell Proteomics 6(8):1318–1330. doi:10.1074/mcp.M600457-MCP200
Xu J, Disteche CM (2006) Sex differences in brain expression of X- and Y-linked genes. Brain Res 1126(1):50–55. doi:10.1016/j.brainres.2006.08.049
Xu J, Burgoyne PS, Arnold AP (2002) Sex differences in sex chromosome gene expression in mouse brain. Hum Mol Genet 11(12):1409–1419
Xu J, Deng X, Disteche CM (2008a) Sex-specific expression of the X-linked histone demethylase gene Jarid1c in brain. PLoS One 3 (7):e2553. doi:10.1371/journal.pone.0002553
Xu J, Deng X, Watkins R, Disteche CM (2008b) Sex-specific differences in expression of histone demethylases Utx and Uty in mouse brain and neurons. J Neurosci 28(17):4521–4527. doi:10.1523/JNEUROSCI.5382-07.2008
Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA et al (2006) Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res 16(8):995–1004. doi:10.1101/gr.5217506
Yang F, Babak T, Shendure J, Disteche CM (2010) Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res 20(5):614–622. doi:10.1101/gr.103200.109
Zechner U, Wilda M, Kehrer-Sawatzki H, Vogel W, Fundele R, Hameister H (2001) A high density of X-linked genes for general cognitive ability: a run-away process shaping human evolution? Trends Genet 17(12):697–701. doi:S0168-9525(01)02446-5[pii]
Zinn AR, Ross JL (2001) Molecular analysis of genes on Xp controlling Turner syndrome and premature ovarian failure (POF). Semin Reprod Med 19(2):141–146
Zinn AR, Roeltgen D, Stefanatos G, Ramos P, Elder FF, Kushner H, Kowal K et al (2007) A Turner syndrome neurocognitive phenotype maps to Xp22.3. Behav Brain Funct 3:24. doi:10.1186/1744-9081-3-24
Acknowledgments
This work was supported by grants from the National Institutes of Health to J. B. B. (HD060402), C. M. D. (GM046883 and GM079537), L. C. (HD056452) and J. X. (MH083949).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Berletch, J.B., Yang, F., Xu, J. et al. Genes that escape from X inactivation. Hum Genet 130, 237–245 (2011). https://doi.org/10.1007/s00439-011-1011-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1011-z