
Abstract
Next-generation sequencing (NGS) will likely facilitate a better understanding of the causes and consequences of human genetic variability. In this context, the validity of NGS-inferred single-nucleotide variants (SNVs) is of paramount importance. We therefore developed a statistical framework to assess the fidelity of three common NGS platforms. Using aligned DNA sequence data from two completely sequenced HapMap samples as included in the 1000 Genomes Project, we unraveled remarkably different error profiles for the three platforms. Compared to confirmed HapMap variants, newly identified SNVs included a substantial proportion of false positives (3–17%). Consensus calling by more than one platform yielded significantly lower error rates (1–4%). This implies that the use of multiple NGS platforms may be more cost-efficient than relying upon a single technology alone, particularly in physically localized sequencing experiments that rely upon small error rates. Our study thus highlights that different NGS platforms suit different practical applications differently well, and that NGS-based studies require stringent data quality control for their results to be valid.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Next-generation sequencing (NGS) may soon become a standard tool in biological and medical research and, before long, may even enter into clinical practice. Moreover, despite the current availability and wide-spread use of various NGS platforms, high-throughput DNA sequencing technologies are expected to develop further still in the future (Metzker 2009, 2010). In human genetics research, NGS appears to be particularly important for the identification of rare variants of moderate to strong phenotypic effect. In fact, low-frequency single-nucleotide variants (SNVs) are presumed to explain many of the hitherto unaccounted statistical associations with common diseases unraveled by genome-wide association studies (GWAS), which focused upon common single-nucleotide polymorphisms (SNPs) and consequently left many researchers with a lot of “missing heritability” for their disorder of interest (Maher 2008; Manolio et al. 2009). It may even be surmised that most of the causal SNVs potentially identifiable by NGS may not have been amenable to GWAS in the first place. Other possible applications of NGS include the characterization of somatic genomes (particularly in cancer genetics), phylogenetic inference, transcriptome analysis, the study of epigenetic phenomena and, eventually, therapeutic decision-making. For all these purposes, the validity of an identified SNV is of paramount importance. Using publicly available data from the 1000 Genomes Project, we therefore set out to assess the error profiles associated with the detection of putative SNVs in humans, using one of three common NGS platforms.
Results
We analyzed the DNA sequence data publicly available at the 1000 Genomes Project web site (http://www.1000genomes.org/) for two HapMap samples of different continental origin: NA12878 (CEPH; Utah resident with ancestry from north-western Europe) and NA19240 (Yoruba in Ibadan, Nigeria). These samples are the offspring members of two trios that have been sequenced with three different technologies, namely SOLiD™ by Applied Biosystems (Foster City, CA, USA) (Valouev et al. 2008), 454 FLX™ by Roche Diagnostics (Branford, CT, USA) (Margulies et al. 2005), and GA IIx™ by Illumina (San Diego, CA, USA) (Bentley et al. 2008). Notably, sequencing in the Trio subproject of the 1000 Genomes Project was carried out at greater depth than in the Low-Coverage and Exon subprojects. We inferred SNVs separately from each technology-specific set of aligned sequence reads, using standard base-calling algorithms as implemented in the SAMtools (Li et al. 2009; see “Materials and methods” for details). Note that the individual read lengths differed between data sets produced not only by different technologies but also by the same technology but at different developmental stages (Durbin et al. 2010).
Since SNV inference is based upon the relative proportion of the four different base calls at a given site, we scrutinized the per-site distribution of these calls for the NGS platforms under study. In so doing, we distinguished between NGS-inferred SNVs that had been reported at least once before in the HapMap phase II + III data (Frazer et al. 2007; International HapMap Consortium 2003, 2005), and which could therefore be deemed ‘known’, and the remaining SNVs that were consequently deemed ‘putative’. Since HapMap SNVs are far better validated than SNVs reported in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), we deliberately refrained from involving dbSNP as a means of confirming SNVs. For ~95% of known SNVs, the heterozygous genotype of NA12878 and/or NA19240 in HapMap matched the NGS-inferred heterozygous genotype, irrespective of the technology used. This subset of SNVs will henceforth be referred to as ‘confirmed’. HapMap genotype data were missing for another 2% of known SNVs, and the remaining 3% of sites inferred as heterozygous by NGS were logged as homozygous in HapMap.
For all three platforms, only a minority of NGS-inferred SNVs had been reported in HapMap at least once before (Table 1). This excess of putative over known NGS-inferred SNVs was much larger when platforms were considered separately, compared to the near parity between putative and known noted for SNVs that were identified on all three platforms (‘consensus’ SNVs). The proportion of consensus SNVs among putative SNVs varied between 23 and 64% (Table 1), depending upon NGS platform and sample.
Both the number of reads per SNV (‘read coverage’) and the SNV-specific quality score provided by SAMtools are often used as quality measures of NGS-derived SNVs. In our study, the read coverage was found to differ substantially between the three technologies (Fig. 1). SNVs identified with 454 FLX™ were covered by the smallest number of reads, followed by SOLiD™ and then GA IIx™ (Table 2). Correspondingly, most 454 FLX™-inferred SNVs (93–100%) were covered by a maximum of 20 reads, whereas most GA IIx™-inferred SNVs (94–97%) were covered by at least 20 reads.

Read coverage of NGS-inferred SNVs on chromosomes 1–22 of two HapMap samples (1000 Genomes Project Pilot 2 data). Histograms are confined to read coverage values between 0 and 100. 454 FLX 454 FLX™ by Roche Diagnostics, GA IIx GA IIx™ by Illumina, SOLiD SOLiD™ by Applied Biosystems, consensus only SNVs that were concordantly inferred by all three platforms
The SNV-specific quality score was also characterized by technology differences, but less so than the read coverage (Fig. 2). Mean scores were again lowest with 454 FLX™, followed by SOLiD™ and then GA IIx™ (Table 2). All three technologies yielded higher quality scores with consensus SNVs, except for sample NA19240 on 454 FLX™. Note that all quality score distributions are left-censored due to filtering of the sequence data prior to base calling (see “Materials and methods” for details).

Quality scores of NGS-inferred SNVs on chromosomes 1–22 of two HapMap samples (1000 Genomes Project Pilot 2 data). Histograms are confined to values between 20 and 220. 454 FLX 454 FLX™ by Roche Diagnostics, GA IIx GA IIx™ by Illumina, SOLiD SOLiD™ by Applied Biosystems, consensus only SNVs that were concordantly inferred by all three platforms
Next, we compared the base-call distributions of putative and confirmed NGS-inferred SNVs, following the idea that any discrepancy between the two signifies the error profile associated with NGS-based SNV identification. To this end, we summarized the individual base-call distribution of each SNV by the Shannon entropy H of the former. Since H = 1.0 corresponds to an equal number of calls of two different bases, this particular H value reflects the ideal result for a heterozygote. In contrast, H > 1.0 indicates calling of a third or fourth base, whereas H < 1.0 implies predominant calling of one base only.
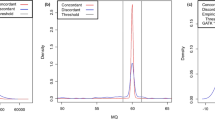
The statistical distribution of base-call entropy H revealed clearly distinct error profiles for the three NGS technologies studied (Fig. 3). Thus, calling in excess of two bases was more evident for GA IIx™ (mean H 1.042–1.076) than for 454 FLX™ (mean H 0.959–0.966) and SOLiD™ (mean H 0.928–0.965). Moreover, with all three technologies, the base-call entropy distribution of putative SNVs was significantly more dispersed around the mean than that of confirmed SNVs (Table 3), particularly when SNVs had been inferred by a single technology alone. The only exception was provided by the consensus SNVs of NA12878 that were identified with SOLiD™. Upon closer inspection, the increased dispersion of H for putative SNVs was found to be due mainly to a larger than expected proportion of H values falling below the first, rather than above the third quartile of H for confirmed SNVs (Table 4). Notably, this downward shift of the distribution of H was significantly less pronounced when only consensus SNVs were considered. Taken together, our results indicate that putative SNVs, particularly when inferred on a single NGS platform alone, are likely to comprise a substantial proportion of false positives, i.e. of erroneous heterozygous calls of homozygous genotypes.

Base-call entropy of NGS-inferred SNVs on chromosomes 1–22 of two HapMap samples (1000 Genomes Project Pilot 2 data). Entropy was calculated for the base calls derived from the platform specified in the left-most column. 454 FLX 454 FLX™ by Roche Diagnostics, GA IIx GA IIx™ by Illumina, SOLiD SOLiD™ by Applied Biosystems, consensus only SNVs that were concordantly inferred by all three platforms. Gray (black) bars confirmed (putative) SNVs, i.e. SNVs that had been reported with a matching genotype (not reported at all) in HapMap
Since consistency with HapMap may be regarded as a gold standard for SNV authenticity, any shift of the distribution of H for putative as compared to confirmed SNVs, particularly with H < 1.0, may serve as an approximation of the false-positive rate among putative SNVs. More specifically, if putative SNVs are assumed to comprise a proportion α of false positives, then the observed density function f putative of H among putative SNVs is a weighted sum of the (unobserved) densities for homozygous and heterozygous genotypes, i.e.
Density f hom is unknown and even though, in principle, it would be possible to estimate f hom from calling truly homozygous sites, such a reference entropy distribution is currently not available. Note that the usual objective of sequencing experiments is the detection of new variants, which implies that putatively homozygous sites are rarely investigated any further. In contrast, f het can be equated to the observed distribution of H among the confirmed SNVs analyzed here. Then a conservative estimate of α is given by
The range of x is restricted to the interval [0,1] in Eq. 2, rather than [0,2], because entropy values larger than unity should reflect sources of error other than the miscalling of homozygous as heterozygous genotypes. Moreover, this restriction renders the estimate of α even more conservative. It should also be noted that the false-positive rate of approximately 3% as observed among known SNVs (see above) is likely to represent a lower limit for α among putative SNVs. This is because the prior probability of homozygosity is much lower for known SNVs than for sites not previously reported to be polymorphic in HapMap, so that the same should apply to the posterior probabilities as long as the conditional error probabilities of NGS are the same for both types of loci.
For all combinations of NGS platform and individual studied, estimates of the false-positive rate α were found to be reduced by up to 90% when only consensus SNVs were considered (Table 5). With the exception of 454 FLX™ in the NA19240 sample, estimates of α ranged from 2.9 to 17.1% for all SNVs, compared to 0.7–4.0% for consensus SNVs only. Estimates of α were also found to be largely concordant between the European (NA12878) and the African sample (NA19240).
In search for systematic errors that might explain the false-positive rate of NGS-based SNV detection as inferred in our study, we first compared the read coverage and quality score distributions of confirmed and putative SNVs. None of these parameters differed significantly between the two types of SNVs (Table 6). Since it may also be that HapMap variants are easier to sequence than non-HapMap variants, we next scrutinized the flanking sequence (±18 bp) of all SNVs studied. Neither GC content, dispersion index nor the proportion of repeat-masked nucleotides were found to differ significantly between confirmed and putative SNVs (Table 7). Therefore, any concerns about systematic sequence differences between HapMap and non-HapMap variants appear to be unwarranted.
Filtering by minimal read coverage or minimal quality score is often used as a means to counteract false-positive SNV detection. To assess the effect of such filtering on α, we repeated our analyses with a minimal coverage of 10 and 20 reads and a minimal quality score of 50 (Durbin et al. 2010). We did not consider higher coverage thresholds because only very few SNVs showed 30-fold of higher coverage with 454 FLX™. Setting the minimal quality score required to 50 substantially decreased the number of SNVs detected by a single platform, and the false-positive rate α either decreased or remained at low levels. Still, consensus calling generally led to smaller α values. Setting the minimum coverage required to 10 or 20 reads yielded less clear results. Although consensus SNVs were again associated with generally smaller error rates than SNVs inferred by a single platform (Supplementary Table 7), the actually observed values of α were found to increase compared to no filtering. Note, however, that the number of SNVs included in the analysis decreased drastically upon filtering by read coverage, in particular for 454 FLX™, and that the base-call entropy distributions became increasingly rugged (Supplementary Figures 4–6), thereby rendering estimation of α less reliable.
In view of the general trend towards a smaller false-positive rate for consensus SNVs, we also assessed error profiles for SNVs detected by pairs of technology platforms. Such pair-wise consensus also reduced the false-positive rate (Table 8) but usually to a smaller extent than all three technologies combined.
Using the same approach as described above, we also analyzed a more recent release of the 1000 Genomes Project data (July 2010). The total numbers of inferred SNVs (Supplementary Table 1) were similar to the Pilot 2 data. The only exception was noted for sample NA12878 on the SOLiD™ platform, where the new data exhibited a massive reduction in SNV number and a remarkably different distribution of both read coverage and quality score (Supplementary Figures 1, 2). Moreover, the base-call entropy was found to follow a more rugged and irregular distribution compared to the Pilot 2 data, in particular for sample NA19240 on both SOLiD™ and 454 FLX™ (Supplementary Figure 3). This notwithstanding, we still observed a statistically significant trend towards a more dispersed and downward shifted distribution of H among putative SNVs than confirmed SNVs, particularly when the former were inferred by a single technology alone (Supplementary Tables 2, 3). This trend was also attenuated when only consensus SNVs were considered. Estimates of α were of the same magnitude as with the Pilot 2 data and were also found to be reduced when the analysis was confined to consensus SNVs (Supplementary Table 4). Differences in read coverage, quality score or flanking sequence characteristics were not sufficient to explain the excess of false-positives among putative SNVs (Supplementary Tables 5, 6). Filtering for minimal coverage failed to consistently reduce α, whereas a minimal quality score of 50 and consensus calling on all three platforms lowered the false-positive rate (Supplementary Table 8; Supplementary Figures 7–9). Thus, an analysis of the July 2010 release led to the same qualitative conclusions as that of the Pilot 2 data.
A variety of SNV calling algorithms have been suggested for practical use, and it might be argued that the results of our study are simply artifacts due to the specific choice of algorithm, i.e. SAMtools. To address this concern, we repeated our analyses using an alternative calling software, namely the Genome Analysis Tool Kit (McKenna et al. 2010). This algorithm has been used to generate the Low-Coverage SNP Call Set of the 1000 Genomes Project (Durbin et al. 2010). In general, GATK identified fewer SNVs (Supplementary Table 10) than SAMtools. The read coverage distribution was similar, but the quality score was always ≥50 (Supplementary Figures 10, 11), apparently because this is a requirement made by GATK. Of the SNVs present in HapMap, virtually all (>99.9%) had a GATK-derived genotype that was concordant with that listed in HapMap. The base-call entropy had a rugged distribution (Supplementary Figure 12) in some cases, e.g. NA19240 on 454 FLX™, that would be hard to explain by the sequencing technology alone. Strikingly, the base-call entropy distribution of NA19240 on SOLiD™ did not even peak at unity. The rugged distribution sometimes led to extreme α values for SNVs identified with a single technology, particularly for 454 FLX™. However, the false-positive rate decreased again when all three platforms (Supplementary Table 11) or pairs of platforms (Supplementary Table 12) were used for consensus calling. Our findings therefore seem to be mostly independent of the algorithm used for SNV calling.
Discussion
Due to its complexity and infrastructural requirements, next-generation sequencing (NGS) is a challenging technology. Many scientists may therefore choose to have their DNA sequence data generated by a service institution, instead of performing the necessary NGS experiments themselves. Furthermore, more and more reference data sets for use in scientific studies are likely to become available in the near future through endeavors such as the 1000 Genomes Project. In both instances, researchers would be put into the position of ‘end users’ with no direct control over the data generation process, including the choice of quality control measures. Estimates of the error rates associated with NGS-inferred SNVs will therefore be of paramount importance to this group of researchers if they wish to assess the validity of such variants.
In terms of data quality, the key question in NGS-based studies is whether a newly discovered SNV is indeed genuine or represents a false-positive result instead. This is true not only for the search for private or low-frequency meiotic mutations at disease gene loci but also for the quest for somatic variants in cancer genomes (Beroukhim et al. 2010; Bignell et al. 2010; Dalgliesh et al. 2010; Lee et al. 2010) and for therapeutic decision-making. Furthermore, some applications such as the assessment of allelic imbalance (Yan et al. 2002) or methylation status require the quantification of allelic ratios and are therefore critically dependent upon the accuracy of the inferred base-call proportions.
In the absence of any sequencing errors, allelic imbalance or somatic mutations, the proportion of calls of either allele at a heterozygous site would follow a binomial distribution with 50% success probability, corresponding to a distribution of the base-call entropy H that peaks at H = 1.0 and decreases monotonously on both sides of this peak. Deviations from such a pattern could indicate problems with sample quality, sequencing chemistry (Metzker 2010) or with the subsequent bioinformatics analysis. With some practical applications, for example, in the analysis of tumor samples (Beerenwinkel et al. 2007; Shah et al. 2009), they could also reflect genetic heterogeneity of the used cell types, but this was clearly not an issue with the 1000 Genomes Project data. In any case, in order for the common SNV detection approaches to be sufficiently sensitive, they have to be tolerant against erroneous base calls, as is evident from the common occurrence of base-call entropies both larger and smaller than unity in the present study, even for SNVs that had been confirmed by HapMap data. However, since putative and HapMap-inferred SNVs did neither differ with respect to neither read coverage, quality score nor flanking sequence, methodological problems should affect the two types of SNV equally. Any discrepancy between the two distributions of H must then point towards the presence of false positives among the newly detected SNVs. The entropy-based approach presented above makes use of this fact and provides a framework for quantifying, at least approximately, the proportion of false-positive SNV detections in NGS-based experiments.
Our analysis highlights that the three NGS platforms under study differ substantially in terms of their error profiles. The consistently lower number of SNVs detected simultaneously by all three platforms, compared to those detected by a single platform alone, may also reflect different bias of the technologies towards particular base calls. However, in view of the above, it appears more likely that a substantial proportion of the technology-specific ‘variants’ actually represented homozygous genotypes erroneously inferred to be heterozygous. The 454 FLX™ platform consistently showed the smallest false-positive rate despite its comparatively low read coverage. This result is likely due to the longer read lengths of this technology because long reads are subject to a lower misalignment probability than short reads. The GAx II™ platform in turn yielded the highest false-positive rate in most instances. While increasing the read length may improve the accuracy of GAx II™ (and SOLiD™) in the future, the problem of a frequently calling more than two alleles on the GAx II™ platform may persist. In any case, we prefer not to speculate any further about the biochemical or computational reasons for the discrepant error profiles observed for the three platforms.
Since we were not ourselves involved in the generation of the 1000 Genomes Project data, we are unaware of the reasons for the striking difference between the two data releases in terms of read coverage, quality score and entropy distribution. It appears to us, however, that there has been no agreement over the filtering and quality control criteria applied within the 1000 Genomes Project, with each data set being generated in a different way. While this is not surprising given the exploratory nature of the project so far, these inconsistencies need to be eliminated because they render any judgement of SNV quality difficult for the ‘end-user’.
An immediate consequence of the technology-specific error profiles revealed by the 1000 Genomes Project data is that any study that combines DNA sequences from different platforms runs a risk of becoming confounded by their different error signatures. In phylogenetic analyses, for example, proximity in the derived phylogenetic tree would then be less indicative of evolutionary relatedness than of shared sequencing technology. One way to reduce the false-positive rate in SNV detection would be to sequence a sample with at least two different NGS technologies, depending upon the required error level, and to consider only those SNVs that are detected by a certain number of platforms. It must be remembered that consensus calling would, of course, increase the false-negative error rate so that the stringency of the consensus needs to be balanced against the power required in a given study. In any case, although costly, such in-depth vetting of potential mutations might be more effective in the long run than the use of a single technology, bearing in mind the resources required for functional follow-up studies.
The NGS technologies are still evolving, and the range of possible applications is constantly expanding. Our analysis has shown that, depending on the envisaged application, the choice of a particular NGS platform should not be based upon coverage and cost alone, but should take the technology-specific error signature into account as well. For instance, exploratory SNV discovery projects may be less dependent upon a high base-calling fidelity than NGS applications, such as SNP-based assessment of allelic imbalance or methylation status, that require precise estimates of allelic proportions. In the latter case, biased allelic ratios or erroneous calling of additional alleles may have a profound impact upon the scientific outcome. In conclusion, we hope that our study serves to stimulate the definition and establishment of quality control criteria for NGS data, thereby enabling full use of the benefits of NGS for a better understanding of human genome variability.
Materials and methods
Sequence data
We used the aligned Pilot 2 DNA sequence reads of samples NA12878 (CEPH; Utah residents with ancestry from northern and western Europe) and NA19240 (Yoruba in Ibadan, Nigeria) that were publicly available at the 1000 Genomes Project web site (http://www.1000genomes.org/). Both individuals represent the offspring members of two of the Trio subproject families that have been sequenced with three different technologies, namely 454 FLX™ by Roche Diagnostics (Branford, CT, USA) (Margulies et al. 2005), GA IIx™ by Illumina (San Diego, CA, USA) (Bentley et al. 2008) and SOLiD™ by Applied Biosystems (Foster City, Ca, USA) (Valouev et al. 2008). For each combination of individual, autosome and sequencing technology, a BAM file with alignment information was downloaded from the 1000 Genomes Project web site (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/pilot_data/data/NA12878/alignment/; ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/pilot_data/data/NA19240/alignment/) in early May 2010. We also analyzed a more recent data release from July 2010, for both samples (genome alignment dates: 20100311 and 20100125, respectively). The relevant files were downloaded from the 1000 Genomes Project web site (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/NA12878/alignment/; ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data/NA19240/alignment/) on 30 June 2010. Sequence reads had been aligned against a reference genome by the project group themselves (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/pilot_data/technical/reference/human_b36_female.fa.gz; ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_g1k_v37.fasta for the July 2010 data).
Identification of heterozygous sites
Heterozygous sites were identified in the DNA sequence data using SAMtools (Li et al. 2009), following standard procedures (http://sourceforge.net/apps/mediawiki/samtools/index.php?title=SAM_protocol). The following default options and filters were applied: a maximum read depth (−D flag) of 100 to exclude variants with excessively high read depth, and a quality score threshold of 20 for nucleotide substitutions. InDels were not considered in the analysis. To assess the effect of more rigorous filtering, we also considered a quality score threshold of 50. Moreover, we also employed an alternative SNV calling algorithm with the July 2010 data release, namely the Genome Analysis Tool Kit (McKenna et al. 2010) to address possible concerns about the software specificity of our results. Calling was done with GATK routines ‘UnifiedGenotyper’, using default parameter values and ‘VariantFiltration’. The later routine was carried out with the same parameters as used by the 1000 Genomes Project (–clusterWindowSize 10; –filterExpression “DP > 360 || (MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1)) || SB > −0.10 || AB > 0.75”).
HapMap confirmation of NGS-inferred SNVs
The SNVs inferred from the 1000 Genomes Project data were distinguished according to whether they had been reported in HapMap at least once before (Frazer et al. 2007; International HapMap Consortium 2003, 2005). To this end, we scrutinized the HapMap phase II + III (release 27) SNP genotype data, NCBI build 36 (ftp://ftp.hapmap.org/hapmap/genotypes/2009-02_phaseII+III; files genotypes_chr*_*_r27_nr.b36_fwd.txt.gz), from which we extracted the genotypes of individuals NA12878 and NA19240. An SNV was then defined as reported in HapMap (‘known’ SNV) if it was logged for NA12878, NA19240 or any other individual in HapMap. Otherwise, the SNV was deemed ‘putative’. Known SNVs for which the NGS-inferred genotype of the respective individual matched the genotype in HapMap (termed ‘confirmed’ SNVs) were used as a ‘gold standard’ in subsequent analyses.
Characterization of the base-call distribution
Since SNVs are inferred from NGS data on the basis of the proportion at which the four different bases are called at a given site, we characterized the site-specific distribution of these base-calls for each NGS technology by means of its Shannon entropy (Shannon 1948), defined as
where p i denotes the relative proportion at which the ith base is called. With only four possible calls, H can assume values in the interval [0,2]. Under the assumption of no errors, the base-calls at a heterozygous site would follow a binomial distribution with 50% success probability. An equal number of calls of both alleles leads to H = 1.0, whereas unequal numbers yield H < 1.0. If calls of three or four different bases occur, H can exceed 1.0.
Statistical analysis
The R software v. 2.11.1 (R Development Core Team 2010) was used for statistical analysis and to create graphs. Histograms of the base-call entropy were generated with 40 equally sized bins covering the interval [0,2]. Estimation of the proportion α of false-positive SNV identifications was based upon only those bins in the interval [0,1] that contained at least 200 observations for both confirmed and putative SNVs. Confidence intervals for α were calculated from 1000 non-parametric bootstrap samples.
Differences between proportions were tested for statistical significance by a χ2 test with one degree of freedom. Means were compared using a two-sided Mann–Whitney U test, whereas differences between standard deviations were tested for statistical significance by a two-sided Siegel–Tukey test. We used the chisq.test and wilcox.test functions in R for these analyses. The Siegel–Tukey test was implemented in R using an in-house script. Differences between α values were tested for statistical significance using a permutation test with 104 replications, using an in-house script. All scripts are available from the authors upon request.
References
Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, Vogelstein B, Nowak MA (2007) Genetic progression and the waiting time to cancer. PLoS Comput Biol 3:e225
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM et al (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53–59
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463:899–905
Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh GL, Teh BT, Deloukas P, Yang F, Campbell PJ, Futreal PA, Stratton MR (2010) Signatures of mutation and selection in the cancer genome. Nature 463:893–898
Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, O’Meara S, Pleasance E, Rajasingham A, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turrell K, Dykema KJ, Khoo SK, Petillo D, Wondergem B, Anema J, Kahnoski RJ, Teh BT, Stratton MR, Futreal PA (2010) Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463:360–363
Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Sun W, Wang H, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T et al (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861
International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796
International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320
Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, Ha C, Johnson S, Kennemer MI, Mohan S, Nazarenko I, Watanabe C, Sparks AB, Shames DS, Gentleman R, de Sauvage FJ, Stern H, Pandita A, Ballinger DG, Drmanac R, Modrusan Z, Seshagiri S, Zhang Z (2010) The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 465:473–477
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Maher B (2008) Personal genomes: the case of the missing heritability. Nature 456:18–21
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Metzker ML (2009) Sequencing in real time. Nat Biotechnol 27:150–151
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Genet 11:31–46
R Development Core Team (2010) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R, Severson T, Taylor GA, Teschendorff AE, Tse K, Turashvili G, Varhol R, Warren RL, Watson P, Zhao Y, Caldas C, Huntsman D, Hirst M, Marra MA, Aparicio S (2009) Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461:809–813
Shannon CE (1948) A mathematical theory of communication. The Bell Syst Tech J 27:379–423, 623–656
Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, Sidow A, Fire A, Johnson SM (2008) A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18:1051–1063
Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW (2002) Allelic variation in human gene expression. Science 297:1143
Acknowledgments
This work was supported by the German National Genome Research Network (NGFNplus; 01GS08182) through the Colon Cancer Network (CCN) and by the German Research Foundation (DFG) excellence cluster ‘‘Inflammation at Interfaces’’. None of the funding organizations had any influence on the design, conduct, or conclusions of the study. The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Nothnagel and A. Herrmann contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Nothnagel, M., Herrmann, A., Wolf, A. et al. Technology-specific error signatures in the 1000 Genomes Project data. Hum Genet 130, 505–516 (2011). https://doi.org/10.1007/s00439-011-0971-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-0971-3