
Abstract
Malignant rhabdoid tumors are highly aggressive neoplasms found primarily in infants and young children. The majority of rhabdoid tumors arise as a result of homozygous inactivating deletions or mutations of the INI1 gene located in chromosome band 22q11.2. Germline mutations of INI1 predispose to the development of rhabdoid tumors of the brain, kidney and extra-renal tissues, consistent with its function as a tumor suppressor gene. We now describe five patients with germline deletions in chromosome band 22q11.2 that included the INI1 gene locus, leading to the development of rhabdoid tumors. Two patients had phenotypic findings that were suggestive but not diagnostic for DiGeorge/Velocardiofacial syndrome (DGS/VCFS). The other three infants had highly aggressive disease with multiple tumors at the time of presentation. The extent of the deletions was determined by fluorescence in situ hybridization and high-density oligonucleotide based single nucleotide polymorphism arrays. The deletions in the two patients with features of DGS/VCFS were distal to the region typically deleted in patients with this genetic disorder. The three infants with multiple primary tumors had smaller but overlapping deletions, primarily involving INI1. The data suggest that the mechanisms underlying the deletions in these patients may be similar to those that lead to DGS/VCFS, as they also appear to be mediated by related, low copy repeats (LCRs) in 22q11.2. These are the first reported cases in which an association has been established between recurrent, interstitial deletions mediated by LCRs in 22q11.2 and a predisposition to cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Malignant rhabdoid tumors (MRT) are rare, highly aggressive neoplasms found most commonly in infants and young children. Although they may present in any location in the body, they are predominantly found in the kidney and central nervous system. Rhabdoid tumors comprise approximately 1–2% of infant brain and renal tumors. Patients may present with apparently sporadic tumors in one anatomic site, or with multiple primary tumors arising in the brain, kidney and/or soft tissues. Isolated cases of adults with rhabdoid tumors have also recently been reported (Raisanen et al. 2005).
Initial cytogenetic studies suggested an association between rhabdoid tumors and alterations of chromosome 22 (Biegel et al. 1990). More recently, the INI1/hSNF5/SMARCB1/BAF47 [MIM 601607] gene has been implicated in the development of MRT (Versteege et al. 1998). The INI1 gene is located in chromosome band 22q11.2. Mutations and deletions of INI1 have been identified in renal and extra-renal rhabdoid tumors as well as central nervous system atypical teratoid/rhabdoid tumors (AT/RT) (Biegel et al. 1999, 2002; Versteege et al. 1998). The INI1 gene codes for one of at least ten subunits of the SWI/SNF complex, an ATP dependent chromatin-remodeling complex. INI1 is an invariant component of all SWI/SNF complexes, and is thus expressed in all normal cells at all stages of development. Studies in model organisms have shown that at least one copy of INI1 is required for normal development. The SWI/SNF complex appears to regulate transcriptional activity, resulting in both repression and activation of a wide variety of target genes (Biggar and Crabtree 1999; Muchardt and Yaniv 1999). The function of INI1 and the SWI/SNF complex and its role in the development of rhabdoid tumors has recently been reviewed (Biegel 2006; Muchardt and Yaniv 1999).
Since the initial reports implicating somatic alterations (mutations or deletions) of INI1 in the development of rhabdoid tumors, more than 30 individuals with germline alterations of INI1 and rhabdoid tumors of the brain, kidney and soft tissues have been reported (Biegel 2006, 1999, 2002; Bourdeaut et al. 2007; Janson et al. 2005; Kusafuka et al. 2004; Sevenet et al. 1999; Taylor et al. 2000; Wieser et al. 2005). The incidence of rhabdoid tumors due to germline mutations is estimated to be between 15 and 30% (Biegel 2006). As with somatically acquired mutations, the vast majority of these alterations are point or frameshift mutations that result in the introduction of a novel stop codon. To date, only three patients with germline deletions of INI1 have been described (Bourdeaut et al. 2007; Kusafuka et al. 2004; Wieser et al. 2005). Families have also been described with multiple affected members, but these reports are rare, likely due to a failure to obtain sustained cures in patients with this disease. The penetrance of germline INI1 mutations is not yet known, as we are aware of only two unaffected carriers of germline INI1 mutations (Janson et al. 2005; Taylor et al. 2000).
DiGeorge/Velocardiofacial syndrome (DGS/VCFS) is a common microdeletion syndrome with an estimated incidence of approximately 1 in 4,000 live births. Patients with DGS/VCFS present with a complex of structural and developmental anomalies that may include congenital cardiac defects; craniofacial abnormalities, such as ear anomalies and cleft palate; hypoplasia or absence of the thymus and parathyroid glands; and neuropsychiatric disorders. Ninety percent of patients with DGS/VCFS have a 3 Mb deletion of chromosome band 22q11.2, which is proximal to the BCR and INI1 genes. DGS/VCFS associated deletions appear to be mediated by low copy repeat (LCR) sequences or segmental duplications (SDs) that map to this region of chromosome 22 (Shaikh et al. 2000). LCRs represent a class of low copy repetitive DNA elements that have evolved by duplication of large segments of genomic DNA ranging in size from 10 to 500 kb (Bailey et al. 2002; Lander et al. 2001). The duplicated segments share >95% of their nucleotide sequence identity with each other, which can lead to aberrant recombination due to misalignment between non-allelic sequences on homologous chromosomes (Shaikh et al. 2000; Shaw and Lupski 2004). There are a total of eight known LCRs within 22q11. Despite the high incidence of deletions in 22q11.2, there are only isolated case reports of malignancies in patients with DGS/VCFS, including renal cell carcinoma, hepatoblastoma, neuroblastoma, and lymphoma (McDonald-McGinn et al. 2006). Only one case of a renal rhabdoid tumor has been described in a patient with features of DGS/VCFS and a complex chromosomal rearrangement of 22q11.2 (Wieser et al. 2005).
Oligonucleotide-based microarrays are slowly emerging as the platform of choice for genome-wide analysis due to their high-throughput and high-resolution (Bignell et al. 2004; Friedman et al. 2006; Janne et al. 2004; Ming et al. 2006; Zhao et al. 2004). Further, single nucleotide polymorphism (SNP) based oligonucleotide microarrays allow for the detection of copy number neutral loss of heterozygosity (LOH) events that are prevalent in cancers (Langdon et al. 2006; Maris et al. 2005). In the present study, we utilized high-density SNP oligonucleotide arrays to determine the extent of 22q11.2 deletions involving INI1 in five patients with rhabdoid tumors. As most array platforms avoid LCR regions when designing probes due to a lack of localizing specificity, we utilized fluorescence in situ hybridization (FISH) as well to further characterize the deletions. Two of the patients had initial genetic evaluations due to phenotypic features seen in DGS/VCFS. The remaining three infants had two or more primary tumors, consistent with a genetic predisposition to malignancy. Such approaches provide a rapid diagnostic tool for patient management. Furthermore, the high density of probes allow for a more precise localization of deletion breakpoints which may lead to a better understanding of the mechanisms responsible for the deletions in patients with congenital disorders and malignancies.
Materials and methods
Tumor tissue, cell lines, and peripheral blood samples were obtained from the five patients for INI1 analysis according to procedures approved by the Institutional Review Board at The Children’s Hospital of Philadelphia. In each case the parents signed consent forms for genetic testing. In two cases parental blood samples were also obtained to rule out an inherited germline deletion.
Fluorescence in situ hybridization
Touch imprints from frozen tissue, formalin fixed tissue sections, or fixed cell pellets from blood or established cell lines were analyzed by FISH. Probe sets were selected based on location relative to LCR regions. The BCR/ABL and TUPLE1 probe sets were purchased from Vysis (Abbott Laboratories, Downer’s Grove, IL). The remaining cosmid (C2C9, 61E11, 45C9, 118D7, 120E12) or BAC clones (20P18 and 297M14) were labeled by nick translation with ChromoTide® AlexaFluor 594-dUTP or ChromoTide®fluorescein-12-dUTP (Molecular Probes, Eugene, OR). Probes for the Ewing’s sarcoma (EWS) region in 22q12 (120E12) were used simultaneously with the probes for 22q11.2 to assess copy number and as an internal control. The probes were applied to slides of the tumor cells or lymphocytes and co-denatured at 75°C on an Isotemp 125D heat block (Fisher Scientific, Pittsburgh, PA). Slides were incubated at 37°C overnight in a moist slide moat (Boekel Scientific, Feasterville, PA). They were then washed in a 0.4X SSC solution at 73°C for 2 min, followed by a 1 min wash in 2X SSC/0.1% NP-40 and counterstained with DAPI (Sigma, St Louis, MO). Fluorescent signals from 100 to 200 cells were evaluated at 100X with a Nikon Eclipse E800 fluorescence microscope equipped with the proper filter sets. An Applied Imaging System (Santa Clara, CA) was used to record images of representative cells.
Mutation analysis
DNA was extracted from tumor tissue, peripheral blood lymphocytes or lymphoblastoid cell lines using a Puregene kit (Gentra Systems, Minneapolis, MN). Oligonucleotide primers for exons 1–9 of the INI1 gene were designed from the intron/exon boundary sequences (GenBank accession nos. AP000349-350) for PCR. PCR products for individual exons were analyzed by the heteroduplex method and/or direct sequencing as previously described (Biegel et al. 2002). Sequencing of the PCR products was performed by the nucleic acid/protein core facility of the Children’s Hospital of Philadelphia.
High-density SNP oligonucleotide array analysis
The oligonucleotide array experiments were performed using the Affymetrix GeneChip 50 K Xba and 250 K Nsp chips as previously described (Ming et al. 2006). About 250 ng of genomic DNA from the tumor or blood samples (as indicated) were processed and labeled using reagents and protocols supplied by Affymetrix. After hybridization, the microarrays were processed in the Affymetrix GeneChip Fluidics Station 450 and the resultant images were analyzed with the Affymetrix GeneChip® Genotyping Analysis Software (GTYPE). The SNP call rate for all experiments was >98%. The median call rate (MCR) for all experiments was >95%. For the 50 K Xba arrays, the GTYPE output file (.CHP) was then analyzed using the Affymetrix Chromosome Copy Number Analysis tool (CNAT 2.0). Deletions and LOH were identified as previously described (Ming et al. 2006). The 250 K Nsp arrays were analyzed using the Copy Number Analyzer for GeneChip (CNAG) version 2.0, a publicly available software package which allows for the detection of copy number alteration using the signal intensities of the probes (http://www.genome.umin.jp) (Nannya et al. 2005).
In order to maintain uniformity in analysis, the 50 K Xba arrays were also re-analyzed with CNAG. The controls used in the CNAG analysis included samples obtained from Affymetrix which had previously been genotyped using the GeneChip Mapping 100 and 500 K SNP arrays (http://www.affymetrix.com/support/technical/sample_data/copy_number_data.affx). The output from CNAG includes a graphical view of the log2 ratios for each chromosome as well as raw output which contains information regarding the SNP IDs, chromosomal location, log2 ratio of the signal intensity and LOH scores. The program outputs suspected regions of copy number variation and LOH. These regions were then checked manually by review of the raw data to ensure that the mean log2 ratio for the region met criteria for loss or gain (defined as −0.3 for loss and 0.3 for gain). Regions that did not meet these criteria were not included.
The proximal breakpoints were considered to be between the last non-deleted and first deleted SNP while the distal breakpoints were considered to be between the last deleted and first non-deleted SNP. These coordinates were then compared to the coordinates of the LCR regions, given in Supplemental Table 1 (http://www.genome.ucsc.edu; Shaikh et al. 2000, 2007).
Results
Clinical findings
Patient 1
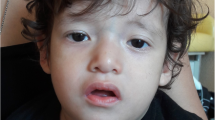
A 3-year-old boy was seen in Genetics clinic due to pervasive developmental delay, submucous cleft palate, and sensorineural hearing loss. Clinical findings are summarized in Table 1. He was noted to have gastroesophageal reflux in the newborn period. His gross motor milestones were normal although his fine motor skills were delayed. He had significant language difficulties. His growth parameters were normal. He had upslanting palpebral fissures, small epicanthal folds bilaterally, and simple ears. There was mild facial asymmetry, with the right ear approximately one-half centimeter smaller than the left ear (Fig. 1a). He had small joint hyper-extensibility and mild tapering to his fingers. Genetic analysis performed at that time did not reveal an underlying diagnosis, as his chromosome analysis and FISH for a probe (TUPLE1) in the DGS/VCFS region were normal.

a Patient 1 at 4 years of age. Note the upslanting palpebral fissures, mild facial asymmetry, and simple ears. Hearing aids are present due to his sensorineural hearing loss. b Patient 2 at 1 year of age. Note the upslanting palpebral fissures similar to patient 1. She also has a down-turned mouth and flat midface, but normal ears
At 5 years of age he presented with weight loss and hematuria. He was diagnosed with a left renal rhabdoid tumor, which was considered metastatic to the right ventricle of the heart and lungs. During these investigations a small muscular ventricular septal defect was found. The child was treated with a left nephrectomy, excision of the cardiac lesion and radiation to the left renal area and lungs. He was treated with a number of courses of chemotherapy, but died of complications due to the lung lesions 15 months following diagnosis.
Patient 2
A female infant was born prematurely at 34 weeks with a V-shaped cleft palate and atrial and ventricular septal defects that subsequently closed. She remained hospitalized for 2 weeks after birth. Her first year of life was complicated by gastroesophageal reflux and difficulty feeding, secondary to her cleft palate. At 12 months, she was hospitalized for a history of weight loss and fussiness. At that time she was noted to have multiple dysmorphic features including short upslanting palpebral fissures, Brushfield spots, malar flatness, mild fifth finger clinodactyly, as well as the V-shaped cleft of her hard and soft palate (Fig. 1b). She acutely developed flaccid diplegia. MRI of her spine revealed a minimally enhancing mass, encasing the spinal cord and nerve roots from T12 to the sacrum. She was taken to the operating room for subtotal resection of the mass. Pathology revealed a malignant rhabdoid tumor. She was treated with radiation and chemotherapy, including etoposide, temozolaminde, cytarabine, methotrexate, dactinomycin and doxorubicin, but succumbed to her disease within a year of diagnosis.
Patient 3
A 5-week-old boy presented to the emergency room with a 2-week history of respiratory distress and a 1-week history of an enlarging axillary mass. On physical examination, he was noted to have difficulty breathing and a palpable four to five centimeter mass in the right axilla. Computed tomography (CT) scan of the chest and abdomen revealed a mediastinal mass with tracheal compression, at least two liver masses with calcification, and a small mass in one kidney. Head CT was within normal limits. He was intubated for respiratory support. A biopsy of the axillary mass was performed. Pathology revealed a monomorphic tumor with features of malignant rhabdoid tumor, confirmed by loss of expression of INI1 by immunohistochemistry. He died shortly after diagnosis.
Patient 4
An 8-month-old boy presented to the hospital with focal seizures and an abdominal mass. Brain MRI revealed a partially cystic and heterogeneously enhancing mass within the right lateral ventricle with associated mass effect and hydrocephalus. Abdominal CT revealed a large right renal mass measuring nine by twelve centimeters. He underwent resection of both the brain and renal masses. Pathology was consistent with CNS AT/RT and malignant rhabdoid tumor of the kidney, and confirmed by INI1 immunohistochemistry. He was treated with chemotherapy using ifosfamide, carboplatin, and etoposide. Three months following diagnosis, he was noted to have pain and swelling of his right elbow. MRI scan of the elbow revealed a soft tissue mass medial and posterior to the right ulna. Biopsy of the mass was consistent with extrarenal rhabdoid tumor. His treatment was changed to an alternative chemotherapy regimen with a plan to administer focal radiotherapy to the right elbow, right renal bed, and tumor cavity in the right cerebral hemisphere. He then developed bilateral pulmonary nodules, consistent with either a fourth primary tumor or metastatic disease. Despite salvage therapy, he subsequently died of progressive disease.
Patient 5
A 6-month-old boy presented to the emergency department with a 2-week history of vomiting with worsening lethargy over the previous day. On examination, he was noted to be macrocephalic with a bulging fontanelle. Head CT revealed a large posterior fossa mass compressing the fourth ventricle with associated hydrocephalus and a separate left lateral ventricle mass. MRI revealed an additional pineal region mass obstructing the cerebral aqueduct. Liver and bilateral renal masses were also identified. A right renal biopsy and third ventriculostomy were performed. The renal mass was diagnosed as a rhabdoid tumor. Following discussion of his prognosis, his parents elected for palliative treatment and he expired approximately 2 weeks later. An autopsy limited to brain tissue was performed, and normal and tumor tissue samples were collected for study.
Deletion and mutation analysis
Patient 1
A diagrammatic representation of the deletion intervals and the FISH results for the five patients are shown in Fig. 3. For patient 1, FISH analysis of a normal tissue cell line established from the kidney revealed a single INI1 (118D7) probe signal with retention of two copies of the control EWS probe, consistent with a germline deletion. Sequence analysis of the tumor DNA revealed an exon 6 mutation with a 2 base pair TG deletion of bases 667–668 (codon 223) or 669–670 (codon 223–224). The mutation is predicted to result in a frameshift and introduce a novel stop codon at position 223 (Fig. 2a). The mutation was not present in the blood or cell line DNA, suggesting an acquired mutation.

a Exon 6 mutation in patient 1 with a 2 base pair TG deletion of bases 667–668 (codon 223) or 669–670 (codon 223–224). Sequence shown is the reverse sequence. The mutation is predicted to result in a frameshift and introduce a novel stop codon at position 223. b Exon 5 mutation in patient 2 with a duplication of 8 bases (either 605–612 or 607–614). This mutation is predicted to result in a frameshift and introduce a novel stop codon at position 211
High-density oligonucleotide array analysis of DNA from the blood using the 50 K Xba SNP chip confirmed the 22q11.2 deletion. Based on the LCR coordinates (Supplemental Table 1), the proximal breakpoint localized to LCR region D, which is the distal end of the typical DGS/VCFS deletion region. The distal breakpoint was in LCR G (Supplemental Table 2). This deletion is approximately 2.7 Mb in size. The oligonucleotide analysis of the blood DNA also revealed a duplication of 20p12.1. There are no known genes or polymorphisms in this region, thus the clinical significance is not known. Additional FISH studies of the cell line confirmed the array results, demonstrating loss of one copy of both 45C9 and 61E11 with retention of both copies of C2C9 and 20P18 (Fig. 3).

Chromosome 22q11.2 map with the location of the LCR regions, cosmid and BAC clones used for FISH and germline deletions indicated. Solid black lines indicate the deleted regions in each blood sample. The typical DGS/CFS deletion is between LCRs A and D
Array analysis was attempted on the tumor DNA, but there were high levels of background noise complicating the analysis of the genome. Nevertheless, the alterations found in the blood were similarly present in the tumor sample.
Patient 2
FISH was initially performed on a blood sample using both a TUPLE1 probe and a BCR probe. Although there was no TUPLE1 deletion, a single BCR probe signal was suggestive of a constitutional 22q11.2 deletion. FISH analysis of the tumor sample revealed loss of one copy of both the INI1 (118D7) and BCR probes. Molecular analysis of the tumor DNA revealed a duplication of 8 bases (either 605–612 or 607–614) in exon 5 of the INI1 gene. This mutation is predicted to result in a frameshift and introduce a novel stop codon at position 211 (Fig. 2b). This mutation was not present in her blood.
Analysis of blood and tumor DNA using the 50 K Xba chips demonstrated a 2.7 Mb germline deletion in chromosome 22q11.2 with breakpoints in LCRs D and G, similar to patient 1 (Supplemental Table 2). In addition to the abnormalities on chromosome 22, the SNP array analysis revealed regions of LOH present in both the tumor and blood in 2p16.3, 3p12.1–12.2, and 3q21.3–22.1. In addition, both a duplication of 6p21.32 and a more proximal deletion in 22q11.21–11.23 were present in both the tumor and blood samples. These abnormalities were in regions with previously described polymorphisms and are thus presumed to be incidental findings (Conrad et al. 2006; Hinds et al. 2006; McCarroll et al. 2006; Sebat et al. 2004; Sharp et al. 2005; Tuzun et al. 2005). Duplications were also found in the tumor DNA in 1p31.1 and 3q25.2. In both instances, although the changes did not meet criteria to be considered significant in the blood sample, there were similar changes present. The only tumor specific change was a deletion in 4q34.3. There are no currently known genes in this deleted region.
As shown in Fig. 3, FISH analysis was consistent with the array results, which demonstrated deletions of 45C9 and 61E11 and retention of both copies of C2C9 and 20P18.
Patient 3
Initial FISH analysis of the tumor from the right axillary mass revealed two populations of cells, with 84% of cells demonstrating homozygous loss of INI1. The remaining 16% of cells demonstrated hybridization for a single copy of INI1, suggesting a possible constitutional deletion. FISH of the blood sample confirmed the germline INI1 deletion with 100% of cells demonstrating hybridization for a single copy of INI1 (Fig. 4).

a Interphase FISH analysis from the tumor of patient 3 demonstrating two populations of cells. One cell has loss of one copy of INI1 (red) and the other cell has loss of both copies of INI1. Both cells demonstrate two copies of the EWS control probe. These two cells likely represent a normal tissue cell with the germline deletion and a tumor cell with loss of INI1. b Interphase FISH analysis from the blood of the same patient demonstrates loss of one copy of INI1 with retention of both copies of EWS, consistent with a germline deletion
High-density oligonucleotide array analysis of the tumor DNA using the 50 K Xba chip revealed a deletion of 22q11.2 in which the region that included INI1 was homozygously deleted. The breakpoints for the tumor deletion were localized to LCR region E and proximal to LCR H with an approximate size of 1.7 Mb. Analysis of the blood DNA on the 250 K Nsp array revealed a smaller 1.5 Mb deletion of chromosome 22 including the INI1 locus. In this instance the breakpoints were localized between LCR regions E and F proximally (proximal to BCR) and between LCR regions G and H distally (proximal to 20P18).
Further FISH analysis of his blood to confirm the array results revealed two copies of 61E11 and 20P18, representing the limits of the germline deletion (Fig. 3). A population of tumor cells (36/100) demonstrated loss of one copy of 20P18 with retention of EWS, suggesting a larger deletion in the tumor. Based on the retention of two copies of 297M14 in the tumor DNA by FISH and the deleted SNP coordinates, the distal extent of the larger tumor deletion was confirmed to be proximal to LCR H.
Oligonucleotide array analysis also revealed multiple abnormalities in addition to chromosome 22. In the blood, deletions were present in both 14q11.2 and 15q11.2, which are regions with previously described polymorphisms (Conrad et al. 2006; Sebat et al. 2004; Tuzun et al. 2005). These were not found in the tumor sample likely due to differences in SNP density coverage for these regions between the 50 and 250 K chips. There were additional regions of LOH in the tumor, including 1p22.2–22.3, 2p11.2–p12, 6p24.1, and 9q21.1. The changes for chromosomes 2 and 9 were seen to some extent in the blood, but were not confirmed statistically, while the changes on chromosomes 1 and 6 were not seen in the blood and are thus felt to be tumor specific.
Patient 4
FISH was performed on a peripheral blood sample and samples of both the renal tumor and brain tumor. Analysis of the blood revealed loss of a single copy of INI1, whereas both copies of INI1 in the tumor samples were homozygously deleted. Although both copies of EWS were retained in the blood and brain tumor, the renal tumor demonstrated only a single signal for the EWS locus, suggestive of monosomy 22. These results confirmed that the brain and renal tumors were both primary malignancies that arose independently of one another. Consistent with the FISH results, PCR analysis revealed homozygous deletions of the INI1 gene in both tumors. The germline deletion was not found in his parents’ blood and was thus felt to be a de novo deletion.
Analysis of the blood DNA with the 250 K Nsp chip revealed a 0.9 Mb deletion with breakpoints within LCR region F and between LCR regions G and H in a known recombination hotspot. These results were confirmed by FISH, which revealed one copy of 20P18 but two copies of 297M14. Both BCR and 61E11 were present in two copies in the blood sample (Fig. 3).
Analysis of the oligonucleotide arrays did not reveal any other areas of the genome with significant changes from normal controls. Furthermore, as the tumors were fixed and embedded in paraffin, high-density oligonucleotide analysis of the tumor DNA was not performed.
Patient 5
FISH and high-density oligonucleotide analysis were performed on both the normal brain and brain tumor tissue from the autopsy. FISH analysis of the tumor DNA revealed homozygous loss of INI1. Analysis of the normal brain tissue revealed a deletion of one copy of INI1 with retention of both BCR and 20P18 (Fig. 3). These results were subsequently confirmed on an established lymphoblast cell line.
The 250 K SNP array demonstrated a germline deletion approximately 1.0 Mb in size in the normal brain with the same 1.0 Mb deletion (homozygous deletion) in the tumor tissue (Fig. 5). The breakpoints were found to be in identical locations for both the normal brain and tumor tissue, within LCR region F and between LCR regions G and H, respectively. Interestingly, analysis of the tumor DNA revealed loss of heterozygosity for the entire chromosome 22 suggestive of loss and duplication of the deleted homologue.

CNAG output for chromosome 22 of patient 5. Plot analysis demonstrates a decrease in the log2R ratio for both the normal brain and tumor samples in the same region of chromosome 22 (including INI1). The magnitude of the decrease in log2R ratio for the tumor DNA is about twice the magnitude of the decrease found in the normal brain DNA, consistent with loss of one allele in the blood and loss of both alleles in the tumor
In addition to chromosome 22, the oligonucleotide array highlighted a region of LOH for 3q13.11–13.13 and a deletion in 14q11.2. Both regions were present in the normal brain and the tumor specimens and have been previously described as regions of copy number variation (Conrad et al. 2006; McCarroll et al. 2006; Sebat et al. 2004; Tuzun et al. 2005).
Discussion
We report here five patients with germline 22q11.2 deletions that include the INI1 gene, which predisposed these children to development of MRT. The extent of the deletions and the nature of the second inactivating event, as determined by SNP array and mutation analysis, appear to be associated with the developmental or phenotypic defects observed in these children.
Several genomic disorders, including DGS/VCFS and cat eye syndrome (CES), have been associated with rearrangements of 22q11. Of the eight previously reported LCRs within 22q11.2, the four proximal LCRs (A to D) have been extensively characterized, due to their involvement in recurrent rearrangements that lead to DGS/VCFS (Edelmann et al. 1999) and CES (McTaggart et al. 1998). LCR A and LCR D mediate the common 3 Mb deletion of DGS/VCFS and are the largest and most complex in their organization (Shaikh et al. 2000). The four distal LCRs, which are referred to as LCRs E to H, are smaller in size and complexity. This distal cluster of LCRs has rarely been associated with deletions of distal 22q11.2. Nevertheless, recent evidence suggests that these distal LCRs may be involved in recurrent, constitutional rearrangements of 22q11.2 associated with novel genomic disorders (Shaikh et al. 2007).
The first two children in the present study were evaluated by a clinical geneticist prior to the diagnosis of rhabdoid tumor. Both children had a cleft palate, cardiac defects (ASD, VSD), and dysmorphic features, as well as developmental delay in patient 1. In both patients, the standard diagnostic FISH test for DGS/VCFS, employing TUPLE1 was normal. In retrospect, these results can be explained by the fact that this gene maps to the region between LCR A and B, which is proximal to their deletions. The deletions in both patients were mediated by LCRs D and G, which are not typically associated with DGS/VCFS. The finding of conotruncal defects and palatal anomalies are similar, however, to a small number of patients with a 22q11.2 deletion syndrome and atypical LCR D to E and LCR D to F deletions (Rauch et al. 1999; Saitta et al. 1999). The variability of phenotypic features in DGS/VCFS patients with different overlapping and non-overlapping deletions of 22q11.2 have made it difficult to correlate the extent of deletion with specific phenotypic features. Determination of which patients with constitutional 22q11.2 rearrangements should be screened for INI1 deletions is therefore unclear. Once high density SNP array analysis is available as a clinical diagnostic assay, it may be possible to make predictions of phenotype based on the extent of the 22q11.2 deletions. Clinical guidelines for screening patients for INI1 alterations could then be established.
The other three patients in this report (patients 3–5) did not have any obvious phenotypic findings consistent with an underlying genomic disorder such as DGS/VCFS, although all three may have been too young to potentially appreciate a developmental delay or other clinical features of this syndrome. Of interest, each of these children had extremely aggressive disease, with multiple primary tumors. Patients 1 and 2 had more extensive deletions than patients 3–5, yet they developed their rhabdoid tumors at a relatively later age. Patient 1, in particular, presented at 5 years of age, which is past the peak incidence of this disease. These findings suggest that the second, acquired event may also influence disease presentation or aggressiveness in children with germline deletions of INI1. Patient 1 had a two base pair deletion, whereas patient 2 had an 8 base pair duplication, which resulted in a frameshift mutation. In contrast, patients 3, 4 and 5, respectively, had loss of the wild-type allele as result of a second interstitial deletion, monosomy 22, or a loss and duplication of the deleted homologue leading to LOH for all of chromosome 22.
Several genetic disorders are reported to be associated with chromosomal deletions or duplications mediated by LCRs. Isochromosomes, specifically for chromosome 17, may also arise as a result of mis-alignment mediated by LCRs near the centromere (Barbouti et al. 2004; Mendrzyk et al. 2006). In contrast to a previous report suggesting that homozygous deletions in cancer are negatively associated with LCRs (Cox et al. 2005), the chromosome 22 and INI1 deletions described in this report appear to be due to LCR mediated rearrangements, and, as such, are the first interstitial chromosomal deletions that have been implicated in cancer development. Low copy repeats have also recently been identified as potential breakpoints in the generation of 11q13.3 amplicons, which have been implicated in a variety of carcinomas (Gibcus et al. 2007). To further elucidate the role of LCR mediated rearrangements in cancer, studies are in progress utilizing high density SNP based oligonucleotide analysis of primary tumors with acquired deletions to determine if somatic deletions in sporadic rhabdoid tumors are also mediated by LCRs in chromosome 22. These studies should also allow us to determine whether the extent or location of the deletions or mutations predict the biological course of disease in patients who develop rhabdoid tumors.
We have described five patients with germline deletions of chromosome 22 that include the INI1 gene. Similar to previously published cases of patients with rhabdoid tumors and INI1 germline deletions (Kusafuka et al. 2004; Wieser et al. 2005), these patients had varying initial presentations. Two of our patients had features of DGS/VCFS, whereas the other three patients had multiple primary and metastatic rhabdoid tumors. Although the second event leading to INI1 inactivation appears to differ among these patients, at least some of these events may be influenced by recombination events at LCR regions, suggesting a possible common underlying mechanism for disease in these patients. Furthermore, it is clear that the use of the SNP arrays provides a sensitive means of assessing both copy number aberrations and LOH to help elucidate underlying mechanisms of disease.
References
Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE (2002) Recent segmental duplications in the human genome. Science 297:1003–1007
Barbouti A, Stankiewicz P, Nusbaum C, Cuomo C, Cook A, Hoglund M, Johansson B, Hagemeijer A, Park SS, Mitelman F, Lupski JR, Fioretos T (2004) The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am J Hum Genet 74:1–10
Biegel JA (1999) Cytogenetics and molecular genetics of childhood brain tumors. Neuro-oncology 1:139–151
Biegel JA (2006) Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 20:E11
Biegel JA, Rorke LB, Packer RJ, Emanuel BS (1990) Monosomy 22 in rhabdoid or atypical tumors of the brain. J Neurosurg 73:710–714
Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59:74–79
Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8:3461–3467
Biggar SR, Crabtree GR (1999) Continuous and widespread roles for the Swi–Snf complex in transcription. EMBO J 18:2254–2264
Bignell GR, Huang J, Greshock J, Watt S, Butler A, West S, Grigorova M, Jones KW, Wei W, Stratton MR, Futreal PA, Weber B, Shapero MH, Wooster R (2004) High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res 14:287–295
Bourdeaut F, Freneaux P, Thuille B, Lellouch-Tubiana A, Nicolas A, Couturier J, Pierron G, Sainte-Rose C, Bergeron C, Bouvier R, Rialland X, Laurence V, Michon J, Sastre-Garau X, Delattre O (2007) hSNF5/INI1-deficient tumours and rhabdoid tumours are convergent but not fully overlapping entities. J Pathol 211:323–330
Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK (2006) A high-resolution survey of deletion polymorphism in the human genome. Nat Genet 38:75–81
Cox C, Bignell G, Greenman C, Stabenau A, Warren W, Stephens P, Davies H, Watt S, Teague J, Edkins S, Birney E, Easton DF, Wooster R, Futreal PA, Stratton MR (2005) A survey of homozygous deletions in human cancer genomes. Proc Natl Acad Sci 102:4542–4547
Edelmann L, Pandita RK, Morrow BE (1999) Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet 64:1076–1086
Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Asano J, Bailey DK, Barber S, Birch P, Brown-John M, Cao M, Chan S, Charest DL, Farnoud N, Fernandes N, Flibotte S, Go A, Gibson WT, Holt RA, Jones SJ, Kennedy GC, Krzywinski M, Langlois S, Li HI, McGillivray BC, Nayar T, Pugh TJ, Rajcan-Separovic E, Schein JE, Schnerch A, Siddiqui A, Van Allen MI, Wilson G, Yong SL, Zahir F, Eydoux P, Marra MA (2006) Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet 79:500–513
Gibcus JH, Kok K, Menkema L, Hermsen MA, Mastik M, Kluin PM, van der Wal JE, Schuuring E (2007) High-resolution mapping identifies a commonly amplified 11q13.3 region containing multiple genes flanked by segmental duplications. Hum Genet 121:187–201
Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA (2006) Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet 38:82–85
Janne PA, Li C, Zhao X, Girard L, Chen TH, Minna J, Christiani DC, Johnson BE, Meyerson M (2004) High-resolution single-nucleotide polymorphism array and clustering analysis of loss of heterozygosity in human lung cancer cell lines. Oncogene 23:2716–2726
Janson K, Nedzi LA, David O, Schorin M, Walsh JW, Bhattacharjee M, Pridjian G, Tan L, Judkins AR, Biegel JA (2005) Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer 47:279–284
Kusafuka T, Miao J, Yoneda A, Kuroda S, Fukuzawa M (2004) Novel germ-line deletion of SNF5/INI1/SMARCB1 gene in neonate presenting with congenital malignant rhabdoid tumor of kidney and brain primitive neuroectodermal tumor. Genes Chromosomes Cancer 40:133–139
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Langdon JA, Lamont JM, Scott DK, Dyer S, Prebble E, Bown N, Grundy RG, Ellison DW, Clifford SC (2006) Combined genome-wide allelotyping and copy number analysis identify frequent genetic losses without copy number reduction in medulloblastoma. Genes Chromosomes Cancer 45:47–60
Maris JM, Hii G, Gelfand CA, Varde S, White PS, Rappaport E, Surrey S, Fortina P (2005) Region-specific detection of neuroblastoma loss of heterozygosity at multiple loci simultaneously using a SNP-based tag-array platform. Genome Res 15:1168–1176
McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, Altshuler DM (2006) Common deletion polymorphisms in the human genome. Nat Genet 38:86–92
McDonald-McGinn DM, Reilly A, Wallgren-Pettersson C, Hoyme HE, Yang SP, Adam MP, Zackai EH, Sullivan KE (2006) Malignancy in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Am J Med Genet A 140:906–909
McTaggart KE, Budarf ML, Driscoll DA, Emanuel BS, Ferreira P, McDermid HE (1998) Cat eye syndrome chromosome breakpoint clustering: identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenet Cell Genet 81:222–228
Mendrzyk F, Korshunov A, Toedt G, Schwarz F, Korn B, Joos S, Hochhaus A, Schoch C, Lichter P, Radlwimmer B (2006) Isochromosome breakpoints on 17p in medulloblastoma are flanked by different classes of DNA sequence repeats. Genes Chromosomes Cancer 45:401–410
Ming JE, Geiger E, James AC, Ciprero KL, Nimmakayalu M, Zhang Y, Huang A, Vaddi M, Rappaport E, Zackai EH, Shaikh TH (2006) Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum Mutat 27:467–473
Muchardt C, Yaniv M (1999) The mammalian SWI/SNF complex and the control of cell growth. Sem Cell Dev Biol 10:189–195
Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, Ogawa S (2005) A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 65:6071–6079
Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A (2005) Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol 15:23–28
Rauch A, Pfeiffer RA, Leipold G, Singer H, Tigges M, Hofbeck M (1999) A novel 22q11.2 microdeletion in DiGeorge syndrome. Am J Hum Genet 64:659–666
Saitta SC, McGrath JM, Mensch H, Shaikh TH, Zackai EH, Emanuel BS (1999) A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am J Hum Genet 65:562–566
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M (2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528
Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O (1999) Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65:1342–1348
Shaikh TH, Kurahashi H, Saitta SC, O’Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS (2000) Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 9:489–501
Shaikh TH, O’Connor RJ, Pierpont ME, McGrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC (2007) Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res 17:482–491
Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE (2005) Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77:78–88
Shaw CJ, Lupski JR (2004) Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 13(Spec No 1):R57–R64
Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT (2000) Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet 66:1403–1406
Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, Olson MV, Eichler EE (2005) Fine-scale structural variation of the human genome. Nat Genet 37:727–732
Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206
Wieser R, Fritz B, Ullmann R, Muller I, Galhuber M, Storlazzi CT, Ramaswamy A, Christiansen H, Shimizu N, Rehder H (2005) Novel rearrangement of chromosome band 22q11.2 causing 22q11 microdeletion syndrome-like phenotype and rhabdoid tumor of the kidney. Hum Mutat 26:78–83
Zhao X, Li C, Paez JG, Chin K, Janne PA, Chen TH, Girard L, Minna J, Christiani D, Leo C, Gray JW, Sellers WR, Meyerson M (2004) An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res 64:3060–3071
Acknowledgments
The authors thank Dr. Sulagna Saitta for her helpful and informative discussions. We would also like to acknowledge Dr. Eric Rappaport, Elizabeth Geiger and Madhavi Vaddi for their technical assistance. Supported by grants from the National Institutes of Health CA46274, CA98543 (J.A.B.) and GM64725 (T.H.S.). Eric Jackson receives grant support from the Neurosurgery Research and Education Foundation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Jackson, E.M., Shaikh, T.H., Gururangan, S. et al. High-density single nucleotide polymorphism array analysis in patients with germline deletions of 22q11.2 and malignant rhabdoid tumor. Hum Genet 122, 117–127 (2007). https://doi.org/10.1007/s00439-007-0386-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-007-0386-3