
Abstract
Africa is the ultimate source of modern humans and as such harbors more genetic variation than any other continent. For this reason, studies of the patterns of genetic variation in African populations are crucial to understanding how genes affect phenotypic variation, including disease predisposition. In addition, the patterns of extant genetic variation in Africa are important for understanding how genetic variation affects infectious diseases that are a major problem in Africa, such as malaria, tuberculosis, schistosomiasis, and HIV/AIDS. Therefore, elucidating the role that genetic susceptibility to infectious diseases plays is critical to improving the health of people in Africa. It is also of note that recent and ongoing social and cultural changes in sub-Saharan Africa have increased the prevalence of non-communicable diseases that will also require genetic analyses to improve disease prevention and treatment. In this review we give special attention to many of the past and ongoing studies, emphasizing those in Sub-Saharan Africans that address the role of genetic variation in human disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
African population history
Africa is a region of considerable genetic, linguistic, and cultural diversity. There are over 2,000 distinct ethno-linguistic groups in Africa, speaking languages that constitute nearly a third of the world’s languages (http://www.ethnologue.com/). These languages have been classified into four major language families: Niger-Kordofanian (spoken predominantly by agriculturalist populations across a broad geographic distribution in Africa), Afro-Asiatic (spoken predominantly by northern and eastern Africa pastoralists and agro-pastoralists), Nilo-Saharan (spoken predominantly by eastern and central African pastoralists), and Khoisan (a language containing click-consonants, spoken by southern and eastern African hunter–gatherer populations) (Fig. 1). These populations live in a diverse set of environments and climates, including tropical forests, savannah, desert, and coastal regions and have diverse subsistence patterns and exposure to infectious disease. These populations also have high levels of genetic and phenotypic diversity (e.g. high HLA diversity, Supplemental Table).

A map of selected migrations and language family distributions in Africa (adapted from Reed and Tishkoff 2006). More recent migrations in historical times are represented by thin arrows and inferred prehistoric migrations are represented by medium arrows
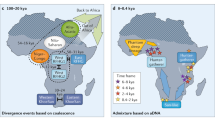
The pattern of genetic variation in modern African populations is influenced by their demographic history, which affects all regions of the genome, as well as by natural selection, which affects specific loci that play a role in adaptation. African populations have a complex demographic history, consisting of ancient and recent population expansion and contraction events, short and long range migrations (e.g. the migration of agricultural Bantu-speakers from West Africa throughout sub-Saharan Africa within the past ~4,000 year and the migration of Khoisan-speakers from eastern to southern Africa within the past ~20,000–40,000 year), and population admixture (Lahr and Foley 1998; Reed and Tishkoff 2006; Tishkoff et al. 2007a). To date the earliest claimed fossil evidence for anatomically modern humans has been identified in Ethiopia (Omo 1) and dated to about 195,000 years ago (McDougall et al. 2005); however, the “modern” characteristics of this specimen have been debated (as it displays important differences in the brow and chin regions) and other records indicate a slightly more recent age of 190,000 years or less (White et al. 2003; Haile-Selassie et al. 2004). Following a period of population differentiation in Africa, one (or more) populations migrated out of East Africa, probably towards Western India, within the past 50,000–100,000 years, resulting in a world wide range expansion of modern humans (Quintana-Murci et al. 1999, reviewed in Tishkoff and Verrelli 2003a). Because African populations are older than non-Africans, there has been more time for genetic diversity to accumulate. Phylogenetic analyses of mtDNA, Y chromosome, and autosomal haplotypes indicate that the deepest lineages are in Africa (reviewed in Tishkoff and Verrelli 2003a). Within Africa, the oldest mtDNA and Y chromosome lineages are found amongst the click speaking hunter–gatherers in southern Africa (i.e. the Khoisan or !Kung San), although there is some evidence for shared ancestry between the southern African and East African click-speaking groups, suggesting a possible East African origin of these populations (Chen et al. 2000; Gonder et al. 2007; Hammer et al. 2001; Passarino et al. 1998; Scozzari et al. 1999; Tishkoff et al. 2003, 2007a).
The migration of modern humans out of Africa resulted in a population bottleneck and concomitant loss of genetic diversity (Liu et al. 2006). Numerous studies have observed higher levels of nucleotide and haplotype diversity in Africans compared to non-Africans, reflecting a larger effective population size in African populations (Tishkoff et al. 2003). Non-African populations appear to have a subset of the genetic diversity present in sub-Saharan Africa, higher levels of linkage disequilibrium, and larger and more uniform haplotype blocks relative to Africans (Tishkoff et al. 2003; Tishkoff and Kidd 2004; Tishkoff and Verrelli 2003b; Tishkoff and Williams 2002). African populations also have a more subdivided population structure relative to non-Africans, with high levels of genetic diversity amongst populations (Tishkoff et al. 1996, 1998; 2000). A recent study of 800 STRPs and 400 In/Dels genotyped in over 3,000 geographically and ethnically diverse African populations indicates at least 12–14 genetically distinct ancestral populations in Africa and high levels of population admixture in many regions (Reed and Tishkoff, unpublished).
Patterns of genetic diversity in humans in general and in African populations in particular are also influenced by natural selection (Barreiro et al. 2008). Because of differences in diet, climate, and exposure to pathogens, ethnically and geographically distinct populations are likely to have experienced distinct selection pressures, resulting in local genetic adaptations (Sabeti et al. 2007). For example, mutations causing G6PD enzyme deficiency that are associated with malarial resistance have risen to high frequency in populations exposed to malarial infection, despite the negative consequences associated with this deficiency (Sabeti et al. 2002b; Saunders et al. 2002; Tishkoff et al. 2001; Verrelli et al. 2002). Another example of local adaptation is the origin and rapid spread of mutations associated with lactase persistence in East African pastoralist populations (Tishkoff et al. 2007b). Lactase persistence (LP) is a classic example of genetic adaptation in humans. The ability to digest milk as adults is a cis-regulated genetic trait. Although a mutation associated with LP was previously identified in Europeans (Enattah et al. 2002), the genetic basis of LP in African populations remained unknown. A study of genotype and phenotype association in a sample of 43 populations from Tanzania, Kenya, and the Sudan identified three novel SNPs located ~14 kbp upstream of the lactase gene (LCT; this and all other gene names are from the HUGO Gene Nomenclature Committee (HGNC) database: http://www.genenames.org/index.html) that are significantly associated with the LP trait in African populations, and enhance transcription from the LCT promoter in vitro (Tishkoff et al. 2007b). These SNPs are located within 100 bp of the European LP-associated variant (C/T −13910). One LP-associated SNP (G/C −14010) is common in Tanzanian and Kenyan pastoralist populations, whereas the other two (T/G −13915, and C/G −13907) are common in northern Sudanese and Kenyans. Genotyping of 123 SNPs across a 3 Mbp region in these populations demonstrated that these African LP-associated mutations exist on haplotype backgrounds that are distinct from the European LP-associated mutation and from each other. In addition, haplotype homozygosity extends >2 Mbp on chromosomes with the LP associated C −14010 SNP, consistent with an ongoing selective sweep over the past 3,000–7,000 years. These data indicate a striking example of convergent evolution and local adaptation due to strong selective pressure resulting from shared cultural traits (e.g. cattle domestication and adult milk consumption) in Europeans and Africans. This study also demonstrates the effect of local adaptation on patterns of genetic variation and the importance of resequencing across geographically and ethnically diverse African populations for studies of disease susceptibility. Another recent study has provided evidence that selective pressure on genes in related pathways can occur; this would be the case of two separate gene regions, containing the LARGE and DMD genes, both having a demonstrated role in Lassa fever infection, that have been subjected to positive selection in West African Yoruba (Sabeti et al. 2007). In conclusion, the amount and patterns of genetic diversity in Africa can make studying African population particularly informative about how genes impact human disease and health.
Susceptibility to infectious disease
In his paper entitled “Observations made during the epidemic of measles on the Faroe Islands in the year 1846” the Danish physician P.L. Panum described an outbreak in which the great majority of the Faroe population developed measles (6,000 of 7,800 people) and more than 200 people died. In this epidemic none of the individuals who had previous exposure during the epidemic of 1781 (many of whom were still alive in 1846) developed disease (Poland 1998). As pointed out by Poland, key questions on disease susceptibility and immunization arise from this epidemic: why did some people survive while others died; why was mortality so high; and why was the protective effect from a past epidemic so high? These are important issues that involve both innate susceptibility and resistance as well as acquired immunity conferred by previous exposure. In essence, these observations integrate clinical, epidemiological and evolutionary approaches that allow an understanding of the diverse functions of immunity genes, affecting survival, and therefore subjected to natural selection (reviewed by Quintana-Murci et al. 2007). The concept of differential susceptibility to infectious disease motivates much of the genetic work in Africa as infectious disease stills plays an important role in the health of most African populations. In these studies different approaches have been and are being employed to assess how genetic variation affects infection status in sub-Saharan Africa. This research involves multiple protocols, including twin studies, where concordance rates are compared between monozygotic and dizygotic twins, case–control association studies, and family-based association and linkage studies where the co-segregation of a marker with the disease phenotype is tested in families (Hill 2006). Because the prevalence of DZ (dizygous) twinning in some parts of West Africa is unusually high, it is feasible to collect large twin cohorts to investigate the effect of host genetics on response to both infection and vaccines (Creinin and Keith 1989). For example, a large cohort of 267 twin pairs (where 60 twin pairs were determined to be MZ (monozygous) by microsatellite typing), has provided unequivocal data that cell proliferation responses to a range of malarial antigens were more highly correlated in MZ than in DZ pairs (Jepson et al. 1997). Another study has shown higher concordance for cellular immune responses to mycobacterial and other antigens in MZ compared to DZ twins, suggesting that genetic factors are important regulators of this immune response (Newport et al. 2004a). Of potential interest is the fact that DZ twinning itself may be under genetic control and possibly under selection (Sirugo et al., in preparation).
As with all such studies a major challenge is the precise and consistent definition of the clinical phenotypes. Therefore, over the past decade a very large effort has been made to ascertain samples, using carefully defined, standardized criteria; as a result of this effort thousands of African samples have been archived, providing a unique resource for studying the complex genetics of susceptibility and resistance to infection. These bio-banking initiatives, collected within appropriate ethical frameworks and supported by dedicated databases, provide unique resources for understanding the genetic risk factors for infectious disease (Sgaier et al. 2007; Sirugo et al. 2004).
Malaria
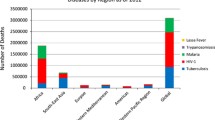
Malaria is a serious health issue in Africa, accounting for one in every five childhood deaths. In 2006 the WHO estimated that almost 74% of the African population lives in areas endemic for malaria, about 19% in epidemic-prone and only 7% in malaria-free areas (http://www.afro.who.int/malaria/publications/annual_reports/africa_malaria_report_2006.pdf). Studies of large populations have begun to elucidate the complex genetics of malaria susceptibility and several genes have already been associated with “malaria” susceptibility (Table 1).
As with all genetic traits there is a real need for accurate and precise definition of the phenotype. “Malaria illness” is a complex phenotype that can be clinically either uncomplicated or severe; the latter, in particular, affects various organs and tissues and takes diverse forms with variable mortality. In addition malaria can be easily over-diagnosed or under-diagnosed and, other than parasitemia, no single specific factor exists that is predictive of the malaria etiology of an illness. Attempts to associate specific acute phase proteins with malaria have not provided conclusive evidence; for example, one acute phase protein proposed as an index of malaria endemicity, haptoglobin (ahaptoglobinaemia), is equivocal and the effect of haptogloblin genotypes on severe malaria has been contradictory (reviewed by Koram and Molyneux 2007).
Despite this complexity, certain phenomena are critical to disease development, and genetic variants that disrupt these processes can protect against disease. The invasion of erythrocytes by malaria parasites is central to the disease process, and the Duffy blood group antigen, a chemokine receptor expressed in many cell types and encoded by the FY gene, is important because Plasmodium vivax cannot infect individuals who do not express the Duffy antigen, resulting in full protection of Duffy (−) individuals. The lack of Duffy expression is due to a promoter SNP that alters a binding site for the GATA-1 transcription factor (Tournamille et al. 1995), resulting in the parasite being unable to invade red blood cells. Over 97% of individuals in West and Central Africa are Duffy (−). The date of emergence of Duffy negativity has been broadly dated, from more than 90,000 to about 6,500 years ago, (Webb 2005). There has been considerable debate whether the spread of Duffy (−) (FY*0) was due to selection in response to P.vivax or if it evolved independently and probably earlier. The latter hypothesis is consistent with a Southeast Asian origin of P. vivax, and the independent evolution of the Duffy null phenotype in Africa. Under this scenario the spread of P.vivax across large areas in Africa would have been prevented. In support of the independent evolution hypothesis is the observation that current P. vivax induced malaria is mild, although the historical strength of selection cannot be known and may have been different than indicated by the present severity of disease (Carter and Mendis 2002; Livingstone 1984). A complicating factor is the suggestion that P. vivax infection could be “beneficial” to humans by conferring some cross-immunity to the more severe P. falciparum-related malaria (Williams et al. 1996). In contrast, evidence of strong recent positive selection on FY*0 is provided by the observation that this variant has a high Fst, the highest ever detected in humans (Hamblin and Di Rienzo 2000; Hamblin et al. 2002). However, no compelling model has yet to be developed to explain how such a high Fst could be produced under a model of such seemingly mild selection.
Other notable examples of genetic protection from malaria in sub-Saharan Africa include G6PD deficiency (Ruwende et al. 1995), HbB sickle-cell trait or HbA/HbS heterozygosity that is associated with a tenfold reduction in malaria risk, HbC (beta6Glu → Lys) (Kwiatkowski 2005; Ntoumi et al. 2007) and alpha thalassemia (HbA gene) that confers protection against severe malaria and malarial anemia (Haldane 1949; Williams et al. 2005a; Wambua et al. 2006; Pasvol 2006). In addition, beta thalassemia, which confers some protection against malaria, occurs only in limited parts of West Africa (Willcox et al. 1983). Sickle cell disease is the classic example for a human balanced polymorphism [a concept introduced by Neel (1953)] and has been studied extensively. In contrast to the heterozygote advantage of HbS, HbC associates with a very strong reduction in risk of clinical malaria of 93% in homozygotes versus 29% in heterozygotes in a large case–control study on more than 4,000 Mossi subjects from Burkina Faso (Modiano et al. 2001b). The conundrum is that despite a very modest pathological load and a very strong protective effect of HbC, the distribution of this allele is limited to central West Africa, while HbS, a quasi-lethal mutation that confers a severe clinical phenotype in homozygotes, has a significantly more cosmopolitan distribution across sub-Saharan Africa. This distribution is quite peculiar given that evidence demonstrates that the “harmless” C is older than S (Modiano et al. 2008). However, while the C allele confers mild protection in heterozygotes (a recessive selection model), the S mutation spread much faster by providing strong protection to heterozygotes (i.e. through over-dominance or heterosis for fitness), despite the cost of causing sickle cell anemia in homozygotes. Experimental data suggest that both HbC and HbS might protect against severe malaria by abnormal cell-surface display of P. falciparum erythrocyte membrane protein-1 (PfEMP-1), which would reduce the effects of parasitized erythrocyte sequestration in post-capillary microvessels, resulting in cerebral malaria (Cholera et al. 2008; Fairhurst et al. 2005).
Clearly, single gene variants affect malaria risk, but the currently known genes do not explain all of the risk. Additionally, it has been shown that interactions among genes can impact malaria. In an elegant study by Williams et al. (2005b), it was shown that the combination α (+) thalassemia homozygosity with HbS trait in the same Kenyan subjects causes loss of protection from severe malaria, a negative epistatic effect that could explain why α (+)-thalassemia did not fix anywhere in sub-Saharan Africa (Williams et al. 2005b). HLA has been suggested to have an important role as well; a study in The Gambia has shown that a class I antigen (HLA-Bw53) and a class II haplotype (DRB1*1302-DQB1*0501) independently associate with protection from severe malaria (Hill et al. 1991). In a subsequent study, malaria morbidity was associated with an overall distribution of Class II haplotypes, but no signals were seen from specific DR-DQ alleles (Bennett et al. 1993). In the same Gambian population, individuals homozygous for a specific TNF promoter SNP (−308) were found to have an increased risk for cerebral malaria, independent of their HLA alleles (McGuire et al. 1994). In Gabonese children this TNF SNP was associated with the rate of symptomatic P. falciparum re-infections (Meyer et al. 2002). Other SNPs in TNF (including −376 and −238) have been associated with susceptibility to severe malaria, severe malaria anemia and control of parasite density (Flori et al. 2005; Kwiatkowski 2005) and TNF variation has also been suggested to explain linkage of malaria fever with MHC Class III (Flori et al. 2003b; Jepson et al. 1997). A linkage study from the holoendemic village of Dielmo (Senegal) has provided evidence that the “asymptomatic parasite density” trait maps to chromosome 5q31, along with suggestive evidence for loci at 5p15 and 13q13 for the “number of clinical malaria attacks” phenotype. Additionally a signal for “maximum parasite density, during asymptomatic infection”, was detected at 12q21 in families from the mesoendemic area of Ndiop (about 5 km SouthEast of Dielmo) (Sakuntabhai et al. 2008). Interestingly, the four chromosomal regions detected in this study overlap with asthma or atopy related loci.
The Th1 pathway seems to have an effect on protection from severe malaria, as suggested by reported associations of IFNG and IL12B regulatory SNPs with protection (IFNG up-regulation) and increased susceptibility (IL12B down-regulation) (Cabantous et al. 2005; Marquet et al. 2008). However, these findings, from hemoglobin defects to polymorphisms of HLA and/or SNPs in the TNF promoter (the latter possibly tagging a neighboring Class III causal gene), or IFNG and IL12B do not explain most inter-individual variation in response to P. falciparum infection, and the distribution of various forms and manifestations of malaria. The current estimate that host genetics accounts for approximately 25% of the risk of infection and contracting malaria implies that there is ample room for gene variant discoveries, explaining differences in disease susceptibility and resistance (Mackinnon et al. 2005).
One way to approach the issue of genetic resistance/susceptibility to malaria is to study individuals stratified by previously known genes. A linkage study from Ghana, using only families with HB and G6PD normal genotypes, has recently identified a locus on chromosome 10p15 that affects malaria fever episodes (Timmann et al. 2007). In a population sample from Burkina Faso, a locus controlling the levels of parasitemia/immune responses to P. falciparum was mapped on 5q31-33 (Rihet et al. 1998), and recent data have shown that SNPs in interferon regulatory factor 1 (IRF1) on 5q31 associate with malaria infection control and with severe disease in Burkinabès (Mangano et al. 2008).
A further approach to study possible host genetic effects in malaria is to compare the susceptibility to the infection and disease among sympatric populations in endemic areas with different genetic backgrounds. This approach revealed the existence of important inter-ethnic differences in the susceptibility to P. falciparum malaria between West African ethnic groups (Modiano et al. 1996). It was clearly shown that such differential resistance was not associated with the classic malaria resistance genes (Modiano et al. 2001a), but could rather be explained by variant genes controlling the immune responses to the parasite (Torcia et al. 2008).
Tuberculosis
Every year more than 8 million people develop tuberculosis (TB) disease and 3 million patients die. The total number of people infected with Mycobacterium tuberculosis is much larger (approximately 2 billion), but the vast majority of those infected never develop clinical disease. In 2005 in Africa there were approximately 3.8 million TB cases, more than 2.5 million of these were new cases, accounting for 29% of the worldwide incidence, and almost 550,000 TB patients died (http://www.who.int/mediacentre/factsheets/fs104/en/). The analyses of TB in Africa is complicated by the parallel epidemic of HIV because co-morbidity is common, making it necessary in studies of the genetics of TB to consider HIV infection, especially in high HIV prevalence areas. Twin studies in Africa, comparing MZ to DZ twins, have provided evidence of a significant role for heritable factors in TB susceptibility (Jepson et al. 2001). In the first genome-wide linkage scan for a major infectious disease in Africans, evidence of linkage was found on chromosomes Xq27 and 15q11 (Bellamy et al. 2000). This report also identified association in these regions of linkage, supporting the conclusion that TB susceptibility loci reside at these chromosomal locations (Bellamy et al. 2000). At 15q a promoter variant of ubiquitin-protein ligase E3A (UBE3A) associates (although not very strongly) with susceptibility (Cervino et al. 2002), but at Xq27 no positional candidate has yet been identified. Using the complementary approach of candidate gene analysis, case–control studies of West African samples have identified associations with variants in several genes; for example SLC11A1 (NRAMP1) (Awomoyi et al. 2002; Bellamy et al. 1998c), IL1B (Awomoyi et al. 2005), vitamin D receptor (Bornman et al. 2004; Lombard et al. 2006; Olesen et al. 2007), CD209 (DC-SIGN), PTX3 (Olesen et al. 2007) and P2X7 genes (Li et al. 2002), to mention a few, have all been associated with TB. In East Africa, a combined linkage and association study of Ugandans has shown that IL10, interferon gamma receptor 1 (IFNGR1), and TNF alpha receptor 1 (TNFR1) variants are linked and associated to TB, but not with susceptibility to latent infection (Stein et al. 2007). Another recent analysis of affected sibling pairs from South Africa (of mixed ancestry) and from Malawi, along with a case–control study in West Africans have identified two putative loci for susceptibility, one at 6p21-q23 and one at 20q13.31-33. At the latter locus, variation in the melanocortin 3 receptor (MC3R) and cathepsin Z (CTSZ) genes were implicated in the pathogenesis of tuberculosis (Cooke et al. 2008).
Importantly, these studies provide additional clues to relevant pathways involved in disease susceptibility. Studies of HLA Class II variation detected an association with increased susceptibility, but these findings await replication (Lombard et al. 2006). As with all candidate gene studies, other reports of association have failed to replicate the original findings (Table 2). In summary, candidate gene studies have indicated the existence of susceptibility loci in specific populations, but without providing evidence of strong effects in Africans. Although some evidence for major TB susceptibility loci has been provided (Baghdadi et al. 2006, Cooke et al. 2008), it is apparent that several loci determine or modulate susceptibility to tuberculosis (Table 2).
Malaria and tuberculosis genome wide association studies
As malaria and tuberculosis are both leading causes of morbidity and mortality in Africa a considerable effort is underway to understand their complex genetic etiology. Genome wide association studies (GWAS) have been launched recently through a consortia of investigators specifically created for this purpose, including the MalariaGen initiative (funded by the Wellcome Trust and the Gates Foundation; http://www.malariagen.net) and the Tuberculosis Gambia–Oxford/African Tuberculosis Genetics Groups, which are part of the Wellcome Trust Case Control Consortium (WTCCC; http://www.wtccc.org.uk). During 2006–2008 the MalariaGen and WTCCC consortia have generated data for up to 500,000 SNPs typed with the Affymetrix GeneChip 500 K mapping array set in thousands of African samples ascertained for both malaria and tuberculosis. The TB study group has been focusing on samples collected in four African countries: The Gambia, Guinea Conakry, Guinea Bissau, and Malawi. From just one West African country (The Gambia) the consortium was granted access to ~1,500 cases and ~2,500 controls. Analysis of the genome-wide association data is in progress and should lead to the identification of SNPs and genes associating with disease susceptibility, thereby providing a wealth of information. These initiatives will also provide important insights into the technical, analytical, methodological and biological aspects of genome-wide association analysis, although it should be noted that the current GWAS platforms may not provide a level of coverage that is complete enough for African samples to detect all important signals. This is of particular interest given the diversity of patterns of LD that probably exist across African populations and that we do not yet fully understand.
HIV/AIDS
Although only 10% of the world’s population lives in Sub-Saharan Africa, 68% of adults and nearly 90% of children infected with HIV-1 live in this region. Overall 22.5 million Africans are estimated to be infected and there are 12 million AIDS orphans, making Africa the worst affected region in the AIDS pandemic. Prevalences in the adult populations (age 15 and more) are more than 16% in South Africa, 23% in Botswana up to a dramatic 34.5% in Swaziland (http://www.who.int/whosis/database/core/core_select.cfm).
In the absence of antiretroviral treatment, the great majority of subjects progress to AIDS and death, following HIV infection. However, although the asymptomatic period averages 10 years, and ranges from a few to 20 years, there are rare instances of infected individuals who do not progress to AIDS at all. Also, some subjects at high risk are apparently resistant and never become infected, e.g. the well-described female sex worker cohort from Nairobi, Kenya (the so called “Majengo” slum women). Some of these women (~5% of the 3,000 sex workers in Nairobi) are persistently sero-negative despite exposure to the virus, although discontinuous exposure seems to lower protection (Kimani et al. 2008). These cases indicate that genetics and the environment interact in determining the resistant phenotype, and several factors, genetic and immune mediated, have been associated with altered susceptibility to HIV (Martin and Carrington 2005; Lama and Planelles 2007) (Table 3). It has recently been reported that in Kenyan sex workers interferon regulatory factor 1 (IRF1) variation and its low gene expression are associated with some resistance to HIV-1 infection. However, the same IRF1 variation does not seem to be linked with differential disease progression (Ball et al. 2007). Given the potential role of IRF1 in supporting HIV-1 transcription and amplifying replication, these results would suggest that the key to protection from infection lies in gene variants that do not support viral transcription and replication.
Variation in human genes that modulate HIV pathogenesis by influencing post-entry steps of the viral life cycle is likely to offer new insights into both protection from infection and modulation of disease course. SNPs in genes affecting viral replication such as the cytidine deaminase enzymes APOBEC3F, APOBEC3G, CUL5 and TRIM5 α have been shown to confer protection in African Americans against disease phenotypes, including infection, accelerated CD4 loss, and faster progression to AIDS (Lama and Planelles 2007). Other studies have indicated significant epistasis between HLA-B and Killer Immunoglobulin-like Receptors (KIRs) in eliciting protection from infection or progression to AIDS (Jennes et al. 2006; Lopez-Vazquez et al. 2005) (Table 4). These results await replication in additional cohorts.
Substantially different from HIV-1 and more benign (less transmissible, slower progression to AIDS), HIV-2 is limited to West Africa and exceeds 5% only in the adult population of Guinea-Bissau. Even in the case of HIV-2, some people die rapidly (within 3 years of infection), but others seem to be able to live with it for decades without immunological or clinical deterioration (Schim van der Loeff 2007). Although a few small studies exist in the literature that suggest possible associations between gene variants and HIV-2 infection, e.g fucosyltransferase 2 (FUT2, “secretor” blood group, Ali et al. 2000) or accelerated disease progression (with HLA B35, Diouf et al. 2002), research on HIV-2 host-genetics might contribute to the understanding of the role genes play in influencing HIV post-entry restriction and disease progression.
Other infections: leishmaniasis, leprosy, schistosomiasis, trachoma
Several other infectious diseases are relatively common in Africa and present serious public health issues. Among those that have been analyzed from a genetic perspective are leishmaniasis, schistosomiasis, leprosy, and trachoma. Although not as much work has been done on these diseases as on malaria, TB and HIV, recent studies have begun to shed light on the genetic susceptibility to them in African populations.
Leishmaniasis
Visceral leishmaniasis or kala-azar is a common disease caused by protozoa of the genus Leishmania carried by sand flies. It is characterized by high fever, dramatic weight loss, swelling of the spleen and liver, and anemia. If untreated, the disease has a fatality rate of nearly 100% within 2 years. In Sudan an epidemic of visceral leishmaniasis caused by Leishmania donovani occurred from 1984 to 1994. The Sudanese populations were highly susceptible, as this was the first such epidemic in the area. It was estimated that the disease caused 100,000 deaths in a population of around 300,000 in the Western Upper Nile area of Sudan; in some villages, more than 50% of the population succumbed to the disease (http://www.who.int/leishmaniasis/en/). The incidence varied among game wardens, working in a reserve in eastern Sudan, who were of different ethnic backgrounds (Ibrahim et al. 1999). Another outbreak occurred in 1996–1997 in a village at the border with Ethiopia, and while most villagers were infected only 30% developed kala-azar (El-Safi et al. 2002).
Candidate gene studies of this population detected linkage of kala-azar with polymorphisms at the SLC11A1 (formerly NRAMP1) locus, on 2q35 (Bucheton et al. 2003a, 2003b). A separate investigation of Sudanese multiplex families, living in the same geographical region, confirmed the linkage of SLC11A1 with kala-azar, mainly detected with a SNP in the fourth intron of the gene. However, a mutation screening of the coding and the SLC11A1 3′ UTR regions in selected patients failed to identify any functionally relevant sequence change. It was therefore hypothesized that the intron 4 SNP could be in disequilibrium with causative variation in the upstream promoter region (Mohamed et al. 2004). An investigation of the same families showed linkage and association of kala-azar with IL4 (Mohamed et al. 2003), while genomic variation of IFNGR1 was associated with post-kala-azar dermal leishmanaisis (Salih et al. 2007). Remarkably, sequence variation in this gene (particularly the promoter region) has been found to modulate susceptibility to other parasitic diseases, including cerebral malaria (Koch et al. 2002) and schistosomal hepatic fibrosis (Blanton et al. 2005).
Linkage studies have reported peaks for kala-azar on 2q22-q23 and 22q12 (Bucheton et al. 2003b; El-Safi et al. 2006) and more recently on 1p22 and 6q27 (Miller et al. 2007), although this latter study did not replicate the earlier linkage peaks on 2q and 22q. However, inconsistency of linkage reports can be explained in part by the fact that both design (and power) and the ethnic groups differed across the two studies. To date, SLC11A1 remains the most impressive of the susceptibility loci. This is reinforced by the known role of SLC11A1 in mouse models infected with L. donovani (Foote and Handman 2005).
Leprosy
In 2005 Mycobacterium leprae caused about 295,000 new cases of leprosy worldwide (http://www.who.int/lep/situation/NCDetection2006.pdf). Of these over 40,000 were in Africa, and more than 10,000 were in the Democratic Republic of Congo alone. However, only ~5% of people exposed to M. leprae develop disease. Leprosy affects the skin, the peripheral nerves, the mucosa of the upper respiratory tract, the eyes, and several other organs. Clinically, leprosy can be differentiated into two forms: (1) a tuberculoid, paucibacillary form, characterized by a low bacterial count, strong cell-mediated immunity, and localized disease and (2) a lepromatous, multibacillary form, characterized by high bacterial count, poor cell-mediated immunity and strong humoral immunity with progressive, disseminated disease. The different forms of the disease do not appear to be complicated by variation in the M. leprae genome since it is surprisingly invariant (Monot et al. 2005). However, family studies, twin studies, and segregation analyses have provided evidence that, in addition to environmental and exposure components, host genetics plays an important role in the disease. Loci/genes affecting differential susceptibility can be subdivided into those influencing infection after exposure, the disease per se, and those related to the paucibacillary or multibacillary type of disease. The risk of developing the lepromatous, multibacillary form can be measured by the extent of skin reactivity to lepromin (Mitsuda reaction; see Ranque et al. 2007 for implicated loci). To date, several genetic studies have identified genes putatively important in leprosy susceptibility, including the PARK2/ PACRG and LTA genes (6q25) in influencing susceptibility to leprosy per se in Indian, Vietnamese and Brazilian subjects, as well as a locus on 10p13 linked to the tuberculoid, paucibacillary form in population samples from India and Vietnam (Alter et al. 2008; Ranque et al. 2008 for a review). Few studies have been carried out on African populations, with exceptions in Nigerians, describing the role of HLA associations (Class II DRB1 leprogenic motifs modulating the clinical outcome of infection, Uko et al. 1999), and in Malians for non-HLA genes (SLC11A1 3' allele associated with lepromatous type, Meisner et al. 2001). More recently, linkage and large-scale candidate gene studies with samples from the Karonga district of northern Malawi have been performed (Wallace et al. 2004; Fitness et al. 2004). These studies have found suggestive evidence for a susceptibility locus on 21q22 influencing leprosy type, as well as associations with the VDR (increased risk of leprosy per se) and with Complement Receptor 1 (CR1) (protection from disease) gene variants, that however require replication in additional African populations.
Schistosomiasis
Schistosomiasis (bilharziosis), is a chronic disease caused by parasites of the genus Schistosoma (trematode flatworms) and, if we exclude the broad category of soil transmitted helminths, the second most frequent parasitic disease in Africa after malaria. Larval forms of the parasites are released by freshwater snails, the parasite’s natural reservoir. As the parasite can penetrate the human skin in the water, the main route of infection is contact with infested water. The larvae migrate into the peripheral vasculature, traverse the lung and settle in the portal or pelvic venous system where they develop into adult parasites. In sub-Saharan Africa S. mansoni causes intestinal schistosomiasis, which cause hepatic granulomas and fibrosis, portal hypertension, splenomegaly, bleeding from esophageal varices, and eventually terminal hepatic failure. Another species, S. haematobium, causes the urinary form of the disease, associated with progressive granulomatosis of the bladder, resulting in obstructive uropathy (http://www.who.int/schistosomiasis/en/). There is a significant association between the urinary infection and squamous cell carcinoma of the bladder, and possibly of the liver infection with hepatocarcinoma, an enlightening example of pathogenetic link between infections and cancer in developing countries (Mostafa et al. 1999). In 2000 it was estimated that approximately 200 million people were affected in the developing world and nearly 85% (170 million) of these were in sub-Saharan Africa (Chitsulo et al. 2000). Schistosomiasis is therefore a very important public health problem in Africa, causing approximately 280,000 deaths per year (150,000 from kidney failure and 130,000 from hematemesis) (van der Werf et al. 2003). During the prepatent period of infection, the first 4–5 weeks following exposure to cercariae (the parasitic larvae), the immune response is primarily of the Th1 type but it becomes progressively polarised towards Th2 about 8 weeks after infection (Pearce et al. 2004); parasite egg antigens seem to inhibit IL12 production and induce IL4 production, promoting a general amplification of the Th2 response.
Host genetic studies have shown that a few gene variants/loci are important in both controlling the infection and in modulating the susceptibility to hepatic and urinary diseases (reviewed in Campino et al. 2006). In 1997 a study of the intestinal form in Senegalese, detected a locus conferring susceptibility to S. mansoni at 5q31-33 (Müller-Myhsok et al. 1997) that had been previously mapped in Brazilian families (Marquet et al. 1996). Immune response genes of the cytokine cluster in the 5q31 region (including Th2 cytokines IL4, IL5, IL13) were assayed in two Dogon population samples (Mali) from a region endemic for S. haematobium, using family based associaton analyses. No association was found with IL4 and IL5 SNPs, but two IL13 5′ variants, IL13-1055C and IL13-591A, were preferentially transmitted to children with the highest infection levels. In contrast, subjects with the IL13-1055T/T genotype appeared to be relatively protected from infection (Kouriba et al. 2005). This “protective” genotype had previously been associated with increased expression of IL13, as well as with elevated IgE levels. Another locus was identified by linkage analysis at 6q23 in Sudanese families from an endemic, irrigated area in the Gezira region (Dessein et al. 1999). This locus is near the gene encoding the α-chain of the interferon gamma receptor 1 (IFNGR1) that seems to control severe hepatic peri-portal fibrosis in S. mansoni infection, a condition affecting 2–10% of subjects infected in the Sudan. A subsequent study in North African families from Egypt confirmed linkage of severe hepatic disease with IFNGR1 and possibly a region on 5q31, encompassing IL4 and IL13, as well as the TGFB1 locus on 19q (Blanton et al. 2005). In a Sudanese population sample two SNPs in the third intron of IFNG were found to produce opposite effects with respect to fibrotic phenotypes: +2109 A/G SNP was associated with a higher risk for fibrosis while +3810 G/A was associated with less severe disease (Chevillard et al. 2003). Associations of aggravation and protection from hepatic fibrosis have also been reported with TNF (Henri et al. 2002).
Other reported associations include HLA Class I alleles with hepatosplenomegaly in Egypt (Abdel-Salam et al. 1986). The urinary form of the disease (by S. haematobium) has been associated with SNPs in the STAT6 gene (on chromosome 12q13) in a Dogon (Mali) population sample. The STAT6 gene is key in Th2 cell differentiation (Shimoda et al. 1996), providing further evidence for an important role of the Th2 cytokine pathway in modulating resistance to schistosomiasis (He et al. 2008). However, recent studies addressing the complex interaction between the immune system and the parasite, indicate contrasting, age-dependent cytokine responses that would suggest that simple Th1/Th2 (or pro-inflammatory/anti-inflammatory) dichotomy is not sufficient to explain susceptibility or resistance to S. haemotobium (Mutapi et al. 2007).
Trachoma
Trachoma is caused by Chlamydia trachomatis, a bacterium that infects the epithelial cells of the conjunctiva. It is transmitted through contact with eye discharge from an infected person or by eye-seeking fly vectors. Repeated infection can result in scarring, distortion and in-turning of the eyelids, with the eyelashes rubbing on the globe (trichiasis), ultimately leading to corneal opacity and irreversible blindness. It has been estimated that more than 2 million people are blind because of trachoma in sub-Saharan Africa (Lewallen and Courtright 2001). The blinding complications of trachoma are thought to be immuno-pathological. Both innate and adaptive immune responses are involved, with cell-mediated immunity playing a dual role in both the resolution of the infection as well as in scarring. In the early stages of infection, pro-inflammatory cytokines (IL1, TNF) are released by the epithelium, and these cytokines attract an initial wave of macrophage and neutrophil infiltration to the site of infection. These cells are soon replaced by lymphocytes that become organized into lymphoid follicles. Repeated or severe inflammatory episodes along with persistent formation and resolution of lymphoid follicles results in tissue remodeling, scar formation and eventual blindness from the mechanical abrasion of the cornea by the eye lashes and rim of the upper eye-lid. The adaptive cellular responses that follow the initial innate response appear greater in individuals who rapidly resolve infection compared to those with persistent clinical disease, implicating an innate or genetic component. Furthermore patients with conjunctival scarring have lower peripheral blood lymphocyte proliferation responses with respect to controls (Burton et al. 2007). IFNG and FOXP3 (and possibly IL10) appear to play an important role in the resolution of the infection (Faal et al. 2006) and genetic polymorphisms in class I HLA, IFNG, TNF, IL10 and MMP9 have been associated with variation in scarring in Gambians (Natividad et al. 2007, 2005, 2006, 2008).
Non-communicable diseases
Although infectious diseases are the most important public health concern in Africa at present, the health landscape is rapidly changing with economic development and urbanization. For example, in South Africa recent studies have suggested that more than 75% of Black Africans have at least one major risk factor for heart disease (Tibazarwa et al. 2008). This scenario is beginning to be the rule for other common, complex diseases of the West, including obesity, diabetes and hypertension that are sensitive to the transition from rural to urban lifestyles (Abubakari et al. 2008; Cooper et al. 1997; Opie and Seedat 2005). In addition, cancers are not uncommon in sub-Saharan Africa (Parkin et al. 2003). Many of these phenotypes have been studied for genetic risk factors over the last few years and a small but rapidly increasing body of literature exists.
Genetics of diabetes and obesity in Africa
Type 2 diabetes (T2D) is currently the most common metabolic disorder in the world. However, there is extremely limited quality data, using standardized criteria for most countries in sub-saharan Africa. The available data indicate great variation in prevalence from 0% in Togo to 4.8–8.0% in South Africa and 10% in Northern Sudan (Motala 2002). To date, there has been only one major systematic effort to study the genetics of diabetes in Africa: this is the Africa America diabetes mellitus (AADM) study, a multi-institutional, multi-country collaboration designed primarily to map T2D susceptibility genes in the ancestral populations of African Americans (Rotimi et al. 2001). One obvious rationale for studying T2D in West Africa, where diabetes is less common than in the US, is that in an environment where caloric intake is lower, cases of T2D might carry a proportionately greater genetic component. As described below the AADM study has been extremely active in linkage analyses of not only diabetes, but multiple related traits.
Genome-wide linkage analysis was performed, using a sample of 343 affected sibships (691 individuals). Although multipoint non parametric linkage analysis showed suggestive linkage on chromosomes 12 and 19 (Rotimi et al. 2004), the strongest evidence of linkage was observed on chromosome 20. Putative linkage to chromosome 20 has been reported by at least ten other studies in multiple ethnic groups (Ghosh et al. 1999; Ji et al. 1997; Mori et al. 2002; Zouali et al. 1997). The linkage peak at 20q in AADM was within 1 cM of the peak reported in Caucasian families (Klupa et al. 2000). The AADM study is noteworthy because it was the first genome scan study to search for susceptibility genes for T2D in sub-Saharan Africa. Secondly, it showed that the same genomic regions are implicated in T2D in both Ghana and Nigeria where environmental risk is low.
Genome wide linkage analysis for T2D related traits
Obesity related traits
Given the central role of obesity as a risk factor for T2D, genome wide linkage analysis was done in AADM (Chen et al. 2005a) to identify linkage signals to three obesity-related traits: body mass index (BMI), fat mass (FM) and percent body fat (PBF). In West Africa, obesity is still relatively uncommon, with a prevalence of approximately 5%, reflecting the high physical activity levels and low caloric intake. A survey in The Gambia showed significant differences between rural and urban areas, with the prevalence of obesity (body mass index > or = 30 kg/m2) at 4.0% in the rural areas but about 33% in urban women 35 years or older (van der Sande et al. 2001). PBF showed the strongest evidence of linkage with a signal on chromosome 2 (location 72.6 cM). Additional signals were found on chromosomes 4 and 5. FM showed suggestive evidence for linkage to chromosome 2 within 10 cM of the signal for PBF. The strongest evidence for linkage to BMI was observed on chromosomes 1 and 4, although in both cases the highest LOD score was below 2. The areas of linkage for the three phenotypes showed significant clustering as all three phenotypes (BMI, FM and PBF) had linkage peaks in the same regions in 2p13, 4q23 and 5q14; however, not all of the peaks reached the thresholds for significant or even suggestive linkage. This study also provided substantial evidence for linkage to QTLs previously reported to be linked to serum leptin and plasma adiponectin levels on chromosome 2 (Comuzzie et al. 1997).
Serum lipids
The AADM study also conducted a genome wide linkage analysis to five serum lipid fractions: total cholesterol, triglycerides, HDL-cholesterol, LDL-cholesterol and VLDL-cholesterol (Adeyemo et al. 2005). Significant linkage of HDL-C to a QTL on chromosome 7 at 7q31 was observed. Other QTL met the criteria for suggestive linkage with three of them (chromosome 7 for TG, chromosome 5 for LDL-C and another locus on chromosome 7 for HDL-C) reaching LOD scores of at least 3.0. Significant or suggestive linkage was found for two of the five traits at the same locus for a QTL on chromosomes 5 and 7.
Several of the linkage signals for these lipid traits overlap linkage regions found in other studies. For example, 7q31 has also been found for lipid levels in Mexican Americans and Pima Indians (Arya et al. 2002). Thus, AADM has found linkage signals very close to those reported for multiple lipid phenotypes in several other major studies. However, two of the linkage regions in AADM are novel: 5q33 for LDL-C and 7p21 for HDL-C (Adeyemo et al. 2005).
Other diabetes related phenotypes
Other linkage analyses, using AADM, have been performed on phenotypes related to diabetes, including intraocular pressure, renal functions and C-peptide concentrations (Chen et al. 2007a, 2007b; Rotimi et al. 2006). For intraocular pressure in diabetics, multipoint linkage analyses showed significant linkage on 5q22 and suggestive evidence of linkage to chromosome 14q22 (Rotimi et al. 2006). The strong signal on chromosome 5 lies in the region implicated in glaucoma susceptibility in previous studies (Monemi et al. 2005). For renal function, linkage to creatinine clearance was observed on chromosomes 7, 16, and 17. Maximum LOD scores for serum creatinine were observed on chromosomes 3 and 10, and for glomerular filtration rate (GFR) on chromosomes 6 and 8 (Chen et al. 2007b). Several of these results are replications of significant findings from other genome scans. In AADM a linkage analysis for C-peptide identified potentially important QTLs on chromosomes 4, 15, and 18 (Chen et al. 2007b). Two positional candidate genes for diabetes (the pituitary adenylate cyclase activating polypeptide (PACAP) on 18p11 and the peroxisome proliferator-activated receptor gamma coactivator 1 (PPARGC1) on 4p15), are located in the genomic regions showing suggestive linkage evidence.
Candidate gene studies in the AADM study
Based on previous findings of association between T2D and three calpain 10 (CAPN10) gene polymorphisms (SNP-43, SNP-56 and SNP-63), these SNPs were investigated in the AADM study (Chen et al. 2005b; Horikawa et al. 2000). Calpain 10 is a nonlysosomal, neutral cysteine protease expressed in skeletal muscle, liver and pancreatic islets reported to be associated with T2D (Horikawa et al. 2000). No association was found between any individual alleles or genotypes of the three and T2D. However, in the Nigerian ethnic groups, one haplotype was significantly associated with type 2 diabetes (OR 3.765 and 95% CI 1.577–8.989). Also, no association was found between the CAPN10 gene polymorphisms and several diabetic-related quantitative traits, including glucose, insulin or other diabetes related quantitative traits such as waist–hip ratio, body mass index (BMI), fast insulin level, fasting C-peptide level, leptin level, glucose level, systolic blood pressure, and diastolic blood pressure. This preliminary observation suggests that the three CAPN10 SNPs tested may play a limited role, if any, in the risk of T2D in the AADM study.
The AADM study also evaluated the association between the functional agouti-related protein (AGRP) promoter SNP −38C/T and weight-related traits, namely BMI, FM and fat-free mass (FFM), as well as diabetes status (Bonilla et al. 2006). Women homozygous for the variant T allele had significantly lower BMI. Also, men with at least one copy of the variant T allele were over two times less likely to be diabetic than subjects without the protective allele. These results replicate previous findings and implicate the AGRP SNP −38C/T in the regulation of body weight in West Africans.
Finally, potential association between polymorphisms of the eNOS gene and diabetes-related phenotypes was investigated in the AADM study (Chen et al. 2007c). The insertion/deletion (4a/b) and the G894T polymorphisms of the eNOS gene were genotyped in cases and controls and the b/b genotype was associated with a 2.4-fold increased risk of diabetic retinopathy. In contrast, no association was observed between the genotypes or alleles of the G894T polymorphism and diabetic retinopathy, hypertension, or nephropathy.
A major contribution of the AADM study to knowledge about T2D is in the area of replication of associations found in other populations and refinement of such associations. A clear instance of this was in the association between risk of type 2 diabetes and variants in the transcription factor 7-like 2 gene (TCF7L2) first reported in populations of European ancestry. The AADM study sample aided in refining the definition of the TCF7L2 type 2 diabetes risk variant, HapB (T2D), to the ancestral T allele of a SNP, rs7903146 (Helgason et al. 2007). This study is a powerful demonstration that populations with shorter LD blocks, such as those of West Africa, provide the means to refine association signals detected in more recent populations (Tishkoff and Williams 2002). It is noteworthy that, to date, the TCF7L2 association has provided the strongest evidence of association of any gene with T2D risk from multiple GWAS and replication studies in multiple population groups. The AADM study also provided replication evidence of a genetic variant in the TCF2 gene that confers protection against type 2 diabetes (Gudmundsson et al. 2007).
In summary, the genetic epidemiology of T2D in Africa is still in its infancy. There have been few genome wide linkage studies and only a handful of association studies. To date, there have been no GWAS conducted in an African population, despite the great utility of using such an approach as has been demonstrated with multiple complex diseases over the last 2 years.
Hypertension
Over the past decade numerous studies have been undertaken in an attempt to identify genetic risk factors for hypertension and blood pressure regulation in Africans. These studies are justified by the observation that blood pressure regulation and the control of several plasma proteins thought to affect blood pressure, such as angiotensinogen (AGT) and angiotensin converting enzyme (ACE), are highly heritable (Adeyemo et al. 2002; Cooper et al. 2000; Rotimi et al. 1999). Of note, the heritability of ACE and AGT was considerably higher in Nigerians (~70–80%) than in African Americans (~20%) most likely reflecting the differential role of environment in these two geographic populations. It is also important to note that the vast majority of work on the genetic basis of hypertension in sub-Saharan Africa is in West Africa.
The genetic studies have included a few linkage studies (Cooper et al. 2002) and many candidate gene studies. As with all linkage studies of hypertension and related phenotypes the results have been uncertain. However, a few regions of the genome do provide evidence for linkage to blood pressure; notably 2p, 3p, 5q, 7p, 7q and 10q provided evidence of linkage to diastolic blood pressure, and 19p and 19q to systolic blood pressure in Nigerians (Cooper et al. 2002). Studies of candidate genes include the renin–angiotensinogen genes (Bouzekri et al. 2004; Fejerman et al. 2006; Nkeh et al. 2003; Robinson and Williams 2004; Tiago et al. 2003; Williams et al. 2004, 2000), barttin (BSND) (Sile et al. 2007), the beta subunit of the epithelial sodium channel (Dong et al. 2001; Nkeh et al. 2003; Rayner et al. 2003), alpha adducin (Barlassina et al. 2000) and G-protein coupled receptor kinase (GRK4) (Williams et al. 2004, 2000). Although several of these studies report positive associations with either hypertension or blood pressure, the data are still not conclusive. One approach that has tried to address the failure to identify replicable results has been to test multilocus genotypes that predispose to hypertension (Williams et al. 2004, 2000). This approach has identified a two locus model with ACE and GRK4 in a Ghanaian population; however, substantial retesting will be required to assess validity of both the results and the approach (Williams et al. 2004).
Cardiovascular disease
As with hypertension there are compelling epidemiological data indicating an increase in prevalence of CVD in African populations as individuals acquire CVD risk factors (Unwin et al. 2001). Despite the prevalence of CVD risk factors in some African populations (Alberts et al. 2005; Steyn et al. 2005), little research has directly addressed the role of genetic variation on the susceptibility to disease. However, recent work has shown that in South Africa, for example, family history confers an odds ratio ~17 (Loock et al. 2006). In addition, studies of African Americans have shown that variation in the leukotriene A4 hydrolase gene increases risk of myocardial infarction more than threefold while the relative risk is only 1.16 in Europeans (Helgadottir et al. 2006). Such studies reinforce the need for significantly more research on this topic in Africa.
A recent set of studies in a Ghanaian population has begun to assess the genetic control of CVD risk factors, plasma levels of serpin peptidase inhibitor (plasminogen activator inhibitor type 1, PAI1) and tissue plasminogen activator (PLAT), that affect the risk of thrombosis because thrombosis is a precursor to CVD (Williams et al. 2007). This study recruited more than 2,000 participants to assess the role that genetic variation plays in regulating plasma levels of these proteins. Preliminary findings indicate that not only does genetic variation in the PAI1 and PLAT genes affect plasma levels of both proteins, but that variants in at least one other gene, renin, does as well. Of note, the effects of the genetic variants differ significantly between males and females, suggesting a complex pattern of genetic regulation via gene–environment interaction (Schoenhard et al., submitted).
Cancer
Cancer is not rare in Africa and based on lifestyle changes its prevalence is expected to increase. However, due to severe deficiencies in health care systems and disease registration, epidemiological data in sub-Saharan Africa are limited. Underdiagnosis and underreporting differentially affect cancer types, gender and age classes, so that it is difficult to assess disease patterns. In general, however, cancer in Africa is characterized by younger age and advanced stage at diagnosis and correspondingly poor prognosis. Demographic and socio-economic factors, including access to medical care, contribute to these features, obscuring intrinsic biological factors (Parkin et al. 2003). Overall, there are few studies on the genetic basis of cancer in Africa (Table 5). Below, we briefly highlight features relevant to the genetics of the most common or characteristic cancers in African populations.
Prostate cancer
It has been well documented that men of African ancestry have higher rates of prostate cancer incidence and mortality compared to men of other ancestries, particularly in the younger age groups (Brawley 1998; Bunker et al. 2002; Delongchamps et al. 2007). This is supported by data from sub-Saharan Africa, where prostate cancer is estimated to be the third most common cancer of males, with rapidly increasing incidence (Magoha 2007; Parkin et al. 2003). Multiple candidates have been suggested, including genes that affect susceptibility to oxidative DNA damage, growth-related pathways, androgen receptor signaling, chronic inflammatory responses, and RNA processing (Rennert et al. 2005; Shand and Gelmann 2006; Sarma et al. 2008; Zabaleta et al., 2008). Several of these candidate genes have been investigated for differential distribution of allelic variants that may affect risk, and cohorts from Africa and those of African descent have the highest frequencies of the putative risk alleles (Esteban et al. 2006; Kittles et al. 2001; Zeigler-Johnson et al. 2002). Of interest is that one study found an association with a CYP3A4 promoter variant in both African Americans and European Americans, but not in Nigerians, indicating the complexity of analyzing stratified data (Kittles et al. 2002; Hainaut and Boyle 2008; Lessells and Cooke 2008) (Table 5). Gene expression profiles of tumors obtained by microarray technology from African–American and European–American patients point to prominent differences in primary prostate cancer immunobiology between African–American and European–American men (Wallace et al. 2008).
Recent genome-wide and linkage scans that have investigated associations between polymorphisms and prostate cancer in multiple ethnic groups provide support for at least five risk-associated chromosomal regions, three of which are at 8q24 (Freedman et al. 2006; Duggan et al. 2007; Haiman et al. 2007; Robbins et al. 2007; Schumacher et al. 2007; Yeager et al. 2007; Zheng et al. 2008). The available evidence compellingly indicates that the reasons for the disparity in prostate cancer risk between African Americans and other ethnic groups involve the differential distribution of 8q24 markers on African chromosomes, although the responsible genes in the chromosomal region remain to be identified. These results have not yet been extended to studies of African populations.
Colorectal cancer
Preliminary data indicate that colorectal cancer exhibits a multimodal distribution, reflecting heterogeneity, with different contributions from genetic and environmental factors. However, the effects of urbanization seem to be increasing the incidence of disease in previously largely rural African populations. Evidence that heritable factors are stronger in any one population is missing; however, it is known that when cancer occurs at a younger age in African populations, it is likely to be more aggressive.
Up to 20% of colorectal cancers in individuals under the age of 50 years appear to be hereditary (a combination of Hereditary Nonpolypotic Colorectal Cancers, i.e. HNPCC, and Familial Adenomatous Polyposis, or FAP). A similar proportion of HNPCC or Lynch syndrome was reported in a small sampling of colorectal cancers in Nigeria (Adebamowo et al. 2000).
A wide range of predisposing mutations has been identified in individuals of various ethnic groups. In one study, a single founder mutation (g.1528C > T in the hMLH1 gene) has been shown to underlie a major burden of disease in the Nama group of the far Northern Cape Province of South Africa (Anderson et al. 2007). In another study null mutations in GSTM1 and GSTT1 were studied in a cohort where all subjects carried an hMLH1 mutation. It was shown that individuals with both null mutations had a threefold increased risk of cancer at an earlier age (Felix et al. 2006) (Table 5). These studies indicate the potential of not only identifying important variants, but in studying their effects on each other.
Breast cancer
Breast cancer (BC) is the most common cancer of women worldwide, and is a major malignancy in African women. The estimated age-standardized rates (ASR) for breast cancer incidence in sub-Saharan Africa ranges from 15 to 53 per 100,000 women (Ferlay et al. 2004). Even though diagnosed breast cancer is less prevalent in Africa than Europe, due to late diagnosis and poor survival, mortality rates estimated for Africa are not lower than those registered in Europe.
Studies that compared extensive series of African–American and European–American breast cancer patients found associations between aggressive estrogen receptor (ER)-negative BC and both young age at diagnosis and black ethnicity (Carey et al. 2006; Porter 2008). These data raise the possibility that genetic factors could contribute to a higher burden of aggressive ER-negative breast cancer in indigenous African populations. However, recent data from Sudan and Nigeria do not support this hypothesis, but suggest that the differences between African and European women reflect stage at diagnosis rather than intrinsic biological characteristics (Adebamowo et al. 2008; Awadelkarim et al. 2008).
As in industrialized countries, strong genetic factors contribute to a subset of breast cancer cases in Africa. Pilot studies in Nigeria, Sudan and Tunisia show that mutations in the two major susceptibility genes, BRCA1 and BRCA2, account for variable but significant fractions of the premenopausal cases (Awadelkarim et al. 2007; Fackenthal et al. 2005; Gao et al. 2000; Troudi et al. 2007) (Table 5). From Nigeria the data suggest that both truncating and non-truncating mutations of BRCA1 or BRCA2 occur more frequently in young women with breast cancer (Fackenthal et al. 2005; Gao et al. 2000). The observations of higher levels of genetic variation in the BRCA genes are supported by other studies (Wagner et al. 1999). Data from Sudan also suggest that BRCA1/2 mutations could represent an important etiological factor in male patients and in young female patients less exposed to pregnancy and lactation (Awadelkarim et al. 2007).
Other cancers
Hepatocellular carcinoma is the second most common cancer of men in sub-Saharan Africa (Parkin et al. 2003). As with other major cancers of Africa, it is associated with environmental factors such as early infection with hepatitis viruses types B and C that interact with dietary exposure to aflatoxins from Aspergillus molds and specific genetic variants in Africa (Kirk et al. 2000, 2005a, 2006; Montesano et al. 1997; Hainaut and Boyle 2008; Lessells and Cooke 2008) (Table 5).
Cancer of the bladder occurs with particularly high frequency in North Africa, where the main histotype is transitional cell carcinoma (as in industrialized countries). With regard to transitional cell carcinoma, studies conducted in Tunisia and Egypt support the view that individual susceptibility is modulated by genetic variation in pathways that control metabolic detoxification, redox cycling, free radical injury, and metabolism of folate and methionine, critical for DNA synthesis/repair and methylation (Ouerhani et al. 2007, 2006; Saad et al. 2005). In Egypt GSTT1, GSTM1 and GSTP1 genotypes all associate with bladder cancer, but the sample sizes in this study were small (Saad et al. 2005). Similar results were found in Tunisia for GSTM1, but not GSTT1 (Ouerhani et al. 2006). Additional work in Tunisia indicates that methylenetetrahydrofolate reductase and methionine synthase genes associate with bladder cancer (Ouerhani et al. 2007). Unfortunately, almost nothing is known about the influence of genetic factors on bladder cancer in sub-Saharan Africa.
Nasopharyngeal carcinoma is an undifferentiated neoplasm with marked lymphocytic infiltration that arises in the squamous epithelium overlying the nasopharyngeal lymphoid tissue. There is evidence suggesting that certain genotypes in HLA class I, TP53, and antigen processing genes influence susceptibility in North Africa (Hadhri-Guiga et al. 2007; Hassen et al. 2007; Li et al. 2007b).
Podoconiosis: a paradigm of genetics and environment interactions
Podoconiosis (non-infectious geochemical elephantiasis) is a chronic tropical disease that phenotypically resembles filariasis (Davey et al. 2007b). Although not widely recognised, prevalence rates of over 5% have been reported in endemic areas, where it is more common than HIV/AIDS or tuberculosis (Destas et al. 2003). Exposure to red alkalic clay soil, in individuals who cannot afford footwear, leads to the absorption of silicate particles. These induce an inflammatory response in some, but not all, individuals, even though silicate particles have been identified in inguinal lymph nodes in unaffected individuals. Without intervention, chronic inflammation leads to lymphatic obstruction and the clinical phenotype of progressive asymmetrical bilateral swelling of the lower leg (Price 1990). Podoconiosis can be differentiated from its phenocopy, infectious filariasis (caused by various nematode species such as Wucheria bancrofti) on clinical grounds since disease is often symmetrical and extends above the knee in filariasis. Furthermore, podoconiosis occurs in high altitude settings that preclude transmission of filariasis by its mosquito vector.
Since only a proportion of exposed individuals develop disease and the disease clusters in families, the hypothesis that genetic factors determine whether an individual is susceptible to disease was tested in the Wolaitta region of Ethiopia. Multiplex family analysis estimated the heritability of podoconiosis to be 0.62 with a single major dominant gene as the most parsimonious model (Davey et al. 2007a). Genetic studies towards gene identification are planned, and compared to other chronic diseases, the genetic basis of podoconiosis appears to be relatively simple.
Genetics and disease prevention/treatment
Vaccine-induced immunity
Given the fact that infectious diseases play such an important role in the health of African populations, it is important to understand how genetic variation affects the efficacy of vaccines. The Extended Program in Immunisation (EPI) introduced by WHO and organizations such as the Global Alliance for Vaccines and Immunisation (GAVI) work towards the prevention of predominantly childhood diseases through vaccination. Vaccines currently delivered on a routine basis across the African continent are: BCG for TB and leprosy, individual or combination vaccination against diphtheria, tetanus and pertussis (DTP), oral poliomyelitis (OVP), and vaccinations against measles, and yellow fever. Additionally, Haemophilus influenzae (Hib) and hepatitis B virus infection (HBV) vaccines are recommended by the WHO but do not form part of the routine program in most countries with exceptions such as The Gambia. More recent immunizations are pneumococcal and meningococcal vaccines (in some instances targeted at high-risk groups) and others are still in a more or less promising experimental phase, such as malaria and HIV vaccines. Many factors that are known to influence immune responses to vaccines will not be discussed here including, the vaccine, adjuvants, age, gender, UV light exposure, smoking, infectious diseases, nutritional factors, etc. (reviewed by van Loveren et al. 2001).
Immune responses induced by vaccination are in part under genetic control, and the degree of heritability varies by vaccine between 35 and 90%, as shown by family and twin studies both within African settings (Lee et al. 2006; Marchant et al. 2006; Newport et al. 2004b, 2005) and across the rest of the world (Alper 1995; De et al. 2001; Hohler et al. 2002; Konradsen et al. 1993, 1994; Kruger et al. 2005; Lin et al. 1989; Musher et al. 2000, Musher et al. 1997 and reviewed by Kimman et al. 2007). Additionally, we know that differences in vaccine efficacy exist between different ethnic groups, also indicating a putative role for genetic factors (Kimman et al. 2007). Vaccine efficacy in a given population can be affected by the frequency of protective alleles, emphasizing the importance of ethnic comparisons for a thorough understanding of the role of genetics in determining or modulating immune responses. However, the heterogeneity in vaccine-induced immunity (also termed vaccinomics) is not well understood and little data are available from genetic studies worldwide, let alone Africa (Kimman et al. 2007; Ovsyannikova et al. 2004a; Poland et al. 2007; Poland and Jacobson 1998). Host genetic variation may affect multiple processes such as antigen presentation and recognition, the magnitude or kinetics of vaccine-induced antibody response, lymphocyte proliferation, and long-term immune memory.
The most exhaustively studied region of the human genome with respect to the correlation of immuno-phenotypic and genotypic data is the HLA region. As noted above, HLA variation has also been studied extensively in terms of susceptibility to the disease themselves (Tables 6, 7, Supplemental Tables). Relatively consistent findings have been reported for HLA associations with immunity induced by HBV vaccination (Kimman et al. 2007; Milich and Leroux-Roels 2003; Thursz 2001); the HLA data on responses to other vaccinations, except measles (Ovsyannikova et al. 2004c, 2006a), are sparse. However, even less is published in relation to variation in non-HLA genes. Table 7 lists existing publications on both HLA and other candidate loci, concentrating on English language reports and vaccines administered routinely across Africa. Most of these studies are hampered by small sample sizes, a limited number of markers/genes screened, poor information on covariates and environmental factors, differences in study design and analysis; therefore we only have a snapshot of what genetic factors are implicated in the control of vaccine induced immunity. Furthermore, genetically distinct populations have been studied, making comparisons difficult. However, even if good data on host genetic variability as well as comprehensive information on clinical, serological, demographic and environmental factors were available several issues would remain unclear. For instance, how accurate is the measurement of currently used correlates of protection, such as vaccine-induced antibody level? What effect does natural boosting through infection have on the evaluation of vaccine efficacy? How relevant is the genetic variability of the infectious agent and will long-term vaccination programs lead to the rise of vaccine-escape mutants? Will functionally relevant variants be identified from family, twin, cohort and case–control studies?
Pharmacogenetics
It has been well documented that genetic differences exist among individuals that impact the efficacy of specific drug treatments. In some cases this is due to the ability to process/metabolize drugs that can occur at ADMET genes that determine drug Absorption and Distribution (transporters and plasma proteins), drug Metabolism and Excretion (metabolising enzymes and transporters) as well as Toxicity. In other situations, such as non-communicable diseases, the efficacy may be more tied to the actual etiological risk. For example, it has been shown in Black South Africans that AGT genotype can affect response to ACE inhibitor therapy for hypertension (Woodiwiss et al. 2006). Pharmacogenetics aims at understanding this genetic diversity underlying the pharmacokinetic and pharmacodynamic variability in drug response among patients, enabling personalized treatment, optimal dosing and minimal adverse effects.
With respect to drug metabolism, the genes most intensively studied so far encode drug transporters and drug metabolising enzymes such as cytochrome P450s (CYPs), glucuronyl transferases (UGTs), N-acetyl transferases (NATs), epoxide hydrolases (EHs), glutathione S-transferases (GSTs), flavin monooxygenases (FMOs) and multidrug transporters (MDR). These genes are highly polymorphic and their variation results in proteins or enzymes with enhanced, normal or reduced capacity, thereby dividing populations into groups of extensive, intermediate or poor metabolisers. Of these the CYP genes show the highest level of variation. For example, 70 alleles of CYP2D6 have been reported so far, yet only a few haplotypes were found to be of functional importance (http://www.cypalleles.ki.se). Whereas most of this diversity is caused by single nucleotide polymorphisms (SNPs), gene copy number variation (CNV) has also been found in CYP2D6, GSTM1 and GSTT1 (Gaedigk et al. 2007; Ingelman-Sundberg et al. 2007; Ouahchi et al. 2006; Rotger et al. 2007). It has been shown that in some cases the CYP genotyping efforts are less predictive of metabolizer status in African Americans than European Americans (Gaedigk et al. 2005). Such findings are suggestive of the role that total genetic variation may play in prediciting drug efficacy and underscore the need to perform analyses in a diversity of populations.
The distribution of drug response alleles shows distinct clusters among the world populations (Aklillu et al. 2007; Sistonen et al. 2007). In addition, there is inter-individual variation that in some cases supersedes population diversity (Sistonen et al. 2007). Due to limited data on African genetics/polymorphisms, no comprehensive patterns of variation are known for African populations so far. African-specific SNPs have been reported for the CYP, NAT, FMO genes (Allabi et al. 2005, 2004; Yasar et al. 2002), but their population or inter-individual variation are not well understood. In addition, phenotypic expression of polymorphisms may differ in individuals of different ethnicities and environments (Aklillu et al. 2002). This suggests an urgent need for population as well as location based genotype–phenotype correlation studies.
The high frequency of reduced-function alleles CYP2D6*17 and *29 in Africans predicts a considerably higher intermediate-to-poor metabolizer status than in people of European descent (Bertilsson et al. 2002; Masimirembwa et al. 1996; Wennerholm et al. 2001). The CYP2C19*2 allele currently accounts for most (60%) of the poor metaboliser phenotype for substrates of the CYP2C19 enzyme in Africans and Europeans (Bathum et al. 1999; Masimirembwa et al. 1995). The commonly known polymorphisms of NAT2 include alleles *5, *6, *7 and the African-specific *14 allele, and all affect acetylator status in carrier individuals (Dandara et al. 2003). They may translate into ultra-rapid or complete absence of metabolism of some substrates, with important implications for dosage adjustments in patients carrying these alleles.
Since few African populations have been studied so far and the documentation of pharmacogenetic information is scarce, attempts to establish biobanking initiatives and pharmacogenetic databases are underway (Matimba et al. 2008; http://www.aibst.com/biobank). Such databases could be a very helpful tool in promoting drug discovery and development in the public and private sectors.
The data collected for both vaccine and drug responses might eventually lead to the implementation of screening protocols, although this is feasible only in a few African environments presently. Pre-prescription genotyping has been recommended for CYP2D6 and CYP2C19 in antipsychotic therapy (Kirchheiner et al. 2001; Masimirembwa and Hasler 1997). In anticoagulant therapy, CYP2C9 and Vitamin K epoxide reductase subunit 1 (VKORC1) genotyping can help to predict the starting dose of the drug warfarin (Wadelius and Pirmohamed 2006). Host genetic factors may also influence HIV treatment efficacy and safety; for example, the human leukocyte antigen HLA-B*5701 allele has been associated with abacavir sensitivity (Lucas et al. 2007), so patient screening for this allele should minimise incidences of adverse reactions or hypersensitivity. “African” polymorphisms need to be incorporated into the development of these applications, as individuals may be at higher risk of dose-related adverse drug reactions or less efficacious treatment when taking doses recommended for Europeans. For example, the CYP2B6 516G > T polymorphism is highly prevalent in Africans and results in reduced enzyme function (Klein et al. 2005). This has implications for drug toxicity due to high plasma concentrations (Rotger et al. 2005). A recent study showed that individuals carrying this mutation can be treated with reduced dosage and still achieve therapeutic outcomes (Nyakutira et al. 2008). Pre-prescription genotyping of patients should, therefore, result in minimal side effects and lower cost of treatment. Such an outcome is particularly relevant in Africa where healthcare cost usually outstrips affordability by individuals or governments.
Conclusions
Nearly 2000 years ago the Roman scholar and natural philosopher Pliny the Elder wrote in his Natural History: “Ex Africa surgit semper aliquid novi” (from Africa there is always something new); this quote beautifully applies to genetic studies of African populations as they provide a critical resource in the study of genetic risk factors of human disease and to new discoveries. By doing studies throughout Africa it will be possible to capture most of the extant genetic risk factors in all human populations. It may also be possible to use simple and relatively inexpensive genetic tests to reduce overall healthcare costs. Finally, as pointed out there are many diseases that are endemic to Africa that carry significant genetic risk, and studying these could improve the health in Africa. However, despite the advantages and importance of these studies there are substantial impediments to performing genetic research in an African setting, most notably lack of resources and infrastructure. In recognition of these factors it has been argued that bio-banks need to be developed to expedite research (Sgaier et al. 2007; Sirugo et al. 2004). There is an increasing awareness that it is not only important to coordinate research efforts, overcome “territorial issues”, and share resources between research teams, but also that there is an essential need for training African scientists who can lead and promote genetic research in Africa. Such efforts are ongoing and form the basis for many of the objectives of the African Society of Human Genetics that was formed in 2003 (Rotimi 2004). Many such efforts are still in their infancy, as evidenced by the lack of research discussed in this review for some diseases or for vaccines and treatment, and although some progress has been made, as stated in the African proverb “thunder is not yet rain”, it is just the beginning and a lot more needs to be done.
References
Abdel-Salam E, Abdel Khalik A, Abdel-Meguid A, Barakat W, Mahmoud AA (1986) Association of HLA class I antigens (A1, B5, B8 and CW2) with disease manifestations and infection in human schistosomiasis mansoni in Egypt. Tissue Antigens 27:142–146
Abubakari AR, Lauder W, Agyemang C, Jones M, Kirk A, Bhopal RS (2008) Prevalence and time trends in obesity among adult West African populations: a meta-analysis. Obes Rev (in press)
Adebamowo CA, Adeyi O, Pyatt R, Prior TW, Chadwick RB, de La Chapelle A (2000) Case report on hereditary non-polyposis colon cancer (HNPCC) in Nigeria. Afr J Med Med Sci 29:71–73
Adebamowo CA, Famooto A, Ogundiran TO, Aniagwu T, Nkwodimmah C, Akang EE (2008) Immunohistochemical and molecular subtypes of breast cancer in Nigeria. Breast Cancer Res Treat (in press)
Adeyemo AA, Omotade OO, Rotimi CN, Luke AH, Tayo BO, Cooper RS (2002) Heritability of blood pressure in Nigerian families. J Hypertens 20:859–863
Adeyemo AA, Johnson T, Acheampong J, Oli J, Okafor G, Amoah A, Owusu S, Agyenim-Boateng K, Eghan BA Jr, Abbiyesuku F, Fasanmade O, Rufus T, Doumatey A, Chen G, Zhou J, Chen Y, Furbert-Harris P, Dunston G, Collins F, Rotimi C (2005) A genome wide quantitative trait linkage analysis for serum lipids in type 2 diabetes in an African population. Atherosclerosis 181:389–397
Adje CA, Bile CE, Kestens L, Koblavi-Deme S, Ghys PD, Maurice C, Kalou-Badirou M, Kabran N, Ekpini RE, Roels TH, Wiktor SZ, Nkengasong JN (2001) Lack of effect of chemokine receptor CCR2b gene polymorphism (64I) on HIV-1 plasma RNA viral load and immune activation among HIV-1 seropositive female workers in Abidjan, Cote d’Ivoire. J Med Virol 64:398–401
Agarwal A, Guindo A, Cissoko Y, Taylor JG, Coulibaly D, Kone A, Kayentao K, Djimde A, Plowe CV, Doumbo O, Wellems TE, Diallo D (2000) Hemoglobin C associated with protection from severe malaria in the Dogon of Mali, a West African population with a low prevalence of hemoglobin S. Blood 96:2358–2363
Aklillu E, Herrlin K, Gustafsson LL, Bertilsson L, Ingelman-Sundberg M (2002) Evidence for environmental influence on CYP2D6-catalysed debrisoquine hydroxylation as demonstrated by phenotyping and genotyping of Ethiopians living in Ethiopia or in Sweden. Pharmacogenetics 12:375–383
Aklillu E, Dandara C, Bertilsson L, Masimirembwa C (2007) Pharmacogenetics of cytochrome P450s in African populations: clinical and molecular evolutionary implications. In: Suarez-Kurtz G (ed) Pharmacogenomics in admixed populations. Landes Bioscience, Austin
Alberts M, Urdal P, Steyn K, Stensvold I, Tverdal A, Nel JH, Steyn NP (2005) Prevalence of cardiovascular diseases and associated risk factors in a rural black population of South Africa. Eur J Cardiovasc Prev Rehabil 12:347–354
Ali S, Niang MA, N’doye I, Critchlow CW, Hawes SE, Hill AV, Kiviat NB (2000) Secretor polymorphism and human immunodeficiency virus infection in Senegalese women. J Infect Dis 181:737–739
Allabi AC, Gala JL, Horsmans Y, Babaoglu MO, Bozkurt A, Heusterspreute M, Yasar U (2004) Functional impact of CYP2C95, CYP2C96, CYP2C98, and CYP2C911 in vivo among black Africans. Clin Pharmacol Ther 76:113–118
Allabi AC, Gala JL, Horsmans Y (2005) CYP2C9, CYP2C19, ABCB1 (MDR1) genetic polymorphisms and phenytoin metabolism in a Black Beninese population. Pharmacogenet Genomics 15:779–786
Alm JS, Sanjeevi CB, Miller EN, Dabadghao P, Lilja G, Pershagen G, Blackwell JM, Scheynius A (2002) Atopy in children in relation to BCG vaccination and genetic polymorphisms at SLC11A1 (formerly NRAMP1) and D2S1471. Genes Immun 3:71–77
Alper CA (1995) The human immune response to hepatitis B surface antigen. Exp Clin Immunogenet 12:171–181
Alter A, Alcais A, Abel L, Schurr E (2008) Leprosy as a genetic model for susceptibility to common infectious diseases. Hum Genet (in press)
Alves S, Rocha J, Amorim A, Prata MJ (2004) Tracing the origin of the most common thiopurine methyltransferase (TPMT) variants: preliminary data from the patterns of haplotypic association with two CA repeats. Ann Hum Genet 68:313–323
Ambrosino DM, Schiffman G, Gotschlich EC, Schur PH, Rosenberg GA, DeLange GG, van Loghem E, Siber GR (1985) Correlation between G2 m(n) immunoglobulin allotype and human antibody response and susceptibility to polysaccharide encapsulated bacteria. J Clin Invest 75:1935–1942
Ameyaw MM, Regateiro F, Li T, Liu X, Tariq M, Mobarek A, Thornton N, Folayan GO, Githang’a J, Indalo A, Ofori-Adjei D, Price-Evans DA, McLeod HL (2001) MDR1 pharmacogenetics: frequency of the C3435T mutation in exon 26 is significantly influenced by ethnicity. Pharmacogenetics 11:217–221
Amodu OK, Adeyemo AA, Ayoola OO, Gbadegesin RA, Orimadegun AE, Akinsola AK, Olumese PE, Omotade OO (2005a) Genetic diversity of the msp-1 locus and symptomatic malaria in south-west Nigeria. Acta Trop 95:226–232
Amodu OK, Gbadegesin RA, Ralph SA, Adeyemo AA, Brenchley PE, Ayoola OO, Orimadegun AE, Akinsola AK, Olumese PE, Omotade OO (2005b) Plasmodium falciparum malaria in south-west Nigerian children: is the polymorphism of ICAM-1 and E-selectin genes contributing to the clinical severity of malaria? Acta Trop 95:248–255
Anderson DW, Goldberg PA, Algar U, Felix R, Ramesar RS (2007) Mobile colonoscopic surveillance provides quality care for hereditary nonpolyposis colorectal carcinoma families in South Africa. Colorectal Dis 9:509–514
Arya R, Blangero J, Williams K, Almasy L, Dyer TD, Leach RJ, O’Connell P, Stern MP, Duggirala R (2002) Factors of insulin resistance syndrome—related phenotypes are linked to genetic locations on chromosomes 6 and 7 in nondiabetic Mexican–Americans. Diabetes 51:841–847
Atkinson SH, Mwangi TW, Uyoga SM, Ogada E, Macharia AW, Marsh K, Prentice AM, Williams TN (2007) The haptoglobin 2-2 genotype is associated with a reduced incidence of Plasmodium falciparum malaria in children on the coast of Kenya. Clin Infect Dis 44:802–809
Aucan C, Walley AJ, Greenwood BM, Hill AV (2002) Haptoglobin genotypes are not associated with resistance to severe malaria in The Gambia. Trans R Soc Trop Med Hyg 96:327–328
Aucan C, Walley AJ, Hennig BJ, Fitness J, Frodsham A, Zhang L, Kwiatkowski D, Hill AV (2003) Interferon-alpha receptor-1 (IFNAR1) variants are associated with protection against cerebral malaria in the Gambia. Genes Immun 4:275–282
Aucan C, Walley AJ, Hill AV (2004) Common apolipoprotein E polymorphisms and risk of clinical malaria in the Gambia. J Med Genet 41:21–24
Awadelkarim KD, Aceto G, Veschi S, Elhaj A, Morgano A, Mohamedani AA, Eltayeb EA, Abuidris D, Di GM, Battista P, Verginelli F, Cama A, Elwali NE, Mariani-Costantini R (2007) BRCA1 and BRCA2 status in a Central Sudanese series of breast cancer patients: interactions with genetic, ethnic and reproductive factors. Breast Cancer Res Treat 102:189–199
Awadelkarim KD, Arizzi C, Elamin EO, Hamad HM, De Blasio P, Mekki SO, Osman I, Biunno I, Elwali NE, Mariani-Costantini R, Barberis MC. (2008) Pathological, clinical and prognostic characteristics of breast cancer in Central Sudan versus Northern Italy: implications for breast cancer in Africa. Histopathology 52:445–456
Awomoyi AA, Marchant A, Howson JM, McAdam KP, Blackwell JM, Newport MJ (2002) Interleukin-10, polymorphism in SLC11A1 (formerly NRAMP1), and susceptibility to tuberculosis. J Infect Dis 186:1808–1814
Awomoyi AA, Nejentsev S, Richardson A, Hull J, Koch O, Podinovskaia M, Todd JA, McAdam KP, Blackwell JM, Kwiatkowski D, Newport MJ (2004) No association between interferon-gamma receptor-1 gene polymorphism and pulmonary tuberculosis in a Gambian population sample. Thorax 59:291–294
Awomoyi AA, Charurat M, Marchant A, Miller EN, Blackwell JM, McAdam KP, Newport MJ (2005) Polymorphism in IL1B: IL1B–511 association with tuberculosis and decreased lipopolysaccharide-induced IL-1beta in IFN-gamma primed ex-vivo whole blood assay. J Endotoxin Res 11:281–286
Babb C, van der Merwe L, Beyers N, Pheiffer C, Walzl G, Duncan K, van Helden P, Hoal EG (2007) Vitamin D receptor gene polymorphisms and sputum conversion time in pulmonary tuberculosis patients. Tuberculosis (Edinb) 87:295–302
Baccar Harrath A, Yacoubi Loueslati B, Troudi W, Hmida S, Sedkaoui S, Dridi A, Jridi A, Ben Ayed F, Ben Rhomdhane K, Ben Ammar Elgaaied A (2006) HLA class II polymorphism: protective or risk factors to breast cancer in Tunisia? Pathol Oncol Res 12:79–81
Baghdadi JE, Orlova M, Alter A, Ranque B, Chentoufi M, Lazrak F, Archane MI, Casanova JL, Benslimane A, Schurr E, Abel L (2006) An autosomal dominant major gene confers predisposition to pulmonary tuberculosis in adults. J Exp Med 203:1679–1684
Ball TB, Ji H, Kimani J, McLaren P, Marlin C, Hill AV, Plummer FA (2007) Polymorphisms in IRF-1 associated with resistance to HIV-1 infection in highly exposed uninfected Kenyan sex workers. AIDS 21:1091–1101
Banus S, Bottema RW, Siezen CL, Vandebriel RJ, Reimerink J, Mommers M, Koppelman GH, Hoebee B, Thijs C, Postma DS, Kimman TG, Stelma FF (2007) Toll-like receptor 4 polymorphism associated with the response to whole-cell pertussis vaccination in children from the KOALA study. Clin Vaccine Immunol 14:1377–1380
Barlassina C, Norton GR, Samani NJ, Woodwiss AJ, Candy GC, Radevski I, Citterio L, Bianchi G, Cusi D (2000) Alpha-adducin polymorphism in hypertensives of South African ancestry. Am J Hypertens 13:719–723
Barreiro LB, Neyrolles O, Babb CL, Tailleux L, Quach H, McElreavey K, Helden PD, Hoal EG, Gicquel B, Quintana-Murci L (2006) Promoter variation in the DC-SIGN-encoding gene CD209 is associated with tuberculosis. PLoS Med 3:e20
Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L (2008) Natural selection has driven population differentiation in modern humans. Nat Genet 40:340–345
Bathum L, Skjelbo E, Mutabingwa TK, Madsen H, Horder M, Brosen K (1999) Phenotypes and genotypes for CYP2D6 and CYP2C19 in a black Tanzanian population. Br J Clin Pharmacol 48:395–401
Baum J, Pinder M, Conway DJ (2003) Erythrocyte invasion phenotypes of Plasmodium falciparum in The Gambia. Infect Immun 71:1856–1863
Baynam G, Khoo SK, Rowe J, Zhang G, Laing I, Hayden C, Kusel M, DeKlerk N, Sly P, Goldblatt J, Holt P, LeSouef P (2007) Parental smoking impairs vaccine responses in children with atopic genotypes. J Allergy Clin Immunol 119:366–374
Bel Hadj Jrad B, Chatti A, Laatiri A, Ahmed SB, Romdhane A, Ajimi S, Chouchane L (2007) Tumor necrosis factor promoter gene polymorphism associated with increased susceptibility to non-Hodgkin’s lymphomas. Eur J Haematol 78:117–122
Bellamy R, Kwiatkowski D, Hill AV (1998a) Absence of an association between intercellular adhesion molecule 1, complement receptor 1 and interleukin 1 receptor antagonist gene polymorphisms and severe malaria in a West African population. Trans R Soc Trop Med Hyg 92:312–316
Bellamy R, Ruwende C, Corrah T, McAdam KP, Whittle HC, Hill AV (1998b) Assessment of the interleukin 1 gene cluster and other candidate gene polymorphisms in host susceptibility to tuberculosis. Tuber Lung Dis 79:83–89
Bellamy R, Ruwende C, Corrah T, McAdam KP, Whittle HC, Hill AV (1998c) Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N Engl J Med 338:640–644
Bellamy R, Ruwende C, McAdam KP, Thursz M, Sumiya M, Summerfield J, Gilbert SC, Corrah T, Kwiatkowski D, Whittle HC, Hill AV (1998d) Mannose binding protein deficiency is not associated with malaria, hepatitis B carriage nor tuberculosis in Africans. QJM 91:13–18
Bellamy R, Ruwende C, Corrah T, McAdam KP, Thursz M, Whittle HC, Hill AV (1999) Tuberculosis and chronic hepatitis B virus infection in Africans and variation in the vitamin D receptor gene. J Infect Dis 179:721–724
Bellamy R, Beyers N, McAdam KP, Ruwende C, Gie R, Samaai P, Bester D, Meyer M, Corrah T, Collin M, Camidge DR, Wilkinson D, Hoal-Van Helden E, Whittle HC, Amos W, van Helden P, Hill AV (2000) Genetic susceptibility to tuberculosis in Africans: a genome-wide scan. Proc Natl Acad Sci USA 97:8005–8009
Ben-Ali M, Barbouche MR, Bousnina S, Chabbou A, Dellagi K (2004) Toll-like receptor 2 Arg677Trp polymorphism is associated with susceptibility to tuberculosis in Tunisian patients. Clin Diagn Lab Immunol 11:625–626
Ben-Ali M, Barreiro LB, Chabbou A, Haltiti R, Braham E, Neyrolles O, Dellagi K, Gicquel B, Quintana-Murci L, Barbouche MR (2007) Promoter and neck region length variation of DC-SIGN is not associated with susceptibility to tuberculosis in Tunisian patients. Hum Immunol 68:908–912
Bendjemana K, Abdennebi M, Gara S, Jmal A, Ghanem A, Touati S, Boussen H, Ladgham A, Guemira F (2006) Genetic polymorphism of gluthation-S transferases and N-acetyl transferases 2 and nasopharyngeal carcinoma: the Tunisia experience. Bull Cancer 93:297–302
Bennett S, Allen SJ, Olerup O, Jackson DJ, Wheeler JG, Rowe PA, Riley EM, Greenwood BM (1993) Human leucocyte antigen (HLA) and malaria morbidity in a Gambian community. Trans R Soc Trop Med Hyg 87:286–287
Bertilsson L, Dahl ML, Dalen P, Al Shurbaji A (2002) Molecular genetics of CYP2D6: clinical relevance with focus on psychotropic drugs. Br J Clin Pharmacol 53:111–122
Bienzle U, Eggelte TA, Adjei LA, Dietz E, Ehrhardt S, Cramer JP, Otchwemah RN, Mockenhaupt FP (2005) Limited influence of haptoglobin genotypes on severe malaria in Ghanaian children. Trop Med Int Health 10:668–671
Black GF, Weir RE, Floyd S, Bliss L, Warndorff DK, Crampin AC, Ngwira B, Sichali L, Nazareth B, Blackwell JM, Branson K, Chaguluka SD, Donovan L, Jarman E, King E, Fine PE, Dockrell HM (2002) BCG-induced increase in interferon-gamma response to mycobacterial antigens and efficacy of BCG vaccination in Malawi and the UK: two randomised controlled studies. Lancet 359:1393–1401
Blanton RE, Salam EA, Ehsan A, King CH, Goddard KA (2005) Schistosomal hepatic fibrosis and the interferon gamma receptor: a linkage analysis using single-nucleotide polymorphic markers. Eur J Hum Genet 13:660–668
Boldt AB, Luty A, Grobusch MP, Dietz K, Dzeing A, Kombila M, Kremsner PG, Kun JF (2006) Association of a new mannose-binding lectin variant with severe malaria in Gabonese children. Genes Immun 7:393–400
Bonilla C, Panguluri RK, Taliaferro-Smith L, Argyropoulos G, Chen G, Adeyemo AA, Amoah A, Owusu S, Acheampong J, Agyenim-Boateng K, Eghan BA, Oli J, Okafor G, Abbiyesuku F, Johnson T, Rufus T, Fasanmade O, Chen Y, Collins FS, Dunston GM, Rotimi C, Kittles RA (2006) Agouti-related protein promoter variant associated with leanness and decreased risk for diabetes in West Africans. Int J Obes (Lond) 30:715–721
Bornman L, Campbell SJ, Fielding K, Bah B, Sillah J, Gustafson P, Manneh K, Lisse I, Allen A, Sirugo G, Sylla A, Aaby P, McAdam KP, Bah-Sow O, Bennett S, Lienhardt C, Hill AV (2004) Vitamin D receptor polymorphisms and susceptibility to tuberculosis in West Africa: a case-control and family study. J Infect Dis 190:1631–1641
Bouzekri N, Zhu X, Jiang Y, McKenzie CA, Luke A, Forrester T, Adeyemo A, Kan D, Farrall M, Anderson S, Cooper RS, Ward R (2004) Angiotensin I-converting enzyme polymorphisms, ACE level and blood pressure among Nigerians, Jamaicans and African–Americans. Eur J Hum Genet 12:460–468
Brawley OW (1998) Prostate cancer and black men. Semin Urol Oncol 16:184–186
Bucheton B, Abel L, Kheir MM, Mirgani A, El-Safi SH, Chevillard C, Dessein A (2003a) Genetic control of visceral leishmaniasis in a Sudanese population: candidate gene testing indicates a linkage to the NRAMP1 region. Genes Immun 4:104–109
Bucheton B, Abel L, El-Safi S, Kheir MM, Pavek S, Lemainque A, Dessein AJ (2003b) A major susceptibility locus on chromosome 22q12 plays a critical role in the control of kala-azar. Am J Hum Genet 73:1052–1060
Bunker CH, Patrick AL, Maharaj G, Keenan HA, Ramnarine S, Belle A, Richard JR, Dhir R (2002) Prostate cancer risk is three-fold higher among men, aged 50–64, of African descent compared with men of Asian–Indian descent in Trinidad and Tobago. Ethn Dis 12:S3
Burgner D, Usen S, Rockett K, Jallow M, Ackerman H, Cervino A, Pinder M, Kwiatkowski DP (2003) Nucleotide and haplotypic diversity of the NOS2A promoter region and its relationship to cerebral malaria. Hum Genet 112:379–386
Burton MJ, Adegbola RA, Kinteh F, Ikumapayi UN, Foster A, Mabey DC, Bailey RL (2007) Bacterial infection and trachoma in The Gambia: a case control study. Invest Ophthalmol Vis Sci 48:4440–4444
Cabantous S, Poudiougou B, Traore A, Keita M, Cisse MB, Doumbo O, Dessein AJ, Marquet S (2005) Evidence that interferon-gamma plays a protective role during cerebral malaria. J Infect Dis 192:854–860
Campbell SJ, Sabeti P, Fielding K, Sillah J, Bah B, Gustafson P, Manneh K, Lisse I, Sirugo G, Bellamy R, Bennett S, Aaby P, McAdam KP, Bah-Sow O, Lienhardt C, Hill AV (2003) Variants of the CD40 ligand gene are not associated with increased susceptibility to tuberculosis in West Africa. Immunogenetics 55:502–507
Campino S, Kwiatkowski D, Dessein A (2006) Mendelian and complex genetics of susceptibility and resistance to parasitic infections. Semin Immunol 18:411–422
Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC (2006) Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295:2492–2502
Carter R, Mendis KN (2002) Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev 15:564–594
Casals-Pascual C, Allen S, Allen A, Kai O, Lowe B, Pain A, Roberts DJ (2001) Short report: codon 125 polymorphism of CD31 and susceptibility to malaria. Am J Trop Med Hyg 65:736–737
Cervino AC, Lakiss S, Sow O, Hill AV (2000) Allelic association between the NRAMP1 gene and susceptibility to tuberculosis in Guinea-Conakry. Ann Hum Genet 64:507–512
Cervino AC, Lakiss S, Sow O, Bellamy R, Beyers N, Hoal-van Helden E, van Helden P, McAdam KP, Hill AV (2002) Fine mapping of a putative tuberculosis-susceptibility locus on chromosome 15q11–13 in African families. Hum Mol Genet 11:1599–1603
Chen YS, Olckers A, Schurr TG, Kogelnik AM, Huoponen K, Wallace DC (2000) mtDNA variation in the South African Kung and Khwe-and their genetic relationships to other African populations. Am J Hum Genet 66:1362–1383
Chen G, Adeyemo A, Zhou J, Chen Y, Huang H, Doumatey A, Lashley K, Agyenim-Boateng K, Eghan BA Jr, Acheampong J, Fasanmade O, Johnson T, Okafor G, Oli J, Amoah A, Rotimi C (2007a) Genome-wide search for susceptibility genes to type 2 diabetes in West Africans: potential role of C-peptide. Diabetes Res Clin Pract 78:e1–e6
Chen G, Adeyemo AA, Johnson T, Zhou J, Amoah A, Owusu S, Acheampong J, Agyenim-Boateng K, Eghan BA, Oli J, Okafor G, Abbiyesuku F, Dunston GM, Chen Y, Collins F, Rotimi C (2005a) A genome-wide scan for quantitative trait loci linked to obesity phenotypes among West Africans. Int J Obes (Lond) 29:255–259
Chen Y, Kittles R, Zhou J, Chen G, Adeyemo A, Panguluri RK, Chen W, Amoah A, Opoku V, Acheampong J, Agyenim-Boateng K, Eghan BA Jr., Nyantaki A, Oli J, Okafor G, Ofoegbu E, Osotimehin B, Abbiyesuku F, Johnson T, Fasanmade O, Rufus T, Furbert-Harris P, Daniel HI, Berg KA, Collins FS, Dunston GM, Rotimi CN (2005b) Calpain-10 gene polymorphisms and type 2 diabetes in West Africans: the Africa America Diabetes Mellitus (AADM) Study. Ann Epidemiol 15:153–159
Chen G, Adeyemo AA, Zhou J, Chen Y, Doumatey A, Lashley K, Huang H, Amoah A, Agyenim-Boateng K, Eghan BA Jr, Okafor G, Acheampong J, Oli J, Fasanmade O, Johnson T, Rotimi C (2007b) A genome-wide search for linkage to renal function phenotypes in West Africans with type 2 diabetes. Am J Kidney Dis 49:394–400
Chen Y, Huang H, Zhou J, Doumatey A, Lashley K, Chen G, Agyenim-Boateng K, Eghan BA, Acheampong J, Fasanmade O, Johnson T, Akinsola FB, Okafor G, Oli J, Ezepue F, Amoah A, Akafo S, Adeyemo A, Rotimi CN (2007c) Polymorphism of the endothelial nitric oxide synthase gene is associated with diabetic retinopathy in a cohort of West Africans. Mol Vis 13:2142–2147
Chevillard C, Moukoko CE, Elwali NE, Bream JH, Kouriba B, Argiro L, Rahoud S, Mergani A, Henri S, Gaudart J, Mohamed-Ali Q, Young HA, Dessein AJ (2003) IFN-gamma polymorphisms (IFN-gamma +2109 and IFN-gamma +3810) are associated with severe hepatic fibrosis in human hepatic schistosomiasis (Schistosoma mansoni). J Immunol 171:5596–5601
Chitsulo L, Engels D, Montresor A, Savioli L (2000) The global status of schistosomiasis and its control. Acta Trop 77:41–51
Cholera R, Brittain NJ, Gillrie MR, Lopera-Mesa TM, Diakite SA, Arie T, Krause MA, Guindo A, Tubman A, Fujioka H, Diallo DA, Doumbo OK, Ho M, Wellems TE, Fairhurst RM (2008) Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proc Natl Acad Sci USA 105:991–996
Chuang WC, Sarkodie F, Brown CJ, Owusu-Ofori S, Brown J, Li C, Navarrete C, Klenerman P, Allain JP (2007) Protective effect of HLA-B57 on HCV genotype 2 infection in a West African population. J Med Virol 79:724–733
Comuzzie AG, Hixson JE, Almasy L, Mitchell BD, Mahaney MC, Dyer TD, Stern MP, MacCluer JW, Blangero J (1997) A major quantitative trait locus determining serum leptin levels and fat mass is located on human chromosome 2. Nat Genet 15:273–276
Cooke GS, Aucan C, Walley AJ, Segal S, Greenwood BM, Kwiatkowski DP, Hill AV (2003) Association of Fcgamma receptor IIa (CD32) polymorphism with severe malaria in West Africa. Am J Trop Med Hyg 69:565–568
Cooke GS, Campbell SJ, Fielding K, Sillah J, Manneh K, Sirugo G, Bennett S, McAdam KP, Lienhardt C, Hill AV (2004) Interleukin-8 polymorphism is not associated with pulmonary tuberculosis in The Gambia. J Infect Dis 189:1545–1546
Cooke GS, Campbell SJ, Sillah J, Gustafson P, Bah B, Sirugo G, Bennett S, McAdam KP, Sow O, Lienhardt C, Hill AV (2006a) Polymorphism within the interferon-gamma/receptor complex is associated with pulmonary tuberculosis. Am J Respir Crit Care Med 174:339–343
Cooke GS, Tosh K, Ramaley PA, Kaleebu P, Zhuang J, Nakiyingi JS, Watera C, Gilks CF, French N, Whitworth JA, Hill AV (2006b) A polymorphism that reduces RANTES expression is associated with protection from death in HIV-seropositive Ugandans with advanced disease. J Infect Dis 194:666–669
Cooke GS, Campbell SJ, Bennett S, Lienhardt C, McAdam KPWJ, Sow O, Gustafson P, Mwangulu F, van Helden P, Fine P, Hoal EG, Hill AV (2008) Mapping of a novel susceptibility locus suggests a role for MC3R and CTSZ in human tuberculosis. Am J Respir Crit Care Med (in press)
Cooper R, Rotimi C, Ataman S, McGee D, Osotimehin B, Kadiri S, Muna W, Kingue S, Fraser H, Forrester T, Bennett F, Wilks R (1997) The prevalence of hypertension in seven populations of west African origin. Am J Public Health 87:160–168
Cooper RS, Guo X, Rotimi CN, Luke A, Ward R, Adeyemo A, Danilov SM (2000) Heritability of angiotensin-converting enzyme and angiotensinogen: a comparison of US blacks and Nigerians. Hypertension 35:1141–1147
Cooper RS, Luke A, Zhu X, Kan D, Adeyemo A, Rotimi C, Bouzekri N, Ward R (2002) Genome scan among Nigerians linking blood pressure to chromosomes 2, 3, and 19. Hypertension 40:629–633
Cox SE, Doherty C, Atkinson SH, Nweneka CV, Fulford AJ, Ghattas H, Rockett KA, Kwiatkowski DP, Prentice AM (2007) Haplotype association between haptoglobin (Hp2) and Hp promoter SNP (A–61C) may explain previous controversy of haptoglobin and malaria protection. PLoS ONE 2:e362
Cox SE, Doherty CP, Atkinson SH, Nweneka CV, Fulford AJ, Sirugo G, Rockett KA, Kwiatkowski DP, Prentice AM (2008) Haptoglobin genotype, anaemia and malaria in Gambian children. Trop Med Int Health 13:76–82
Cramer JP, Mockenhaupt FP, Ehrhardt S, Burkhardt J, Otchwemah RN, Dietz E, Gellert S, Bienzle U (2004) iNOS promoter variants and severe malaria in Ghanaian children. Trop Med Int Health 9:1074–1080
Creinin M, Keith LG (1989) The Yoruba contribution to our understanding of the twinning process. J Reprod Med 34:379–387
Dandara C, Masimirembwa CM, Magimba A, Kaaya S, Sayi J, Sommers DK, Snyman JR, Hasler JA (2003) Arylamine N-acetyltransferase (NAT2) genotypes in Africans: the identification of a new allele with nucleotide changes 481C > T and 590G > A. Pharmacogenetics 13:55–58
Dandara C, Li DP, Walther G, Parker MI (2006) Gene–environment interaction: the role of SULT1A1 and CYP3A5 polymorphisms as risk modifiers for squamous cell carcinoma of the oesophagus. Carcinogenesis 27:791–797
Dardari R, Khyatti M, Jouhadi H, Benider A, Ettayebi H, Kahlain A, Mansouri A, El GB, Benslimane A (2001) Study of human leukocyte antigen class I phenotypes in Moroccan patients with nasopharyngeal carcinoma. Int J Cancer 92:294–297
Davies EG, Riddington C, Lottenberg R, Dower N (2004) Pneumococcal vaccines for sickle cell disease. Cochrane Database Syst Rev 1:CD003885
Davey G, Gebrehanna E, Adeyemo A, Rotimi C, Newport M, Desta K (2007a) Podoconiosis: a tropical model for gene-environment interactions? Trans R Soc Trop Med Hyg 101:91–96
Davey G, Tekola F, Newport MJ (2007b) Podoconiosis: non-infectious geochemical elephantiasis. Trans R Soc Trop Med Hyg 101:1175–1180
De SA, Pasi A, Martinetti M, Belloni C, Tinelli C, Rondini G, Salvaneschi L, Cuccia M (2001) Family study of non-responsiveness to hepatitis B vaccine confirms the importance of HLA class III C4A locus. Genes Immun 2:367–372
Delahaye NF, Barbier M, Fumoux F, Rihet P (2007) Association analyses of NCR3 polymorphisms with P. falciparum mild malaria. Microbes Infect 9:160–166
Delongchamps NB, Singh A, Haas GP (2007) Epidemiology of prostate cancer in Africa: another step in the understanding of the disease? Curr Probl Cancer 31:226–236
Demotz S, Barbey C, Corradin G, Amoroso A, Lanzavecchia A (1993) The set of naturally processed peptides displayed by DR molecules is tuned by polymorphism of residue 86. Eur J Immunol 23:425–432
Dessein AJ, Hillaire D, Elwali NE, Marquet S, Mohamed-Ali Q, Mirghani A, Henri S, Abdelhameed AA, Saeed OK, Magzoub MM, Abel L (1999) Severe hepatic fibrosis in Schistosoma mansoni infection is controlled by a major locus that is closely linked to the interferon-gamma receptor gene. Am J Hum Genet 65:709–721
Destas K, Ashine M, Davey G (2003) Prevalence of podoconiosis (endemic non-filarial elephantiasis) in Wolaitta, Southern Ethiopia. Trop Doct 33:217–220
Dhiman N, Ovsyannikova IG, Pinsky NA, Vierkant RA, Jacobsen SJ, Jacobson RM, Poland GA (2003) Lack of association between transporter associated with antigen processing (TAP) and HLA-DM gene polymorphisms and antibody levels following measles vaccination. Eur J Immunogenet 30:195–200
Dhiman N, Ovsyannikova IG, Cunningham JM, Vierkant RA, Kennedy RB, Pankratz VS, Poland GA, Jacobson RM (2007a) Associations between measles vaccine immunity and single-nucleotide polymorphisms in cytokine and cytokine receptor genes. J Infect Dis 195:21–29
Dhiman N, Poland GA, Cunningham JM, Jacobson RM, Ovsyannikova IG, Vierkant RA, Wu Y, Pankratz VS (2007b) Variations in measles vaccine-specific humoral immunity by polymorphisms in SLAM and CD46 measles virus receptors. J Allergy Clin Immunol 120:666–672
Diouf K, Sarr AD, Eisen G, Popper S, Mboup S, Kanki P (2002) Associations between MHC class I and susceptibility to HIV-2 disease progression. J Hum Virol 5:1–7
Donaldson IJ, Shefta J, Hill AV, Boylston AW (2002) Malaria is not responsible for the selection of TCR beta-subunit variable gene haplotypes in The Gambia. Immunogenetics 53:894–899
Dong YB, Zhu HD, Baker EH, Sagnella GA, MacGregor GA, Carter ND, Wicks PD, Cook DG, Cappuccio FP (2001) T594 M and G442 V polymorphisms of the sodium channel beta subunit and hypertension in a black population. J Hum Hypertens 15:425–430
Duggan D, Zheng SL, Knowlton M, Benitez D, Dimitrov L, Wiklund F, Robbins C, Isaacs SD, Cheng Y, Li G, Sun J, Chang BL, Marovich L, Wiley KE, Bälter K, Stattin P, Adami HO, Gielzak M, Yan G, Sauvageot J, Liu W, Kim JW, Bleecker ER, Meyers DA, Trock BJ, Partin AW, Walsh PC, Isaacs WB, Grönberg H, Xu J, Carpten JD (2007) Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst 99:1836–1844
El-Safi SH, Bucheton B, Kheir MM, Musa HA, EL-Obaid M, Hammad A, Dessein A (2002) Epidemiology of visceral leishmaniasis in Atbara River area, eastern Sudan: the outbreak of Barbar El Fugara village (1996–1997). Microbes Infect 4:1439–1447
El-Safi S, Kheir MM, Bucheton B, Argiro L, Abel L, Dereure J, Dedet JP, Dessein A (2006) Genes and environment in susceptibility to visceral leishmaniasis. C R Biol 329:863–870
Elloumi-Zghal H, Barbouche MR, Chemli J, Bejaoui M, Harbi A, Snoussi N, Abdelhak S, Dellagi K (2002) Clinical and genetic heterogeneity of inherited autosomal recessive susceptibility to disseminated Mycobacterium bovis bacille calmette-guerin infection. J Infect Dis 185:1468–1475
Emonts M, Veenhoven RH, Wiertsema SP, Houwing-Duistermaat JJ, Walraven V, de Groot R, Hermans PW, Sanders EA (2007) Genetic polymorphisms in immunoresponse genes TNFA, IL6, IL10, and TLR4 are associated with recurrent acute otitis media. Pediatrics 120:814–823
Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Jarvela I (2002) Identification of a variant associated with adult-type hypolactasia. Nat Genet 30:233–237
Enevold A, Lusingu JP, Mmbando B, Alifrangis M, Lemnge MM, Bygbjerg IC, Theander TG, Vestergaard LS (2008) Reduced risk of uncomplicated malaria episodes in children with alpha + -thalassemia in northeastern Tanzania. Am J Trop Med Hyg 78:714–720
Esteban E, Rodon N, Via M, Gonzalez-Perez E, Santamaria J, Dugoujon JM, Chennawi FE, Melhaoui M, Cherkaoui M, Vona G, Harich N, Moral P (2006) Androgen receptor CAG and GGC polymorphisms in Mediterraneans: repeat dynamics and population relationships. J Hum Genet 51:129–136
Estefania E, Gomez-Lozano N, Portero F, de Pablo R, Solis R, Sepulveda S, Vaquero M, Gonzalez MA, Suarez E, Roustan G, Vilches C (2007) Influence of KIR gene diversity on the course of HSV-1 infection: resistance to the disease is associated with the absence of KIR2DL2 and KIR2DS2. Tissue Antigens 70:34–41
Ezzikouri S, El Feydi AE, El Kihal L, Afifi R, Benazzouz M, Hassar M, Chafik A, Pineau P, Benjelloun S (2008) Prevalence of common HFE and SERPINA1 mutations in patients with hepatocellular carcinoma in a Moroccan population. Arch Med Res 39:236–241
Faal N, Bailey RL, Jeffries D, Joof H, Sarr I, Laye M, Mabey DC, Holland MJ (2006) Conjunctival FOXP3 expression in trachoma: do regulatory T cells have a role in human ocular Chlamydia trachomatis infection? PLoS Med 3:e266
Fackenthal JD, Sveen L, Gao Q, Kohlmeir EK, Adebamowo C, Ogundiran TO, Adenipekun AA, Oyesegun R, Campbell O, Rotimi C, Akang EE, Das S, Olopade OI (2005) Complete allelic analysis of BRCA1 and BRCA2 variants in young Nigerian breast cancer patients. J Med Genet 42:276–281
Fairhurst RM, Baruch DI, Brittain NJ, Ostera GR, Wallach JS, Hoang HL, Hayton K, Guindo A, Makobongo MO, Schwartz OM, Tounkara A, Doumbo OK, Diallo DA, Fujioka H, Ho M, Wellems TE (2005) Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature 435:1117–1121
Fejerman L, Wu X, Adeyemo A, Luke A, Zhu X, Hicks C, Cooper RS (2006) The effect of genetic variation in angiotensinogen on serum levels and blood pressure: a comparison of Nigerians and US blacks. J Hum Hypertens 20:882–887
Felix R, Bodmer W, Fearnhead NS, van der Merwe L, Goldberg P, Ramesar RS (2006) GSTM1 and GSTT1 polymorphisms as modifiers of age at diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC) in a homogeneous cohort of individuals carrying a single predisposing mutation. Mutat Res 602:175–181
Ferlay J, Bray F, Pisani P, Parkin DM (2004) GLOBOCAN 2002: Cancer Incidence. Mortality and Prevalence Worldwide. IARC PRESS, Lyon
Fitness J, Floyd S, Warndorff DK, Sichali L, Malema S, Crampin AC, Fine PE, Hill AV (2004) Large-scale candidate gene study of tuberculosis susceptibility in the Karonga district of northern Malawi. Am J Trop Med Hyg 71:341–349
Flori L, Kumulungui B, Aucan C, Esnault C, Traore AS, Fumoux F, Rihet P (2003a) Linkage and association between Plasmodium falciparum blood infection levels and chromosome 5q31-q33. Genes Immun 4:265–268
Flori L, Sawadogo S, Esnault C, Delahaye NF, Fumoux F, Rihet P (2003b) Linkage of mild malaria to the major histocompatibility complex in families living in Burkina Faso. Hum Mol Genet 12:375–378
Flori L, Delahaye NF, Iraqi FA, Hernandez-Valladares M, Fumoux F, Rihet P (2005) TNF as a malaria candidate gene: polymorphism-screening and family-based association analysis of mild malaria attack and parasitemia in Burkina Faso. Genes Immun 6:472–480
Foote SJ, Handman E (2005) Genetics of murine leishmaniasis. Brief Funct Genomic Proteomic 4:270–276
Freedman ML, Haiman CA, Patterson N, McDonald GJ, Tandon A, Waliszewska A, Penney K, Steen RG, Ardlie K, John EM, Oakley-Girvan I, Whittemore AS, Cooney KA, Ingles SA, Altshuler D, Henderson BE, Reich D (2006) Admixture mapping identifies 8q24 as a prostate cancer risk locus in African–American men. Proc Natl Acad Sci USA 103:14068–14073
Fry AE, Griffiths MJ, Auburn S, Diakite M, Forton JT, Green A, Richardson A, Wilson J, Jallow M, Sisay-Joof F, Pinder M, Peshu N, Williams TN, Marsh K, Molyneux ME, Taylor TE, Rockett KA, Kwiatkowski DP (2008) Common variation in the ABO glycosyltransferase is associated with susceptibility to severe Plasmodium falciparum malaria. Hum Mol Genet 17:567–576
Gaedigk A, Bhathena A, Ndjountche L, Pearce RE, Abdel-Rahman SM, Alander SW, Bradford LD, Rogan PK, Leeder JS (2005) Identification and characterization of novel sequence variations in the cytochrome P4502D6 (CYP2D6) gene in African Americans. Pharmacogenomics J 5:173–182
Gaedigk A, Ndjountche L, Divakaran K, Dianne BL, Zineh I, Oberlander TF, Brousseau DC, McCarver DG, Johnson JA, Alander SW, Wayne RK, Steven LJ (2007) Cytochrome P4502D6 (CYP2D6) gene locus heterogeneity: characterization of gene duplication events. Clin Pharmacol Ther 81:242–251
Gao Q, Adebamowo CA, Fackenthal J, Das S, Sveen L, Falusi AG, Olopade OI (2000) Protein truncating BRCA1 and BRCA2 mutations in African women with pre-menopausal breast cancer. Hum Genet 107:192–194
Garcia A, Marquet S, Bucheton B, Hillaire D, Cot M, Fievet N, Dessein AJ, Abel L (1998) Linkage analysis of blood Plasmodium falciparum levels: interest of the 5q31–q33 chromosome region. Am J Trop Med Hyg 58:705–709
Garner CP, Ding YC, John EM, Ingles SA, Olopade OI, Huo D, Adebamowo C, Ogundiran T, Neuhausen SL (2008) Genetic variation in IGFBP2 and IGFBP5 is associated with breast cancer in populations of African descent. Hum Genet 123:247–255
Ghosh S, Watanabe RM, Hauser ER, Valle T, Magnuson VL, Erdos MR, Langefeld CD, Balow J Jr., Ally DS, Kohtamaki K, Chines P, Birznieks G, Kaleta HS, Musick A, Te C, Tannenbaum J, Eldridge W, Shapiro S, Martin C, Witt A, So A, Chang J, Shurtleff B, Porter R, Kudelko K, Unni A, Segal L, Sharaf R, Blaschak-Harvan J, Eriksson J, Tenkula T, Vidgren G, Ehnholm C, Tuomilehto-Wolf E, Hagopian W, Buchanan TA, Tuomilehto J, Bergman RN, Collins FS, Boehnke M (1999) Type 2 diabetes: evidence for linkage on chromosome 20 in 716 Finnish affected sib pairs. Proc Natl Acad Sci USA 96:2198–2203
Gonder MK, Mortensen HM, Reed FA, de Sousa A, Tishkoff SA (2007) Whole-mtDNA genome sequence analysis of ancient African lineages. Mol Biol Evol 24:757–768
Govan VA, Constant D, Hoffman M, Williamson AL (2006) The allelic distribution of −308 Tumor Necrosis Factor-alpha gene polymorphism in South African women with cervical cancer and control women. BMC Cancer 6:24
Govan VA, Loubser S, Saleh D, Hoffman M, Williamson AL (2007) No relationship observed between human p53 codon–72 genotype and HPV-associated cervical cancer in a population group with a low arginine-72 allele frequency. Int J Immunogenet 34:213–217
Granoff DM, Cates KL (1985) Haemophilus influenzae type b polysaccharide vaccines. J Pediatr 107:330–336
Granoff DM, Holmes SJ (1992) G2 m(23) immunoglobulin allotype and immunity to Haemophilus influenzae type b. J Infect Dis 165(Suppl 1):S66–S69
Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson JT, Thorleifsson G, Manolescu A, Rafnar T, Gudbjartsson D, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Blondal T, Stacey SN, Helgason A, Gunnarsdottir S, Olafsdottir A, Kristinsson KT, Birgisdottir B, Ghosh S, Thorlacius S, Magnusdottir D, Stefansdottir G, Kristjansson K, Bagger Y, Wilensky RL, Reilly MP, Morris AD, Kimber CH, Adeyemo A, Chen Y, Zhou J, So WY, Tong PC, Ng MC, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Tres A, Fuertes F, Ruiz-Echarri M, Asin L, Saez B, van Boven E, Klaver S, Swinkels DW, Aben KK, Graif T, Cashy J, Suarez BK, van Vierssen TO, Frigge ML, Ober C, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Palmer CN, Rotimi C, Chan JC, Pedersen O, Sigurdsson G, Benediktsson R, Jonsson E, Einarsson GV, Mayordomo JI, Catalona WJ, Kiemeney LA, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet 39:977–983
Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA (2007) X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med 4:e66
Gyan BA, Goka B, Cvetkovic JT, Kurtzhals JL, Adabayeri V, Perlmann H, Lefvert AK, Akanmori BD, Troye-Blomberg M (2004) Allelic polymorphisms in the repeat and promoter regions of the interleukin-4 gene and malaria severity in Ghanaian children. Clin Exp Immunol 138:145–150
Hacker UT, Erhardt S, Tschop K, Jelinek T, Endres S (2001) Influence of the IL-1Ra gene polymorphism on in vivo synthesis of IL-1Ra and IL-1beta after live yellow fever vaccination. Clin Exp Immunol 125:465–469
Hadhri-Guiga B, Toumi N, Khabir A, Sellami-Boudawara T, Ghorbel A, Daoud J, Frikha M, Gargouri A, Mokdad-Gargouri R (2007) Proline homozygosity in codon 72 of TP53 is a factor of susceptibility to nasopharyngeal carcinoma in Tunisia. Cancer Genet Cytogenet 178:89–93
Haile-Selassie Y, Asfaw B, White TD (2004) Hominid cranial remains from upper Pleistocene deposits at Aduma, Middle Awash, Ethiopia. Am J Phys Anthropol 123:1–10
Haiman CA, Patterson N, Freedman ML, Myers SR, Pike MC, Waliszewska A, Neubauer J, Tandon A, Schirmer C, McDonald GJ, Greenway SC, Stram DO, Le Marchand L, Kolonel LN, Frasco M, Wong D, Pooler LC, Ardlie K, Oakley-Girvan I, Whittemore AS, Cooney KA, John EM, Ingles SA, Altshuler D, Henderson BE, Reich D (2007) Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 39:638–644
Hainaut P, Boyle P (2008) Curbing the liver cancer epidemic in Africa. Lancet 371:367–368
Haldane J (1949) The rate of mutation of human genes. Proceedings of the 8th international congress of genetics Hereditas 35(Suppl):267–273
Hamblin MT, Di Rienzo A (2000) Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am J Hum Genet 66:1669–1679
Hamblin MT, Thompson EE, Di Rienzo A (2002) Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet 70:369–383
Hammer MF, Karafet TM, Redd AJ, Jarjanazi H, Santachiara-Benerecetti S, Soodyall H, Zegura SL (2001) Hierarchical patterns of global human Y-chromosome diversity. Mol Biol Evol 18:1189–1203
Hansen DS, D’Ombrain MC, Schofield L (2007) The role of leukocytes bearing Natural Killer Complex receptors and Killer Immunoglobulin-like Receptors in the immunology of malaria. Curr Opin Immunol 19:416–423
Hassen E, Farhat K, Gabbouj S, Jalbout M, Bouaouina N, Chouchane L (2007) TAP1 gene polymorphisms and nasopharyngeal carcinoma risk in a Tunisian population. Cancer Genet Cytogenet 175:41–46
Hayney MS, Poland GA, Jacobson RM, Schaid DJ, Lipsky JJ (1996) The influence of the HLA-DRB1*13 allele on measles vaccine response. J Investig Med 44:261–263
Hayney MS, Poland GA, Dimanlig P, Schaid DJ, Jacobson RM, Lipsky JJ (1997) Polymorphisms of the TAP2 gene may influence antibody response to live measles vaccine virus. Vaccine 15:3–6
He H, Isnard A, Kouriba B, Cabantous S, Dessein A, Doumbo O, Chevillard C (2008) A STAT6 gene polymorphism is associated with high infection levels in urinary schistosomiasis. Genes Immun 9(3):195–206
Heath PT (1998) Haemophilus influenzae type b conjugate vaccines: a review of efficacy data. Pediatr Infect Dis J 17:S117–S122
Helgadottir A, Manolescu A, Helgason A, Thorleifsson G, Thorsteinsdottir U, Gudbjartsson DF, Gretarsdottir S, Magnusson KP, Gudmundsson G, Hicks A, Jonsson T, Grant SF, Sainz J, O’Brien SJ, Sveinbjornsdottir S, Valdimarsson EM, Matthiasson SE, Levey AI, Abramson JL, Reilly MP, Vaccarino V, Wolfe ML, Gudnason V, Quyyumi AA, Topol EJ, Rader DJ, Thorgeirsson G, Gulcher JR, Hakonarson H, Kong A, Stefansson K (2006) A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat Genet 38:68–74
Helgason A, Palsson S, Thorleifsson G, Grant SF, Emilsson V, Gunnarsdottir S, Adeyemo A, Chen Y, Chen G, Reynisdottir I, Benediktsson R, Hinney A, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Schafer H, Faruque M, Doumatey A, Zhou J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Sigurdsson G, Hebebrand J, Pedersen O, Thorsteinsdottir U, Gulcher JR, Kong A, Rotimi C, Stefansson K (2007) Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet 39:218–225
Hennig BJ, Fielding K, Broxholme J, Diatta M, Mendy M, Moore C, Pollard AJ, Rayco-Solon P, Sirugo G, van der Sande MA, Waight P, Whittle HC, Zaman SM, Hill AV, Hall AJ (2008) Host genetic factors and vaccine-induced immunity to hepatitis B virus infection. PLoS One 3:e1898
Henri S, Chevillard C, Mergani A, Paris P, Gaudart J, Camilla C, Dessein H, Montero F, Elwali NE, Saeed OK, Magzoub M, Dessein AJ (2002) Cytokine regulation of periportal fibrosis in humans infected with Schistosoma mansoni: IFN-gamma is associated with protection against fibrosis and TNF-alpha with aggravation of disease. J Immunol 169:929–936
Herb F, Thye T, Niemann S, Browne EN, Chinbuah MA, Gyapong J, Osei I, Owusu-Dabo E, Werz O, Rüsch-Gerdes S, Horstmann RD, Meyer CG (2008) ALOX5 variants associated with susceptibility to human pulmonary tuberculosis. Hum Mol Genet 17:1052–1060
Hill AV (2006) Aspects of genetic susceptibility to human infectious diseases. Annu Rev Genet 40:469–486
Hill AV, Allsopp CE, Kwiatkowski D, Anstey NM, Twumasi P, Rowe PA, Bennett S, Brewster D, McMichael AJ, Greenwood BM (1991) Common west African HLA antigens are associated with protection from severe malaria. Nature 352:595–600
Hoal EG, Lewis LA, Jamieson SE, Tanzer F, Rossouw M, Victor T, Hillerman R, Beyers N, Blackwell JM, Van Helden PD (2004) SLC11A1 (NRAMP1) but not SLC11A2 (NRAMP2) polymorphisms are associated with susceptibility to tuberculosis in a high-incidence community in South Africa. Int J Tuberc Lung Dis 8:1464–1471
Hobbs MR, Udhayakumar V, Levesque MC, Booth J, Roberts JM, Tkachuk AN, Pole A, Coon H, Kariuki S, Nahlen BL, Mwaikambo ED, Lal AL, Granger DL, Anstey NM, Weinberg JB (2002) A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. Lancet 360:1468–1475
Hohler T, Reuss E, Evers N, Dietrich E, Rittner C, Freitag CM, Vollmar J, Schneider PM, Fimmers R (2002) Differential genetic determination of immune responsiveness to hepatitis B surface antigen and to hepatitis A virus: a vaccination study in twins. Lancet 360:991–995
Hohler T, Reuss E, Freitag CM, Schneider PM (2005) A functional polymorphism in the IL-10 promoter influences the response after vaccination with HBsAg and hepatitis A. Hepatology 42:72–76
Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Horikawa Y, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175
Ibrahim ME, Lambson B, Yousif AO, Deifalla NS, Alnaiem DA, Ismail A, Yousif H, Ghalib HW, Khalil EA, Kadaro A, Barker DC, El Hassan AM (1999) Kala-azar in a high transmission focus: an ethnic and geographic dimension. Am J Trop Med Hyg 61:941–944
Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C (2007) Influence of cytochrome P450 polymorphisms on drug therapies: Pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther 116:496–526
Jacobson RM, Poland GA (2004) The genetic basis for measles vaccine failure. Acta Paediatr Suppl 93:43–46
Jennes W, Verheyden S, Demanet C, Dje-Toure CA, Vuylsteke B, Nkengasong JN, Kestens L (2006) Cutting edge: resistance to HIV-1 infection among African female sex workers is associated with inhibitory KIR in the absence of their HLA ligands. J Immunol 177:6588–6592
Jepson A, Sisay-Joof F, Banya W, Hassan-King M, Frodsham A, Bennett S, Hill AV, Whittle H (1997) Genetic linkage of mild malaria to the major histocompatibility complex in Gambian children: study of affected sibling pairs. BMJ 315:96–97
Jepson A, Fowler A, Banya W, Singh M, Bennett S, Whittle H, Hill AV (2001) Genetic regulation of acquired immune responses to antigens of Mycobacterium tuberculosis: a study of twins in West Africa. Infect Immun 69:3989–3994
Ji L, Malecki M, Warram JH, Yang Y, Rich SS, Krolewski AS (1997) New susceptibility locus for NIDDM is localized to human chromosome 20q. Diabetes 46:876–881
Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, Cheng J, Goedert JJ, Vlahov D, Hilgartner M, Cox S, Little AM, Alexander GJ, Cramp ME, O’Brien SJ, Rosenberg WM, Thomas DL, Carrington M (2004) HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305:872–874
Khedhaier A, Remadi S, Corbex M, Ahmed SB, Bouaouina N, Mestiri S, Azaiez R, Helal AN, Chouchane L (2003) Glutathione S-transferases (GSTT1 and GSTM1) gene deletions in Tunisians: susceptibility and prognostic implications in breast carcinoma. Br J Cancer 89:1502–1507
Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, Frodsham AJ, Walley AJ, Kyrieleis O, Khan A, Aucan C, Segal S, Moore CE, Knox K, Campbell SJ, Lienhardt C, Scott A, Aaby P, Sow OY, Grignani RT, Sillah J, Sirugo G, Peshu N, Williams TN, Maitland K, Davies RJ, Kwiatkowski DP, Day NP, Yala D, Crook DW, Marsh K, Berkley JA, O’Neill LA, Hill AV (2007a) A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet 39:523–528
Khor CC, Vannberg FO, Chapman SJ, Walley A, Aucan C, Loke H, White NJ, Peto T, Khor LK, Kwiatkowski D, Day N, Scott A, Berkley JA, Marsh K, Peshu N, Maitland K, Williams TN, Hill AV (2007b) Positive replication and linkage disequilibrium mapping of the chromosome 21q22.1 malaria susceptibility locus. Genes Immun 8:570–576
Kimani J, Kaul R, Nagelkerke NJ, Luo M, MacDonald KS, Ngugi E, Fowke KR, Ball BT, Kariri A, Ndinya-Achola J, Plummer FA (2008) Reduced rates of HIV acquisition during unprotected sex by Kenyan female sex workers predating population declines in HIV prevalence. AIDS 22:131–137
Kimman TG, Vandebriel RJ, Hoebee B (2007) Genetic variation in the response to vaccination. Community Genet 10:201–217
Kirchheiner J, Brosen K, Dahl ML, Gram LF, Kasper S, Roots I, Sjoqvist F, Spina E, Brockmoller J (2001) CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 104:173–192
Kirk GD, Camus-Randon AM, Mendy M, Goedert JJ, Merle P, Trepo C, Brechot C, Hainaut P, Montesano R (2000) Ser-249 p53 mutations in plasma DNA of patients with hepatocellular carcinoma from The Gambia. J Natl Cancer Inst 92:148–153
Kirk GD, Lesi OA, Mendy M, Szymanska K, Whittle H, Goedert JJ, Hainaut P, Montesano R (2005a) 249(ser) TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene 24:5858–5867
Kirk GD, Turner PC, Gong Y, Lesi OA, Mendy M, Goedert JJ, Hall AJ, Whittle H, Hainaut P, Montesano R, Wild CP (2005b) Hepatocellular carcinoma and polymorphisms in carcinogen-metabolizing and DNA repair enzymes in a population with aflatoxin exposure and hepatitis B virus endemicity. Cancer Epidemiol Biomarkers Prev 14:373–379
Kirk GD, Bah E, Montesano R (2006) Molecular epidemiology of human liver cancer: insights into etiology, pathogenesis and prevention from The Gambia, West Africa. Carcinogenesis 27:2070–2082
Kittles RA, Young D, Weinrich S, Hudson J, Argyropoulos G, Ukoli F, Adams-Campbell L, Dunston GM (2001) Extent of linkage disequilibrium between the androgen receptor gene CAG and GGC repeats in human populations: implications for prostate cancer risk. Hum Genet 109:253–261
Kittles RA, Chen W, Panguluri RK, Ahaghotu C, Jackson A, Adebamowo CA, Griffin R, Williams T, Ukoli F, Adams-Campbell L, Kwagyan J, Isaacs W, Freeman V, Dunston GM (2002) CYP3A4-V and prostate cancer in African Americans: causal or confounding association because of population stratification? Hum Genet 110:553–560
Klein K, Lang T, Saussele T, Barbosa-Sicard E, Schunck WH, Eichelbaum M, Schwab M, Zanger UM (2005) Genetic variability of CYP2B6 in populations of African and Asian origin: allele frequencies, novel functional variants, and possible implications for anti-HIV therapy with efavirenz. Pharmacogenet Genomics 15:861–873
Klupa T, Malecki MT, Pezzolesi M, Ji L, Curtis S, Langefeld CD, Rich SS, Warram JH, Krolewski AS (2000) Further evidence for a susceptibility locus for type 2 diabetes on chromosome 20q13.1–q13.2. Diabetes 49:2212–2216
Knight JC, Udalova I, Hill AV, Greenwood BM, Peshu N, Marsh K, Kwiatkowski D (1999) A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nat Genet 22:145–150
Koch O, Awomoyi A, Usen S, Jallow M, Richardson A, Hull J, Pinder M, Newport M, Kwiatkowski D (2002) IFNGR1 gene promoter polymorphisms and susceptibility to cerebral malaria. J Infect Dis 185:1684–1687
Kohle C, Mohrle B, Munzel PA, Schwab M, Wernet D, Badary OA, Bock KW (2003) Frequent co-occurrence of the TATA box mutation associated with Gilbert’s syndrome (UGT1A1*28) with other polymorphisms of the UDP-glucuronosyltransferase-1 locus (UGT1A6*2 and UGT1A7*3) in Caucasians and Egyptians. Biochem Pharmacol 65:1521–1527
Konradsen HB, Henrichsen J, Wachmann H, Holm N (1993) The influence of genetic factors on the immune response as judged by pneumococcal vaccination of mono- and dizygotic Caucasian twins. Clin Exp Immunol 92:532–536
Konradsen HB, Oxelius VA, Hahn-Zoric M, Hanson LA (1994) The importance of G1 m and 2 allotypes for the IgG2 antibody levels and avidity against pneumococcal polysaccharide type 1 within mono- and dizygotic twin-pairs. Scand J Immunol 40:251–256
Koram KA, Molyneux ME (2007) When is “malaria” malaria? The different burdens of malaria infection, malaria disease, and malaria-like illnesses. Am J Trop Med Hyg 77:1–5
Kouriba B, Chevillard C, Bream JH, Argiro L, Dessein H, Arnaud V, Sangare L, Dabo A, Beavogui AH, Arama C, Traoré HA, Doumbo O, Dessein A (2005) Analysis of the 5q31–q33 locus shows an association between IL13–1055C/T IL-13–591A/G polymorphisms and Schistosoma haematobium infections. J Immunol 174:6274–6281
Kruger A, Adams P, Hammer J, Bocher WO, Schneider PM, Rittner C, Hoehler T (2005) Hepatitis B surface antigen presentation and HLA-DRB1*-lessons from twins and peptide binding studies. Clin Exp Immunol 140:325–332
Kuhn L, Schramm DB, Donninger S, Meddows-Taylor S, Coovadia AH, Sherman GG, Gray GE, Tiemessen CT (2007) African infants’ CCL3 gene copies influence perinatal HIV transmission in the absence of maternal nevirapine. AIDS 21:1753–1761
Kun JF, Mordmuller B, Perkins DJ, May J, Mercereau-Puijalon O, Alpers M, Weinberg JB, Kremsner PG (2001) Nitric oxide synthase 2(Lambarene) (G-954C), increased nitric oxide production, and protection against malaria. J Infect Dis 184:330–336
Kwiatkowski DP (2005) How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet 77:171–192
Lahr MM, Foley RA (1998) Towards a theory of modern human origins: geography, demography and diversity on recent human evolution. Yearbook of Physical Anthropology 41:137–176
Lama J, Planelles V (2007) Host factors influencing susceptibility to HIV infection and AIDS progression. Retrovirology 4:52
Lee YC, Newport MJ, Goetghebuer T, Siegrist CA, Weiss HA, Pollard AJ, Marchant A (2006) Influence of genetic and environmental factors on the immunogenicity of Hib vaccine in Gambian twins. Vaccine 24:5335–5340
Lessells RJ, Cooke GS (2008) Effect of the HIV epidemic on liver cancer in Africa. Lancet 371:1504
Levesque MC, Hobbs MR, Anstey NM, Vaughn TN, Chancellor JA, Pole A, Perkins DJ, Misukonis MA, Chanock SJ, Granger DL, Weinberg JB (1999) Nitric oxide synthase type 2 promoter polymorphisms, nitric oxide production, and disease severity in Tanzanian children with malaria. J Infect Dis 180:1994–2002
Lewallen S, Courtright P (2001) Blindness in Africa: present situation and future needs. Br J Ophthalmol 85:897–903
Li CM, Campbell SJ, Kumararatne DS, Bellamy R, Ruwende C, McAdam KP, Hill AV, Lammas DA (2002) Association of a polymorphism in the P2X7 gene with tuberculosis in a Gambian population. J Infect Dis 186:1458–1462
Li D, Dandara C, Parker MI (2005) Association of cytochrome P450 2E1 genetic polymorphisms with squamous cell carcinoma of the oesophagus. Clin Chem Lab Med 43:370–375
Li J, Cowden LG, King JD, Briles DA, Schroeder HW Jr, Stevens AB, Perry RT, Chen Z, Simmons MS, Wiener HW, Tiwari HK, Harrell LE, Go RC (2007a) Effects of chronic stress and interleukin-10 gene polymorphisms on antibody response to tetanus vaccine in family caregivers of patients with Alzheimer’s disease. Psychosom Med 69:551–559
Li X, Ghandri N, Piancatelli D, Adams S, Chen D, Robbins FM, Wang E, Monaco A, Selleri S, Bouaouina N, Stroncek D, Adorno D, Chouchane L, Marincola FM (2007b) Associations between HLA class I alleles and the prevalence of nasopharyngeal carcinoma (NPC) among Tunisians. J Transl Med 5:22
Lin T, Chen C, Wu M, Yang C, Chen J, Lin C, Kwang T, Hsu S, Lin S, Hsu L (1989) Hepatitis B virus markers in Chinese twins. Anticancer Res 9:737–741
Lin P, Koutsky LA, Critchlow CW, Apple RJ, Hawes SE, Hughes JP, Toure P, Dembele A, Kiviat NB (2001) HLA class II DR-DQ and increased risk of cervical cancer among Senegalese women. Cancer Epidemiol Biomarkers Prev 10:1037–1045
Lindemann M, Barsegian V, Siffert W, Ferencik S, Roggendorf M, Grosse-Wilde H (2002) Role of G protein beta3 subunit C825T and HLA class II polymorphisms in the immune response after HBV vaccination. Virology 297:245–252
Liu H, Prugnolle F, Manica A, Balloux F (2006) A geographically explicit genetic model of worldwide human-settlement history. Am J Hum Genet 79:230–237
Livingstone FB (1984) The Duffy blood groups, vivax malaria, and malaria selection in human populations: a review. Hum Biol 56:413–425
Lombard Z, Dalton DL, Venter PA, Williams RC, Bornman L (2006) Association of HLA-DR, -DQ, and vitamin D receptor alleles and haplotypes with tuberculosis in the Venda of South Africa. Hum Immunol 67:643–654
Loock M, Steyn K, Becker P, Fourie J (2006) Coronary heart disease and risk factors in Black South Africans: a case-control study. Ethn Dis 16:872–879
Lopez-Vazquez A, Mina-Blanco A, Martinez-Borra J, Njobvu PD, Suarez-Alvarez B, Blanco-Gelaz MA, Gonzalez S, Rodrigo L, Lopez-Larrea C (2005) Interaction between KIR3DL1 and HLA-B*57 supertype alleles influences the progression of HIV-1 infection in a Zambian population. Hum Immunol 66:285–289
Louagie H, Delanghe J, Desombere I, De Buyzere M, Hauser P, Leroux-Roels G (1993) Haptoglobin polymorphism and the immune response after hepatitis B vaccination. Vaccine 11:1188–1190
Lucas A, Nolan D, Mallal S (2007) HLA-B*5701 screening for susceptibility to abacavir hypersensitivity. J Antimicrob Chemother 59:591–593
Luoni G, Verra F, Arca B, Sirima BS, Troye-Blomberg M, Coluzzi M, Kwiatkowski D, Modiano D (2001) Antimalarial antibody levels and IL4 polymorphism in the Fulani of West Africa. Genes Immun 2:411–414
Ma L, Marmor M, Zhong P, Ewane L, Su B, Nyambi P (2005) Distribution of CCR2–64I and SDF1–3’A alleles and HIV status in 7 ethnic populations of Cameroon. J Acquir Immune Defic Syndr 40:89–95
MacDonald KS, Fowke KR, Kimani J, Dunand VA, Nagelkerke NJ, Ball TB, Oyugi J, Njagi E, Gaur LK, Brunham RC, Wade J, Luscher MA, Krausa P, Rowland-Jones S, Ngugi E, Bwayo JJ, Plummer FA (2000) Influence of HLA supertypes on susceptibility and resistance to human immunodeficiency virus type 1 infection. J Infect Dis 181:1581–1589
Mackinnon MJ, Mwangi TW, Snow RW, Marsh K, Williams TN (2005) Heritability of malaria in Africa. PLoS Med 2:e340
Magoha GA (2007) Overview of prostate cancer in indigenous black Africans and blacks of African ancestry in diaspora 1935–2007. East Afr Med J 84:S3–S11
Malik S, Greenwood CM, Eguale T, Kifle A, Beyene J, Habte A, Tadesse A, Gebrexabher H, Britton S, Schurr E (2006) Variants of the SFTPA1 and SFTPA2 genes and susceptibility to tuberculosis in Ethiopia. Hum Genet 118:752–759
Mangano VD, Luoni G, Rockett KA, Sirima BS, Konate A, Forton J, Clark TG, Bancone G, Akha ES, Kwiatkowski DP, Modiano D (2008) Interferon regulatory factor-1 polymorphisms are associated with the control of Plasmodium falciparum infection. Genes Immun (in press)
Marchant A, Pihlgren M, Goetghebuer T, Weiss HA, Ota MO, Schlegel-Hauter SE, Whittle H, Lambert PH, Newport MJ, Siegrist CA (2006) Predominant influence of environmental determinants on the persistence and avidity maturation of antibody responses to vaccines in infants. J Infect Dis 193:1598–1605
Marquet S, Abel L, Hillaire D, Dessein H, Kalil J, Feingold J, Weissenbach J, Dessein AJ (1996) Genetic localization of a locus controlling the intensity of infection by Schistosoma mansoni on chromosome 5q31–q33. Nat Genet 14:181–184
Marquet S, Doumbo O, Cabantous S, Poudiougou B, Argiro L, Safeukui I, Konate S, Sissoko S, Chevereau E, Traore A, Keita MM, Chevillard C, Abel L, Dessein AJ (2008) A functional promoter variant in IL12B predisposes to cerebral malaria. Hum Mol Genet (in press)
Martin MP, Carrington M (2005) Immunogenetics of viral infections. Curr Opin Immunol 17:510-516
Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Trowsdale J, Wilson M, O’Brien SJ, Carrington M (2002) Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet 31:429–434
Masimirembwa CM, Hasler JA (1997) Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull 44:561–571
Masimirembwa C, Bertilsson L, Johansson I, Hasler JA, Ingelman-Sundberg M (1995) Phenotyping and genotyping of S-mephenytoin hydroxylase (cytochrome P450 2C19) in a Shona population of Zimbabwe. Clin Pharmacol Ther 57:656–661
Masimirembwa C, Persson I, Bertilsson L, Hasler J, Ingelman-Sundberg M (1996) A novel mutant variant of the CYP2D6 gene (CYP2D6*17) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br J Clin Pharmacol 42:713–719
Matimba A, Oluka MN, Ebeshi BU, Sayi J, Bolaji OO, Guantai AN, Masimirembwa CM (2008) Establishment of a biobank and pharmacogenetics database of African populations. Eur J Hum Genet (in press)
Matte C, Lajoie J, Lacaille J, Zijenah LS, Ward BJ, Roger M (2004) Functionally active HLA-G polymorphisms are associated with the risk of heterosexual HIV-1 infection in African women. AIDS 18:427–431
McDougall I, Brown FH, Fleagle JG (2005) Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature 433:733–736
McGuire W, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D (1994) Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature 371:508–510
Meisner SJ, Mucklow S, Warner G, Sow SO, Lienhardt C, Hill AV (2001) Association of NRAMP1 polymorphism with leprosy type but not susceptibility to leprosy per se in west Africans. Am J Trop Med Hyg 65:733–735
Meyer CG, May J, Luty AJ, Lell B, Kremsner PG (2002) TNFalpha-308A associated with shorter intervals of Plasmodium falciparum reinfections. Tissue Antigens 59:287–292
Migot-Nabias F, Mombo LE, Luty AJ, Dubois B, Nabias R, Bisseye C, Millet P, Lu CY, Deloron P (2000) Human genetic factors related to susceptibility to mild malaria in Gabon. Genes Immun 1:435–441
Migot-Nabias F, Pelleau S, Watier L, Guitard J, Toly C, De AC, Ngom MI, Chevillard C, Gaye O, Garcia A (2006) Red blood cell polymorphisms in relation to Plasmodium falciparum asymptomatic parasite densities and morbidity in Senegal. Microbes Infect 8:2352–2358
Milich DR, Leroux-Roels GG (2003) Immunogenetics of the response to HBsAg vaccination. Autoimmun Rev 2:248–257
Miller EN, Fadl M, Mohamed HS, Elzein A, Jamieson SE, Cordell HJ, Peacock CS, Fakiola M, Raju M, Khalil EA, Elhassan A, Musa AM, Ibrahim ME, Blackwell JM (2007) Y chromosome lineage- and village-specific genes on chromosomes 1p22 and 6q27 control visceral leishmaniasis in Sudan. PLoS Genet 3:e71
Mockenhaupt FP, Cramer JP, Hamann L, Stegemann MS, Eckert J, Oh NR, Otchwemah RN, Dietz E, Ehrhardt S, Schroder NW, Bienzle U, Schumann RR (2006) Toll-like receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. J Commun Dis 38:230–245
Modiano D, Petrarca V, Sirima BS, Nebie I, Diallo D, Esposito F, Coluzzi M (1996) Different response to Plasmodium falciparum malaria in west African sympatric ethnic groups. Proc Natl Acad Sci USA 93:13206–13211
Modiano D, Luoni G, Sirima BS, Lanfrancotti A, Petrarca V, Cruciani F, Simpore J, Ciminelli BM, Foglietta E, Grisanti P, Bianco I, Modiano G, Coluzzi M (2001a) The lower susceptibility to Plasmodium falciparum malaria of Fulani of Burkina Faso (west Africa) is associated with low frequencies of classic malaria-resistance genes. Trans R Soc Trop Med Hyg 95:149–152
Modiano D, Luoni G, Sirima BS, Simpore J, Verra F, Konate A, Rastrelli E, Olivieri A, Calissano C, Paganotti GM, D’Urbano L, Sanou I, Sawadogo A, Modiano G, Coluzzi M (2001b) Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature 414:305–308
Modiano D, Bancone G, Ciminelli BM, Pompei F, Blot I, Simpore J, Modiano G (2008) Haemoglobin S and haemoglobin C: ‘quick but costly’ versus ‘slow but gratis’ genetic adaptations to Plasmodium falciparum malaria. Hum Mol Genet (in press)
Mohamed HS, Ibrahim ME, Miller EN, Peacock CS, Khalil EA, Cordell HJ, Howson JM, El Hassan AM, Bereir RE, Blackwell JM (2003) Genetic susceptibility to visceral leishmaniasis in The Sudan: linkage and association with IL4 and IFNGR1. Genes Immun 4:351–355
Mohamed HS, Ibrahim ME, Miller EN, White JK, Cordell HJ, Howson JM, Peacock CS, Khalil EA, El Hassan AM, Blackwell JM (2004) SLC11A1 (formerly NRAMP1) and susceptibility to visceral leishmaniasis in The Sudan. Eur J Hum Genet 12:66–74
Mokni-Baizig N, Ayed K, Ayed FB, Ayed S, Sassi F, Ladgham A, Bel Hadj O, El MA (2001) Association between HLA-A/-B antigens and -DRB1 alleles and nasopharyngeal carcinoma in Tunisia. Oncology 61:55–58
Moller M, Kwiatkowski R, Nebel A, Van Helden PD, Hoal EG, Schreiber S (2007a) Allelic variation in BTNL2 and susceptibility to tuberculosis in a South African population. Microbes Infect 9:522–528
Moller M, Nebel A, Kwiatkowski R, Van Helden PD, Hoal EG, Schreiber S (2007b) Host susceptibility to tuberculosis: CARD15 polymorphisms in a South African population. Mol Cell Probes 21:148–151
Mombo LE, Lu CY, Ossari S, Bedjabaga I, Sica L, Krishnamoorthy R, Lapoumeroulie C (2003a) Mannose-binding lectin alleles in sub-Saharan Africans and relation with susceptibility to infections. Genes Immun 4:362–367
Mombo LE, Ntoumi F, Bisseye C, Ossari S, Lu CY, Nagel RL, Krishnamoorthy R (2003b) Human genetic polymorphisms and asymptomatic Plasmodium falciparum malaria in Gabonese schoolchildren. Am J Trop Med Hyg 68:186–190
Monemi S, Spaeth G, DaSilva A, Popinchalk S, Ilitchev E, Liebmann J, Ritch R, Heon E, Crick RP, Child A, Sarfarazi M (2005) Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum Mol Genet 14:725–733
Monot M, Honoré N, Garnier T, Araoz R, Coppée JY, Lacroix C, Sow S, Spencer JS, Truman RW, Williams DL, Gelber R, Virmond M, Flageul B, Cho SN, Ji B, Paniz-Mondolfi A, Convit J, Young S, Fine PE, Rasolofo V, Brennan PJ, Cole ST (2005) On the origin of leprosy. Science 308:1040–1042
Montesano R, Hainaut P, Wild CP (1997) Hepatocellular carcinoma: from gene to public health. J Natl Cancer Inst 89:1844–1851
Morahan G, Boutlis CS, Huang D, Pain A, Saunders JR, Hobbs MR, Granger DL, Weinberg JB, Peshu N, Mwaikambo ED, Marsh K, Roberts DJ, Anstey NM (2002) A promoter polymorphism in the gene encoding interleukin-12 p40 (IL12B) is associated with mortality from cerebral malaria and with reduced nitric oxide production. Genes Immun 3:414–418
Mori Y, Otabe S, Dina C, Yasuda K, Populaire C, Lecoeur C, Vatin V, Durand E, Hara K, Okada T, Tobe K, Boutin P, Kadowaki T, Froguel P (2002) Genome-wide search for type 2 diabetes in Japanese affected sib-pairs confirms susceptibility genes on 3q, 15q, and 20q and identifies two new candidate loci on 7p and 11p. Diabetes 51:1247–1255
Mostafa MH, Sheweita SA, O’Connor PJ (1999) Relationship between schistosomiasis and bladder cancer. Clin Microbiol Rev 12:97–111
Motala AA (2002) Diabetes trends in Africa. Diabetes Metab Res Rev 18(Suppl 3):S14–S20
Müller-Myhsok B, Stelma FF, Gissé-Sow F, Muntau B, Thye T, Burchard GD, Gryseels B, Horstmann RD (1997) Further evidence suggesting the presence of a locus, on human chromosome 5q31–q33, influencing the intensity of infection with Schistosoma mansoni. Am J Hum Genet 61:452–4
Musher DM, Groover JE, Watson DA, Pandey JP, Rodriguez-Barradas MC, Baughn RE, Pollack MS, Graviss EA, de Andrade M, Amos CI (1997) Genetic regulation of the capacity to make immunoglobulin G to pneumococcal capsular polysaccharides. J Investig Med 45:57–68
Musher DM, Watson DA, Baughn RE (2000) Genetic control of the immunologic response to pneumococcal capsular polysaccharides. Vaccine 19:623–627
Mutapi F, Winborn G, Midzi N, Taylor M, Mduluza T, Maizels RM (2007) Cytokine responses to Schistosoma haematobium in a Zimbabwean population: contrasting profiles for IFN-γ, IL-4, IL-5 and IL-10 with age. BMC Infect Dis 7:139
Nadel B, Tang A, Lugo G, Love V, Escuro G, Feeney AJ (1998) Decreased frequency of rearrangement due to the synergistic effect of nucleotide changes in the heptamer and nonamer of the recombination signal sequence of the V kappa gene A2b, which is associated with increased susceptibility of Navajos to Haemophilus influenzae type b disease. J Immunol 161:6068–6073
Nasr A, Iriemenam NC, Troye-Blomberg M, Giha HA, Balogun HA, Osman OF, Montgomery SM, ElGhazali G, Berzins K (2007) Fc gamma receptor IIa (CD32) polymorphism and antibody responses to asexual blood-stage antigens of Plasmodium falciparum malaria in Sudanese patients. Scand J Immunol 66:87–96
Natividad A, Wilson J, Koch O, Holland MJ, Rockett K, Faal N, Jallow O, Joof HM, Burton MJ, Alexander ND, Kwiatkowski DP, Mabey DC, Bailey RL (2005) Risk of trachomatous scarring and trichiasis in Gambians varies with SNP haplotypes at the interferon-gamma and interleukin-10 loci. Genes Immun 6:332–340
Natividad A, Cooke G, Holland MJ, Burton MJ, Joof HM, Rockett K, Kwiatkowski DP, Mabey DC, Bailey RL (2006) A coding polymorphism in matrix metalloproteinase 9 reduces risk of scarring sequelae of ocular Chlamydia trachomatis infection. BMC Med Genet 7:40
Natividad A, Hanchard N, Holland MJ, Mahdi OS, Diakite M, Rockett K, Jallow O, Joof HM, Kwiatkowski DP, Mabey DC, Bailey RL (2007) Genetic variation at the TNF locus and the risk of severe sequelae of ocular Chlamydia trachomatis infection in Gambians. Genes Immun 8:288–295
Natividad A, Holland MJ, Rockett KA, Forton J, Faal N, Joof HM, Mabey DC, Bailey RL, Kwiatkowski DP (2008) Susceptibility to sequelae of human ocular chlamydial infection associated with allelic variation in IL10 cis-regulation. Hum Mol Genet 17:323–329
Nejentsev S, Thye T, Szeszko JS, Stevens H, Balabanova Y, Chinbuah AM, Hibberd M, van de Vosse E, Alisjahbana B, van Crevel R, Ottenhoff TH, Png E, Drobniewski F, Todd JA, Seielstad M, Horstmann RD (2008) Analysis of association of the TIRAP (MAL) S180L variant and tuberculosis in three populations. Nat Genet 40:261–262
Ndiaye R, Sakuntabhai A, Casademont I, Rogier C, Tall A, Trape JF, Spiegel A, Dieye A, Julier C (2005) Genetic study of ICAM1 in clinical malaria in Senegal. Tissue Antigens 65:474–480
Ndung’u T, Gaseitsiwe S, Sepako E, Doualla-Bell F, Peter T, Kim S, Thior I, Novitsky VA, Essex M (2005) Major histocompatibility complex class II (HLA-DRB and -DQB) allele frequencies in Botswana: association with human immunodeficiency virus type 1 infection. Clin Diagn Lab Immunol 12:1020–1028
Neel JV (1953) Data pertaining to the population dynamics of sickle cell disease. Am J Hum Genet 5:154–167
Newport MJ, Allen A, Awomoyi AA, Dunstan SJ, McKinney E, Marchant A, Sirugo G (2004a) The toll-like receptor 4 Asp299Gly variant: no influence on LPS responsiveness or susceptibility to pulmonary tuberculosis in The Gambia. Tuberculosis (Edinb) 84:347–352
Newport MJ, Goetghebuer T, Weiss HA, Whittle H, Siegrist CA, Marchant A (2004b) Genetic regulation of immune responses to vaccines in early life. Genes Immun 5:122–129
Newport MJ, Goetghebuer T, Marchant A (2005) Hunting for immune response regulatory genes: vaccination studies in infant twins. Expert Rev Vaccines 4:739–746
Niesporek S, Meyer CG, Kremsner PG, May J (2005) Polymorphisms of transporter associated with antigen processing type 1 (TAP1), proteasome subunit beta type 9 (PSMB9) and their common promoter in African children with different manifestations of malaria. Int J Immunogenet 32:7–11
Nkeh B, Samani NJ, Badenhorst D, Libhaber E, Sareli P, Norton GR, Woodiwiss AJ (2003) T594 M variant of the epithelial sodium channel beta-subunit gene and hypertension in individuals of African ancestry in South Africa. Am J Hypertens 16:847–852
Novitsky V, Flores-Villanueva PO, Chigwedere P, Gaolekwe S, Bussman H, Sebetso G, Marlink R, Yunis EJ, Essex M (2001) Identification of most frequent HLA class I antigen specificities in Botswana: relevance for HIV vaccine design. Hum Immunol 62:146–156
Ntoumi F, Kwiatkowski DP, Diakite M, Mutabingwa TK, Duffy PE (2007) New interventions for malaria: mining the human and parasite genomes. Am J Trop Med Hyg 77:270–275
Nyakutira C, Roshammar D, Chigutsa E, Chonzi P, Ashton M, Nhachi C, Masimirembwa C (2008) High prevalence of the CYP2B6 516G– > T(*6) variant and effect on the population pharmacokinetics of efavirenz in HIV/AIDS outpatients in Zimbabwe. Eur J Clin Pharmacol 64:357–365
Olesen R, Wejse C, Velez DR, Bisseye C, Sodemann M, Aaby P, Rabna P, Worwui A, Chapman H, Diatta M, Adegbola RA, Hill PC, Ostergaard L, Williams SM, Sirugo G (2007) DC-SIGN (CD209), pentraxin 3 and vitamin D receptor gene variants associate with pulmonary tuberculosis risk in West Africans. Genes Immun 8:456–467
Opie LH, Seedat YK (2005) Hypertension in sub-Saharan African populations. Circulation 112:3562–3568
Osafo-Addo AD, Koram KA, Oduro AR, Wilson M, Hodgson A, Rogers WO (2008) HLA-DRB1*04 allele is associated with severe malaria in northern Ghana. Am J Trop Med Hyg 78:251–255
Ottenhoff TH, Torres P, de las Aguas JT, Fernandez R, van Eden W, de Vries RR, Stanford JL (1986) Evidence for an HLA-DR4-associated immune-response gene for Mycobacterium tuberculosis. A clue to the pathogenesis of rheumatoid arthritis? Lancet 2:310–313
Ouahchi K, Lindeman N, Lee C (2006) Copy number variants and pharmacogenomics. Pharmacogenomics 7:25–29
Ouerhani S, Tebourski F, Ben Slama MR, Marrakchi R, Rabeh M, Hassine LB, Ayed M, El Gaaied AB (2006) The role of glutathione transferases M1 and T1 in individual susceptibility to bladder cancer in a Tunisian population. Ann Hum Biol 33:529–535
Ouerhani S, Oliveira E, Marrakchi R, Ben Slama MR, Sfaxi M, Ayed M, Chebil M, Amorim A, El Gaaied AB, Prata MJ (2007) Methylenetetrahydrofolate reductase and methionine synthase polymorphisms and risk of bladder cancer in a Tunisian population. Cancer Genet Cytogenet 176:48–53
Ouerhani S, Marrakchi R, Bouhaha R, Ben Slama MR, Sfaxi M, Ayed M, Chebil M, El Gaaied AB (2008) The role of CYP2D6*4 variant in bladder cancer susceptibility in Tunisian patients. Bull Cancer 95:E1–E4
Ouma C, Keller CC, Opondo DA, Were T, Otieno RO, Otieno MF, Orago AS, Ong’echa JM, Vulule JM, Ferrell RE, Perkins DJ (2006) Association of FCgamma receptor IIA (CD32) polymorphism with malarial anemia and high-density parasitemia in infants and young children. Am J Trop Med Hyg 74:573–577
Ovsyannikova IG, Jacobson RM, Poland GA (2004a) Variation in vaccine response in normal populations. Pharmacogenomics 5:417–427
Ovsyannikova IG, Jacobson RM, Vierkant RA, Shane Pankratz V, Jacobsen SJ, Poland GA (2004b) Associations between human leukocyte antigen (HLA) alleles and very high levels of measles antibody following vaccination. Vaccine 22:1914–1920
Ovsyannikova IG, Johnson KL, Muddiman DC, Vierkant RA, Poland GA (2004c) Identification and characterization of novel, naturally processed measles virus class II HLA-DRB1 peptides. J Virol 78:42–51
Ovsyannikova IG, Poland GA, Easler NJ, Vierkant RA (2004d) Influence of HLA-DRB1 alleles on lymphoproliferative responses to a naturally processed and presented measles virus phosphoprotein in measles immunized individuals. Hum Immunol 65:209–217
Ovsyannikova IG, Jacobson RM, Ryan JE, Vierkant RA, Pankratz VS, Jacobsen SJ, Poland GA (2005a) HLA class II alleles and measles virus-specific cytokine immune response following two doses of measles vaccine. Immunogenetics 56:798–807
Ovsyannikova IG, Jacobson RM, Vierkant RA, Jacobsen SJ, Pankratz VS, Poland GA (2005b) Human leukocyte antigen class II alleles and rubella-specific humoral and cell-mediated immunity following measles–mumps–rubella-II vaccination. J Infect Dis 191:515–519
Ovsyannikova IG, Ryan JE, Vierkant RA, Pankratz VS, Jacobson RM, Poland GA (2005c) Immunologic significance of HLA class I genes in measles virus-specific IFN-gamma and IL-4 cytokine immune responses. Immunogenetics 57:828–836
Ovsyannikova IG, Dhiman N, Jacobson RM, Poland GA (2006a) Human leukocyte antigen polymorphisms: variable humoral immune responses to viral vaccines. Expert Rev Vaccines 5:33–43
Ovsyannikova IG, Pankratz VS, Vierkant RA, Jacobson RM, Poland GA (2006b) Human leukocyte antigen haplotypes in the genetic control of immune response to measle–mumps–rubella vaccine. J Infect Dis 193:655–663
Ovsyannikova IG, Vierkant RA, Poland GA (2006c) Importance of HLA-DQ and HLA-DP polymorphisms in cytokine responses to naturally processed HLA-DR-derived measles virus peptides. Vaccine 24:5381–5389
Ovsyannikova IG, Jacobson RM, Dhiman N, Vierkant RA, Pankratz VS, Poland GA (2007a) HLA homozygosity does not adversely affect measles vaccine-induced cytokine responses. Virology 364:87–94
Ovsyannikova IG, Jacobson RM, Vierkant RA, Pankratz VS, Poland GA (2007b) HLA supertypes and immune responses to measles–mumps–rubella viral vaccine: findings and implications for vaccine design. Vaccine 25:3090–3100
Pain A, Urban B, Kai O, Casals-Pascual C, Shafi J, Marsh K, Roberts JM (2001) A non-sense mutation in Cd36 gene is associated with protection from severe malaria. Lancet 357:1502–1503
Parikh S, Dorsey G, Rosenthal PJ (2004) Host polymorphisms and the incidence of malaria in Ugandan children. Am J Trop Med Hyg 71:750–753
Parkin D, Ferlay J, Hamdi-Cherif M, Sistas F, Thomas J, Wabinga H (2003) Cancer in Africa:epidemiology and prevention. IARC PRESS, Lyon
Passarino G, Semino O, Quintana-Murci L, Excoffier L, Hammer M, Santachiara-Benerecetti AS (1998) Different genetic components in the Ethiopian population, identified by mtDNA and Y-chromosome polymorphisms. Am J Hum Genet 62:420–434
Pasvol G (2006) Does alpha+-thalassaemia protect against malaria? PLoS Med 3:e235
Patel M, Wadee AA, Galpin J, Gavalakis C, Fourie AM, Kuschke RH, Philip V (2002) HLA class I and class II antigens associated with multiple myeloma in southern Africa. Clin Lab Haematol 24:215–219
Pearce EJ, Kane CM, Sun J, Taylor JJ, McKee AS, Cervi L (2004) Th2 response polarization during infection with the helminth parasite Schistosoma mansoni. Immunol Rev 201:117–126
Petersen DC, Glashoff RH, Shrestha S, Bergeron J, Laten A, Gold B, van Rensburg EJ, Dean M, Hayes VM (2005) Risk for HIV-1 infection associated with a common CXCL12 (SDF1) polymorphism and CXCR4 variation in an African population. J Acquir Immune Defic Syndr 40:521–526
Poland GA (1998) Variability in immune response to pathogens: using measles vaccine to probe immunogenetic determinants of response. Am J Hum Genet 62:215–220
Poland GA, Jacobson RM (1998) The genetic basis for variation in antibody response to vaccines. Curr Opin Pediatr 10:208–215
Poland GA, Jacobson RM, Colbourne SA, Thampy AM, Lipsky JJ, Wollan PC, Roberts P, Jacobsen SJ (1999) Measles antibody seroprevalence rates among immunized Inuit, Innu and Caucasian subjects. Vaccine 17:1525–1531
Poland GA, Ovsyannikova IG, Jacobson RM, Smith DI (2007) Heterogeneity in vaccine immune response: the role of immunogenetics and the emerging field of vaccinomics. Clin Pharmacol Ther 82:653–664
Poland GA, Ovsyannikova IG, Jacobson RM, Vierkant RA, Jacobsen SJ, Pankratz VS, Schaid DJ (2001a) Identification of an association between HLA class II alleles and low antibody levels after measles immunization. Vaccine 20:430–438
Poland GA, Sohni Y, Domanico M, Kroning CM, DeGoey SR, Jimale M, Jacobson RM, Moore SB (2001b) High frequency of HLA-A*0103 allele in a Somali population. Hum Immunol 62:197–200
Porter P (2008) “Westernizing” women’s risks? Breast cancer in lower-income countries. N Engl J Med 358:213–216
Price EW (1990) Podoconiosis. Non-filarial elephantiasis. Oxford University Press, Oxford
Puumalainen T, Dagan R, Wuorimaa T, Zeta-Capeding R, Lucero M, Ollgren J, Kayhty H, Nohynek H (2003) Greater antibody responses to an eleven valent mixed carrier diphtheria- or tetanus-conjugated pneumococcal vaccine in Filipino than in Finnish or Israeli infants. Pediatr Infect Dis J 22:141–149
Quintana-Murci L, Semino O, Bandelt HJ, Passarino G, McElreavey K, Santachiara-Benerecetti AS (1999) Genetic evidence of an early exit of Homo sapiens sapiens from Africa through eastern Africa. Nat Genet 23:437–41
Quintana-Murci L, Alcaïs A, Abel L, Casanova JL (2007) Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nat Immunol 8:1165–1171
Rager-Zisman B, Bazarsky E, Skibin A, Tam G, Chamney S, Belmaker I, Shai I, Kordysh E, Griffin DE (2004) Differential immune responses to primary measles–mumps–rubella vaccination in Israeli children. Clin Diagn Lab Immunol 11:913–918
Ramaley PA, French N, Kaleebu P, Gilks C, Whitworth J, Hill AV (2002) HIV in Africa (Communication arising): chemokine-receptor genes and AIDS risk. Nature 417:140
Ranque B, Alter A, Mira M, Thuc NV, Thai VH, Huong NT, Ba NN, Khoa PX, Schurr E, Abel L, Alcais A (2007) Genomewide linkage analysis of the granulomatous mitsuda reaction implicates chromosomal regions 2q35 and 17q21. J Infect Dis 196:1248–1252
Ranque B, Alter A, Schurr E, Abel L, Alcais A (2008) Leprosy: a paradigm for the study of human genetic susceptibility to infectious diseases. Med Sci 24:491–498
Rayner BL, Owen EP, King JA, Soule SG, Vreede H, Opie LH, Marais D, Davidson JS (2003) A new mutation, R563Q, of the beta subunit of the epithelial sodium channel associated with low-renin, low-aldosterone hypertension. J Hypertens 21:921–926
Reed FA, Tishkoff SA (2006) African human diversity, origins and migrations. Curr Opin Genet Dev 16:597–605
Remus N, El BJ, Fieschi C, Feinberg J, Quintin T, Chentoufi M, Schurr E, Benslimane A, Casanova JL, Abel L (2004) Association of IL12RB1 polymorphisms with pulmonary tuberculosis in adults in Morocco. J Infect Dis 190:580–587
Rennert H, Zeigler-Johnson CM, Addya K, Finley MJ, Walker AH, Spangler E, Leonard DG, Wein A, Malkowicz SB, Rebbeck TR (2005) Association of susceptibility alleles in ELAC2/HPC2, RNASEL/HPC1, and MSR1 with prostate cancer severity in European American and African American men. Cancer Epidemiol Biomarkers Prev 14:949–957
Rihet P, Traore Y, Abel L, Aucan C, Traore-Leroux T, Fumoux F (1998) Malaria in humans: Plasmodium falciparum blood infection levels are linked to chromosome 5q31–q33. Am J Hum Genet 63:498–505
Rihet P, Flori L, Tall F, Traore AS, Fumoux F (2004) Hemoglobin C is associated with reduced Plasmodium falciparum parasitemia and low risk of mild malaria attack. Hum Mol Genet 13:1–6
Robbins C, Torres JB, Hooker S, Bonilla C, Hernandez W, Candreva A, Ahaghotu C, Kittles R, Carpten J (2007) Confirmation study of prostate cancer risk variants at 8q24 in African Americans identifies a novel risk locus. Genome Res 17:1717–1722
Robinson M, Williams SM (2004) Role of two angiotensinogen polymorphisms in blood pressure variation. J Hum Hypertens 18:865–869
Rossouw M, Nel HJ, Cooke GS, Van Helden PD, Hoal EG (2003) Association between tuberculosis and a polymorphic NFkappaB binding site in the interferon gamma gene. Lancet 361:1871–1872
Rotger M, Colombo S, Furrer H, Bleiber G, Buclin T, Lee BL, Keiser O, Biollaz J, Decosterd L, Telenti A (2005) Influence of CYP2B6 polymorphism on plasma and intracellular concentrations and toxicity of efavirenz and nevirapine in HIV-infected patients. Pharmacogenet Genomics 15:1–5
Rotger M, Saumoy M, Zhang K, Flepp M, Sahli R, Decosterd L, Telenti A (2007) Partial deletion of CYP2B6 owing to unequal crossover with CYP2B7. Pharmacogenet Genomics 17:885–890
Rotimi CN (2004) Inauguration of the African society of human genetics. Nat Genet 36:544
Rotimi CN, Cooper RS, Cao G, Ogunbiyi O, Ladipo M, Owoaje E, Ward R (1999) Maximum-likelihood generalized heritability estimate for blood pressure in Nigerian families. Hypertension 33:874–8
Rotimi CN, Dunston GM, Berg K, Akinsete O, Amoah A, Owusu S, Acheampong J, Boateng K, Oli J, Okafor G, Onyenekwe B, Osotimehin B, Abbiyesuku F, Johnson T, Fasanmade O, Furbert-Harris P, Kittles R, Vekich M, Adegoke O, Bonney G, Collins F (2001) In search of susceptibility genes for type 2 diabetes in West Africa: the design and results of the first phase of the AADM study. Ann Epidemiol 11:51–58
Rotimi CN, Chen G, Adeyemo AA, Furbert-Harris P, Parish-Gause D, Zhou J, Berg K, Adegoke O, Amoah A, Owusu S, Acheampong J, Agyenim-Boateng K, Eghan BA Jr., Oli J, Okafor G, Ofoegbu E, Osotimehin B, Abbiyesuku F, Johnson T, Rufus T, Fasanmade O, Kittles R, Daniel H, Chen Y, Dunston G, Collins FS (2004) A genome-wide search for type 2 diabetes susceptibility genes in West Africans: the Africa America Diabetes Mellitus (AADM) Study. Diabetes 53:838–841
Rotimi CN, Chen G, Adeyemo AA, Jones LS, Agyenim-Boateng K, Eghan BA Jr., Zhou J, Doumatey A, Lashley K, Huang H, Fasanmade O, Akinsola FB, Ezepue F, Amoah A, Akafo S, Chen Y, Oli J, Johnson T (2006) Genomewide scan and fine mapping of quantitative trait loci for intraocular pressure on 5q and 14q in West Africans. Invest Ophthalmol Vis Sci 47:3262–3267
Rowe JA, Raza A, Diallo DA, Baby M, Poudiougo B, Coulibaly D, Cockburn IA, Middleton J, Lyke KE, Plowe CV, Doumbo OK, Moulds JM (2002) Erythrocyte CR1 expression level does not correlate with a HindIII restriction fragment length polymorphism in Africans; implications for studies on malaria susceptibility. Genes Immun 3:497–500
Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, Warn P, Allsopp CE, Gilbert SC, Peschu N (1995) Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature 376:246–249
Ryan JR, Stoute JA, Amon J, Dunton RF, Mtalib R, Koros J, Owour B, Luckhart S, Wirtz RA, Barnwell JW, Rosenberg R (2006) Evidence for transmission of Plasmodium vivax among a duffy antigen negative population in Western Kenya. Am J Trop Med Hyg 75:575–581
Saad AA, O’Connor PJ, Mostafa MH, Metwalli NE, Cooper DP, Povey AC, Margison GP (2005) Glutathione S-transferase M1, T1 and P1 polymorphisms and bladder cancer risk in Egyptians. Int J Biol Markers 20:69–72
Sabeti P, Usen S, Farhadian S, Jallow M, Doherty T, Newport M, Pinder M, Ward R, Kwiatkowski D (2002a) CD40L association with protection from severe malaria. Genes Immun 3:286–291
Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES (2002b) Detecting recent positive selection in the human genome from haplotype structure. Nature 419:832–837
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA et al (2007) Genome-wide detection and characterization of positive selection in human populations. Nature 18:913–918
Sakuntabhai A, Ndiaye R, Casademont I, Peerapittayamonkol C, Rogier C, Tortevoye P, Tall A, Paul R, Turbpaiboon C, Phimpraphi W, Trape JF, Spiegel A, Heath S, Mercereau-Puijalon O, Dieye A, Julier C (2008) Genetic determination and linkage mapping of Plasmodium falciparum malaria related traits in Senegal. PLoS ONE 3:e2000
Salih MA, Ibrahim ME, Blackwell JM, Miller EN, Khalil EA, ElHassan AM, Musa AM, Mohamed HS (2007) IFNG and IFNGR1 gene polymorphisms and susceptibility to post-kala-azar dermal leishmaniasis in Sudan. Genes Immun 8:75–78
Sarma AV, Dunn RL, Lange LA, Ray A, Wang Y, Lange EM, Cooney KA (2008) Genetic polymorphisms in CYP17, CYP3A4, CYP19A1, SRD5A2, IGF-1, and IGFBP-3 and prostate cancer risk in African–American men: The Flint Men’s Health Study. Prostate 68:296–305
Saunders MA, Hammer MF, Nachman MW (2002) Nucleotide variability at G6pd and the signature of malarial selection in humans. Genetics 162:1849–1861
Schim van der Loeff (2007) In: Volberding P, Sande MA, Lange J, Greene WC, Gallant JE (eds) Global HIV/AIDS medicine. Elsevier, Amsterdam
Schumacher FR, Feigelson HS, Cox DG, Haiman CA, Albanes D, Buring J, Calle EE, Chanock SJ, Colditz GA, Diver WR, Dunning AM, Freedman ML, Gaziano JM, Giovannucci E, Hankinson SE, Hayes RB, Henderson BE, Hoover RN, Kaaks R, Key T, Kolonel LN, Kraft P, Le Marchand L, Ma J, Pike MC, Riboli E, Stampfer MJ, Stram DO, Thomas G, Thun MJ, Travis R, Virtamo J, Andriole G, Gelmann E, Willett WC, Hunter DJ (2007) A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res 67:2951–2956
Scozzari R, Cruciani F, Santolamazza P, Malaspina P, Torroni A, Sellitto D, Arredi B, Destro-Bisol G, De Stefano G, Rickards O, Martinez-Labarga C, Modiano D, Biondi G, Moral P, Olckers A, Wallace DC, Novelletto A (1999) Combined use of biallelic and microsatellite Y-chromosome polymorphisms to infer affinities among African populations. Am J Hum Genet 65:829–846
Sgaier SK, Jha P, Mony P, Kurpad A, Lakshmi V, Kumar R, Ganguly NK (2007) Public health. Biobanks in developing countries: needs and feasibility. Science 318:1074–1075
Shand RL, Gelmann EP (2006) Molecular biology of prostate-cancer pathogenesis. Curr Opin Urol 16:123–131
Shi YP, Nahlen BL, Kariuki S, Urdahl KB, McElroy PD, Roberts JM, Lal AA (2001) Fcgamma receptor IIa (CD32) polymorphism is associated with protection of infants against high-density Plasmodium falciparum infection. VII. Asembo Bay Cohort Project. J Infect Dis 184:107–111
Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DA, Doherty PC, Grosveld G, Paul WE, Ihle JN (1996) Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 380:630–633
Siber GR, Santosham M, Reid GR, Thompson C, Meido-Hill J, Morell A, de Lange G, Ketcham JK, Callahan EH (1990) Impaired antibody response to Haemophilus influenzae type b polysaccharide and low IgG2 and IgG4 concentrations in Apache children. N Engl J Med 323:1387–1392
Sile S, Gillani NB, Velez DR, Vanoye CG, Yu C, Byrne LM, Gainer JV, Brown NJ, Williams SM, George AL Jr. (2007) Functional BSND variants in essential hypertension. Am J Hypertens 20:1176–1182
Single RM, Martin MP, Gao X, Meyer D, Yeager M, Kidd JR, Kidd KK, Carrington M (2007) Global diversity and evidence for coevolution of KIR and HLA. Nat Genet 39:1114–1119
Sirugo G, van der Loeff SLM, Sam O, Nyan O, Pinder M, Hill AV, Kwiatkowski D, Prentice A, de Toma C, Cann HM, Diatta M, Jallow M, Morgan G, Clarke M, Corrah T, Whittle H, McAdam K (2004) A national DNA bank in The Gambia, West Africa, and genomic research in developing countries. Nat Genet 36:785–786
Sistonen J, Sajantila A, Lao O, Corander J, Barbujani G, Fuselli S (2007) CYP2D6 worldwide genetic variation shows high frequency of altered activity variants and no continental structure. Pharmacogenet Genomics 17:93–101
Sleijffers A, Yucesoy B, Kashon M, Garssen J, De Gruijl FR, Boland GJ, van Hattum J, Luster MI, van Loveren H (2003) Cytokine polymorphisms play a role in susceptibility to ultraviolet B-induced modulation of immune responses after hepatitis B vaccination. J Immunol 170:3423–3428
Snoussi K, Mahfoudh W, Bouaouina N, Ahmed SB, Helal AN, Chouchane L (2006) Genetic variation in IL-8 associated with increased risk and poor prognosis of breast carcinoma. Hum Immunol 67:13–21
Soborg C, Andersen AB, Range N, Malenganisho W, Friis H, Magnussen P, Temu MM, Changalucha J, Madsen HO, Garred P (2007) Influence of candidate susceptibility genes on tuberculosis in a high endemic region. Mol Immunol 44:2213–2220
St Sauver JL, Ovsyannikova IG, Jacobson RM, Jacobsen SJ, Vierkant RA, Schaid DJ, Pankratz VS, Green EM, Poland GA (2002) Associations between human leukocyte antigen homozygosity and antibody levels to measles vaccine. J Infect Dis 185:1545–1549
St Sauver JL, Dhiman N, Ovsyannikova IG, Jacobson RM, Vierkant RA, Pankratz VS, Jacobsen SJ, Poland GA (2005) Extinction of the human leukocyte antigen homozygosity effect after two doses of the measles–mumps–rubella vaccine. Hum Immunol 66:788–798
Stanton T, Boxall S, Bennett A, Kaleebu P, Watera C, Whitworth J, French N, Dawes R, Hill AV, Bodmer W, Beverley PC, Tchilian EZ (2004) CD45 variant alleles: possibly increased frequency of a novel exon 4 CD45 polymorphism in HIV seropositive Ugandans. Immunogenetics 56:107–110
Stein CM, Zalwango S, Chiunda AB, Millard C, Leontiev DV, Horvath AL, Cartier KC, Chervenak K, Boom WH, Elston RC, Mugerwa RD, Whalen CC, Iyengar SK (2007) Linkage and association analysis of candidate genes for TB and TNFalpha cytokine expression: evidence for association with IFNGR1, IL-10, and TNF receptor 1 genes. Hum Genet 121:663–673
Steyn K, Sliwa K, Hawken S, Commerford P, Onen C, Damasceno A, Ounpuu S, Yusuf S (2005) Risk factors associated with myocardial infarction in Africa: the INTERHEART Africa study. Circulation 112:3554–3561
Tanara G, Falugi C, Cesario A, Margaritora S, Russo P, Cosimi A (2003) TP53 codon 72 polymorphism does not affect risk of cervical cancer in patients from The Gambia. Int J Biol Markers 18:280–283
Tang J, Tang S, Lobashevsky E, Myracle AD, Fideli U, Aldrovandi G, Allen S, Musonda R, Kaslow RA (2002) Favorable and unfavorable HLA class I alleles and haplotypes in Zambians predominantly infected with clade C human immunodeficiency virus type 1. J Virol 76:8276–8284
Tang J, Penman-Aguilar A, Lobashevsky E, Allen S, Kaslow RA (2004a) HLA-DRB1 and -DQB1 alleles and haplotypes in Zambian couples and their associations with heterosexual transmission of HIV type 1. J Infect Dis 189:1696–1704
Tang J, Tang S, Lobashevsky E, Zulu I, Aldrovandi G, Allen S, Kaslow RA (2004b) HLA allele sharing and HIV type 1 viremia in seroconverting Zambians with known transmitting partners. AIDS Res Hum Retroviruses 20:19–25
Thathy V, Moulds JM, Guyah B, Otieno W, Stoute JA (2005) Complement receptor 1 polymorphisms associated with resistance to severe malaria in Kenya. Malar J 4:54
Thursz M (2001) MHC and the viral hepatitides. QJM 94:287–291
Thursz MR, Kwiatkowski D, Allsopp CE, Greenwood BM, Thomas HC, Hill AV (1995) Association between an MHC class II allele and clearance of hepatitis B virus in the Gambia. N Engl J Med 332:1065–1069
Thye T, Browne EN, Chinbuah MA, Gyapong J, Osei I, Owusu-Dabo E, Niemann S, Rusch-Gerdes S, Horstmann RD, Meyer CG (2006) No associations of human pulmonary tuberculosis with Sp110 variants. J Med Genet 43:e32
Tiago AD, Badenhorst D, Nkeh B, Candy GP, Brooksbank R, Sareli P, Libhaber E, Samani NJ, Woodiwiss AJ, Norton GR (2003) Impact of renin–angiotensin–aldosterone system gene variants on the severity of hypertension in patients with newly diagnosed hypertension. Am J Hypertens 16:1006–1010
Tibazarwa K, Ntyintyane L, Sliwa K, Gerntholtz T, Carrington M, Wilkinson D, Stewart S (2008) A time bomb of cardiovascular risk factors in South Africa: Results from the Heart of Soweto Study “Heart Awareness Days”. Int J Cardiol (in press)
Timmann C, Evans JA, Konig IR, Kleensang A, Ruschendorf F, Lenzen J, Sievertsen J, Becker C, Enuameh Y, Kwakye KO, Opoku E, Browne EN, Ziegler A, Nurnberg P, Horstmann RD (2007) Genome-wide linkage analysis of malaria infection intensity and mild disease. PLoS Genet 3:e48
Tishkoff SA, Kidd KK (2004) Implications of biogeography of human populations for ‘race’ and medicine. Nat Genet 36:S21–S27
Tishkoff SA, Williams SM (2002) Genetic analysis of African populations: human evolution and complex disease. Nat Rev Genet 3:611–621
Tishkoff SA, Verrelli BC (2003a) Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu Rev Genomics Hum Genet 4:293–340
Tishkoff SA, Verrelli BC (2003b) Role of evolutionary history on haplotype block structure in the human genome: implications for disease mapping. Curr Opin Genet Dev 13:569–575
Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonne-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M (1996) Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science 271:1380–1387
Tishkoff SA, Goldman A, Calafell F, Speed WC, Deinard AS, Bonne-Tamir B, Kidd JR, Pakstis AJ, Jenkins T, Kidd KK (1998) A global haplotype analysis of the myotonic dystrophy locus: implications for the evolution of modern humans and for the origin of myotonic dystrophy mutations. Am J Hum Genet 62:1389–1402
Tishkoff SA, Pakstis AJ, Stoneking M, Kidd JR, Stro-Bisol G, Sanjantila A, Lu RB, Deinard AS, Sirugo G, Jenkins T, Kidd KK, Clark AG (2000) Short tandem-repeat polymorphism/alu haplotype variation at the PLAT locus: implications for modern human origins. Am J Hum Genet 67:901–925
Tishkoff SA, Varkonyi R, Cahinhinan N, Abbes S, Argyropoulos G, Stro-Bisol G, Drousiotou A, Dangerfield B, Lefranc G, Loiselet J, Piro A, Stoneking M, Tagarelli A, Tagarelli G, Touma EH, Williams SM, Clark AG (2001) Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science 293:455–462
Tishkoff SA, Gonder MK, Hirbo JB, Mortensen HM, Powell K, Knight A, Mountain JL (2003) The genetic history of linguistically diverse Tanzanian populations: a multilocus analysis. American Association of Physical Anthropology, Tempe
Tishkoff SA, Gonder MK, Henn BM, Mortensen H, Knight A, Gignoux C, Fernandopulle N, Lema G, Nyambo TB, Ramakrishnan U, Reed FA, Mountain JL (2007a) History of click-speaking populations of Africa inferred from mtDNA and Y chromosome genetic variation. Mol Biol Evol 24:2180–2195
Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, Ibrahim M, Omar SA, Lema G, Nyambo TB, Ghori J, Bumpstead S, Pritchard JK, Wray GA, Deloukas P (2007b) Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet 39:31–40
Torcia MG, Santarlasci V, Cosmi L, Clemente A, Maggi L, Mangano VD, Verra F, Bancone G, Nebie I, Sirima BS, Liotta F, Frosali F, Angeli R, Severini C, Sannella AR, Bonini P, Lucibello M, Maggi E, Garaci E, Coluzzi M, Cozzolino F, Annunziato F, Romagnani S, Modiano D (2008) Functional deficit of T regulatory cells in Fulani, an ethnic group with low susceptibility to Plasmodium falciparum malaria. Proc Natl Acad Sci USA 105:646–651
Tornesello ML, Waddell KM, Duraturo ML, Biryahwaho B, Downing R, Lucas SB, Giani U, Buonaguro L, Buonaguro FM (2005) TP53 codon 72 polymorphism and risk of conjunctival squamous cell carcinoma in Uganda. Cancer Detect Prev 29:501–508
Tosh K, Campbell SJ, Fielding K, Sillah J, Bah B, Gustafson P, Manneh K, Lisse I, Sirugo G, Bennett S, Aaby P, McAdam KP, Bah-Sow O, Lienhardt C, Kramnik I, Hill AV (2006) Variants in the SP110 gene are associated with genetic susceptibility to tuberculosis in West Africa. Proc Natl Acad Sci USA 103:10364–10368
Tournamille C, Colin Y, Cartron JP, Le Van Kim C (1995) Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet 10:224–228
Troudi W, Uhrhammer N, Sibille C, Dahan C, Mahfoudh W, Bouchlaka Souissi C, Jalabert T, Chouchane L, Bignon YJ, Ben Ayed F, Ben Ammar Elgaaied A (2007) Contribution of the BRCA1 and BRCA2 mutations to breast cancer in Tunisia. J Hum Genet 52:915–920
Ukaejiofo EO, Nubila T (2006) Association between ABO, Rhesus blood group systems and haemoglobin genotype among confirmed HIV/AIDS-TB co-infected patients in Enugu Urban, Nigeria. West Afr J Med 25:61–64
Uko GP, Lu LY, Asuquo MA, Fici D, Mahan S, Awdeh Z, Udim ER, Ding W, Umana U, Adewole T, Fraser PA (1999) HLA-DRB1 leprogenic motifs in nigerian population groups. Clin Exp Immunol 118:56–62
Unwin N, Setel P, Rashid S, Mugusi F, Mbanya JC, Kitange H, Hayes L, Edwards R, Aspray T, Alberti KG (2001) Noncommunicable diseases in sub-Saharan Africa: where do they feature in the health research agenda? Bull World Health Organ 79:947–953
Vafa M, Maiga B, Berzins K, Hayano M, Bereczky S, Dolo A, Daou M, Arama C, Kouriba B, Farnert A, Doumbo OK, Troye-Blomberg M (2007) Associations between the IL-4-590 T allele and Plasmodium falciparum infection prevalence in asymptomatic Fulani of Mali. Microbes Infect 9:1043–1048
Van der Sande MA, Ceesay SM, Milligan PJ, Nyan OA, Banya WA, Prentice A, McAdam KP, Walraven GE (2001) Obesity and undernutrition and cardiovascular risk factors in rural and urban Gambian communities. Am J Public Health 91:1641–1644
van der Werf MJ, de Vlas SJ, Brooker S, Looman CW, Nagelkerke NJ, Habbema JD, Engels D (2003) Quantification of clinical morbidity associated with schistosome infection in sub-Saharan Africa. Acta Trop 86:125–139
van Eden W, de Vries RR, Stanford JL, Rook GA (1983) HLA-DR3 associated genetic control of response to multiple skin tests with new tuberculins. Clin Exp Immunol 52:287–292
van Loveren H, Van Amsterdam JG, Vandebriel RJ, Kimman TG, Rumke HC, Steerenberg PS, Vos JG (2001) Vaccine-induced antibody responses as parameters of the influence of endogenous and environmental factors. Environ Health Perspect 109:757–764
Vannberg FO, Chapman SJ, Khor CC, Tosh K, Floyd S, Jackson-Sillah D, Crampin A, Sichali L, Bah B, Gustafson P, Aaby P, McAdam KP, Bah-Sow O, Lienhardt C, Sirugo G, Fine P, Hill AV (2008) CD209 genetic polymorphism and tuberculosis disease. PLoS ONE 3:e1388
Verra F, Luoni G, Calissano C, Troye-Blomberg M, Perlmann P, Perlmann H, Arca B, Sirima BS, Konate A, Coluzzi M, Kwiatkowski D, Modiano D (2004) IL4–589C/T polymorphism and IgE levels in severe malaria. Acta Trop 90:205–209
Verrelli BC, McDonald JH, Argyropoulos G, stro-Bisol G, Froment A, Drousiotou A, Lefranc G, Helal AN, Loiselet J, Tishkoff SA (2002) Evidence for balancing selection from nucleotide sequence analyses of human G6PD. Am J Hum Genet 71:1112–1128
Wadelius M, Pirmohamed M (2006) Pharmacogenetics of warfarin: current status and future challenges. Pharmacogenomics J 7:99–111
Wagner TM, Hirtenlehner K, Shen P, Moeslinger R, Muhr D, Fleischmann E, Concin H, Doeller W, Haid A, Lang AH, Mayer P, Petru E, Ropp E, Langbauer G, Kubista E, Scheiner O, Underhill P, Mountain J, Stierer M, Zielinski C, Oefner P (1999) Global sequence diversity of BRCA2: analysis of 71 breast cancer families and 95 control individuals of worldwide populations. Hum Mol Genet 8:413–423
Wallace C, Fitness J, Hennig B, Sichali L, Mwaungulu L, Ponnighaus JM, Warndorff DK, Clayton D, Fine PE, Hill AV (2004) Linkage analysis of susceptibility to leprosy type using an IBD regression method. Genes Immun 5:221–225
Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, Stephens RM, Caporaso NE, Loffredo CA, Ambs S (2008) Tumor immunobiological differences in prostate cancer between African–American and European–American men. Cancer Res 68:927–936
Walley AJ, Aucan C, Kwiatkowski D, Hill AV (2004) Interleukin-1 gene cluster polymorphisms and susceptibility to clinical malaria in a Gambian case-control study. Eur J Hum Genet 12:132–138
Wambua S, Mwangi TW, Kortok M, Uyoga SM, Macharia AW, Mwacharo JK, Weatherall DJ, Snow RW, Marsh K, Williams TN (2006) The effect of alpha + -thalassaemia on the incidence of malaria and other diseases in children living on the coast of Kenya. PLoS Med 3:e158
Willcox M, Bjorkman A, Brohult J (1983) Falciparum malaria and beta-thalassaemia trait in northern Liberia. Ann Trop Med Parasitol 77:335–347
Wang C, Tang J, Song W, Lobashevsky E, Wilson CM, Kaslow RA (2004) HLA and cytokine gene polymorphisms are independently associated with responses to hepatitis B vaccination. Hepatology 39:978–988
Webb JLA (2005) Malaria and the Peopling of Early Tropical Africa. J World History 16:270–291
Wennerholm A, Johansson I, Hidestrand M, Bertilsson L, Gustafsson LL, Ingelman-Sundberg M (2001) Characterization of the CYP2D6*29 allele commonly present in a black Tanzanian population causing reduced catalytic activity. Pharmacogenetics 11:417–427
White TD, Asfaw B, DeGusta D, Gilbert H, Richards GD, Suwa G, Howell FC (2003) Pleistocene Homo sapiens from Middle Awash, Ethiopia. Nature 12:742–747
Wiertsema SP, Veenhoven RH, Walraven V, Uiterwaal CS, Schilder AG, Rijkers GT, Sanders EA (2006) Pneumococcal vaccine efficacy for mucosal pneumococcal infections depends on Fcgamma receptor IIa polymorphism. Vaccine 24:792–797
Wiertsema SP, Baynam G, Khoo SK, Veenhoven RH, van Heerbeek N, Zhang G, Laing IA, Rijkers GT, Goldblatt J, Sanders EA, Le Souef PN (2007) Impact of genetic variants in IL-4, IL-4 RA and IL-13 on the anti-pneumococcal antibody response. Vaccine 25:306–313
Williams TN, Maitland K, Bennett S, Ganczakowski M, Peto TE, Newbold CI, Bowden DK, Weatherall DJ, Clegg JB (1996) High incidence of malaria in alpha-thalassaemic children. Nature 10:522–525
Williams SM, Addy JH, Phillips JA3, Dai M, Kpodonu J, Afful J, Jackson H, Joseph K, Eason F, Murray MM, Epperson P, Aduonum A, Wong LJ, Jose PA, Felder RA (2000) Combinations of variations in multiple genes are associated with hypertension. Hypertension 36:2–6
Williams SM, Ritchie MD, Phillips JA, Dawson E, Prince M, Dzhura E, Willis A, Semenya A, Summar M, White BC, Addy JH, Kpodonu J, Wong L-J, Felder RA, Jose PA, Moore JH (2004) Multilocus analysis of hypertension: a hierarchical approach. Human Heredity 57:28–38
Williams TN, Wambua S, Uyoga S, Macharia A, Mwacharo JK, Newton CR, Maitland K (2005a) Both heterozygous and homozygous alpha + thalassemias protect against severe and fatal Plasmodium falciparum malaria on the coast of Kenya. Blood 106:368–371
Williams TN, Mwangi TW, Wambua S, Peto TE, Weatherall DJ, Gupta S, Recker M, Penman BS, Uyoga S, Macharia A, Mwacharo JK, Snow RW, Marsh K (2005b) Negative epistasis between the malaria-protective effects of alpha + -thalassemia and the sickle cell trait. Nat Genet 37:1253–1257
Williams SM, Stocki S, Jiang L, Brew K, Gordon S, Vaughan DE, Brown NJ, Poku KA, Moore JH (2007) A population-based study in Ghana to investigate inter-individual variation in plasma t-PA and PAI–1. Ethn Dis 17:492–497
Woodiwiss AJ, Nkeh B, Samani NJ, Badenhorst D, Maseko M, Tiago AD, Candy GP, Libhaber E, Sareli P, Brooksbank R, Norton GR (2006) Functional variants of the angiotensinogen gene determine antihypertensive responses to angiotensin-converting enzyme inhibitors in subjects of African origin. J Hypertens 24:1057–1064
Yasar U, Aklillu E, Canaparo R, Sandberg M, Sayi J, Roh HK, Wennerholm A (2002) Analysis of CYP2C9*5 in Caucasian, Oriental and black-African populations. Eur J Clin Pharmacol 58:555–558
Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G (2007) Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 39:645–649
Yee AM, Phan HM, Zuniga R, Salmon JE, Musher DM (2000) Association between Fc gamma RIIa-R131 allotype and bacteremic pneumococcal pneumonia. Clin Infect Dis 30:25–28
Yucesoy B, Sleijffers A, Kashon M, Garssen J, De Gruijl FR, Boland GJ, van Hattum J, Simeonova PP, Luster MI, van Loveren H (2002) IL-1beta gene polymorphisms influence hepatitis B vaccination. Vaccine 20:3193–3196
Zaahl MG, Warnich L, Victor TC, Kotze MJ (2005) Association of functional polymorphisms of SLC11A1 with risk of esophageal cancer in the South African colored population. Cancer Genet Cytogenet 159:48–52
Zabaleta J, Lin HY, Sierra RA, Hall MC, Clark PE, Sartor OA, Hu JJ, Ochoa AC (2008) Interactions of cytokine gene polymorphisms in prostate cancer risk. Carcinogenesis 29:573–8
Zeigler-Johnson CM, Walker AH, Mancke B, Spangler E, Jalloh M, McBride S, Deitz A, Malkowicz SB, Ofori-Adjei D, Gueye SM, Rebbeck TR (2002) Ethnic differences in the frequency of prostate cancer susceptibility alleles at SRD5A2 and CYP3A4. Hum Hered 54:13–21
Zheng SL, Sun J, Wiklund F, Smith S, Stattin P, Li G, Adami HO, Hsu FC, Zhu Y, Bälter K, Kader AK, Turner AR, Liu W, Bleecker ER, Meyers DA, Duggan D, Carpten JD, Chang BL, Isaacs WB, Xu J, Grönberg H (2008) Cumulative association of five genetic variants with prostate cancer. N Engl J Med 358:910–919
Zijenah LS, Hartogensis WE, Katzenstein DA, Tobaiwa O, Mutswangwa J, Mason PR, Louie LG (2002) Association of high HIV-1 RNA levels and homozygosity at HLA class II DRB1 in adults coinfected with Mycobacterium tuberculosis in Harare, Zimbabwe. Hum Immunol 63:1026–1032
Zimmerman PA, Fitness J, Moulds JM, McNamara DT, Kasehagen LJ, Rowe JA, Hill AV (2003) CR1 Knops blood group alleles are not associated with severe malaria in the Gambia. Genes Immun 4:368–373
Zouali H, Hani EH, Philippi A, Vionnet N, Beckmann JS, Demenais F, Froguel P (1997) A susceptibility locus for early-onset non-insulin dependent (type 2) diabetes mellitus maps to chromosome 20q, proximal to the phosphoenolpyruvate carboxykinase gene. Hum Mol Genet 6:1401–1408
Acknowledgments
We thank Drs. Philip Hill, Martin Holland, Gerard Morris, Maarten Schim Van Der Loeff and Christian Wejse for critical reading of the manuscript and Profs. Phillip Tobias and Ian Tattersall for advice. We acknowledge the important role that the African Society of Human Genetics (http://www.AfSHG.org) played in bringing together the group of scientists that contributed to this manuscript. The high sense of purpose displayed during the development of this review is a testament to the wonderful atmosphere created by the AfSHG, from its small inaugural meeting (Accra, Ghana 2003) to the most recent and much larger meeting held in Cairo in November 2007, to facilitate discussions of genetics, genomics and health within the larger context of the cultural experiences of the peoples of Africa.
Author information
Authors and Affiliations
Corresponding authors
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00439-008-0534-4
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sirugo, G., Hennig, B.J., Adeyemo, A.A. et al. Genetic studies of African populations: an overview on disease susceptibility and response to vaccines and therapeutics. Hum Genet 123, 557–598 (2008). https://doi.org/10.1007/s00439-008-0511-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-008-0511-y