
Abstract
Wilson disease is an autosomal recessive inherited disorder of copper metabolism. The Wilson disease gene codes for a copper transporting P-type ATPase (ATP7B). Molecular genetic analysis reveals at least 300 distinct mutations. While most reported mutations occur in single families, a few are more common. The most common mutation in patients from Central, Eastern, and Northern Europe is the point mutation H1069Q (exon 14). About 50–80% of Wilson disease (WD) patients from these countries carry at least one allele with this mutation with an allele frequency ranging between 30 and 70%. Other common mutations in Central and Eastern Europe are located on exon 8 (2299insC, G710S), exon 15 (3400delC) and exon 13 (R969Q). The allele frequency of these mutations is lower than 10%. In Mediterranean countries there is a wide range of mutations, the frequency of each of them varies considerably from country to country. In Sardinia, a unique deletion in the 5′ UTR (−441/−427 del) is very frequent. In mainland Spain the missense mutation M645R in exon 6 is particularly common. Data from non-European countries are scarce. Most data from Asia are from Far Eastern areas (China, South Korea and Japan) where the R778L missense mutation in exon 8 is found with an allele frequency of 14–49%. In summary, given the constant improvement of analytic tools genetic testing will become an integral part for the diagnosis of WD. Knowledge of the differences in the worldwide distribution of particular mutations will help to design shortcuts for genetic diagnosis of WD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wilson disease (WD) is an autosomal recessive inherited disorder of copper metabolism resulting in pathological accumulation of copper in many organs and tissues. The hallmarks of the disease are the presence of liver disease, neurologic symptoms and Kayser–Fleischer corneal rings. The basic defect in WD is the impaired biliary excretion of copper resulting in the accumulation of copper in various organs including the liver, the cornea and the brain. The consequence of copper accumulation is the development of severe hepatic and neurological disease.
The Wilson disease gene
The WD gene (Petrukhin et al. 1993; Tanzi et al. 1993) codes for a copper-transporting CPx-type ATPase (ATP7B) (Petrukhin et al. 1994). In hepatocytes, ATP7B delivers copper to apoceruloplasmin and mediates the excretion of excess copper into bile. ATP7B contains the following functional domains: six copper binding domains, a transduction domain (amino acid residues 837–864; containing a Thr-Gly-Glu motif) involved in the transduction of the energy of ATP hydrolysis to cation transport, a cation channel and phosphorylation domain (amino acid residues 971–1,035; containing the highly conserved Asp-Lys-Thr-Gly-Thr motif), a nucleotide-binding domain (the N-domain; amino acid residues 1,240–1,291) and eight hydrophobic transmembrane sequences (1–8), in one of which (region 6) is the Cys-Pro-Cys sequence found in all P-type ATPases. The six N-terminal metal-binding sites (MBS) are required for trafficking and are essential for the copper transport function, however it was suggested that the first three N-terminal motifs were not required for copper-dependent intracellular trafficking and could not functionally replace sites 4–6 when placed in the same sequence position (Cater et al. 2004). Structural analysis of the N-terminus has revealed that both secondary and tertiary structural changes take place after the binding of copper (DiDonato et al. 2000). Furthermore, it was demonstrated that copper co-ordination induces the phosphorylation of ATP7B, which coincides with the trafficking of the protein to vesicular compartments (Vanderwerf et al. 2001).
ATP7B mutations in Wilson disease
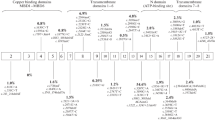
Molecular genetic analysis reveals at least 300 distinct mutations (database maintained at the University of Alberta -http://www.medgen.med.ualberta.ca) include missense and nonsense mutations, deletions and insertions. Some mutations are associated with a severe impairment of copper transport resulting in severe liver disease very early in life; other mutations appear to be less severe with disease appearance in mid adulthood. Several WD mutations are clustered within the nucleotide-binding domain (N-domain), where they are predicted to disrupt ATP binding (see Fig. 1). The mechanism by which the N-domain coordinates ATP is presently unknown. Mutations of the invariant WD protein residues E1064A and H1069Q drastically reduce nucleotide affinities, pointing to the likely role of these residues in nucleotide coordination. In contrast, the R1151H mutant exhibits only a 1.3-fold reduction in affinity for ATP. The C1104F mutation significantly alters protein folding, whereas C1104A does not affect the structure or function of the N-domain (Morgan et al. 2004). These results directly demonstrate the phenotypic diversity of WD mutations within the N-domain.

Schematic presentation of the WD gene showing the site of common mutations
Regional differences in the frequency of various mutations
While most reported mutations occur in single families, a few are more common. Knowledge of the regional distribution of mutations of the WD gene is important to design appropriate screening strategies. Limitations of the available data in certain populations include a selection bias based on how patients are recruited for the study, whether all patients or just the index cases are tested and finally the proportion of patients with WD within a country available for testing. A study of the frequency of mutation is virtually impossible in countries with a highly mixed population like the USA or the United Kingdom.
The most important considerations when assessing the regional distribution of WD mutations are:
Diagnosis of Wilson disease
The diagnosis of WD is usually made on the basis of clinical findings and laboratory abnormalities if two of the following symptoms are present: Kayser–Fleischer rings, typical neurologic symptoms and low serum ceruloplasmin levels (Scheinberg and Sternlieb 1984). In a patient presenting with typical neurologic symptoms and having Kayser–Fleischer rings the diagnosis is straightforward. No further diagnostic procedures are necessary to establish the diagnosis. Kayser–Fleischer rings are rarely absent in neurologically symptomatic patients.
Diagnosis is far more complex in patients presenting with liver diseases (Ferenci 2005b). None of the commonly used parameters alone allows a certain diagnosis of WD. Whereas, serum ceruloplasmin is decreased in most patients with neurologic WD, it may be in the low normal range in upto 45% of patients with hepatic disease. Kayser–Fleischer rings may be absent in upto 50% of patients with Wilsonian liver disease and even in a higher proportion in fulminant WD (Steindl et al. 1997). Urine copper excretion is markedly increased in patients with WD; however, its usefulness in clinical practice is limited. Hepatic copper content is increased in 82% of patients with WD and usually exceeds 250 μg/g dry weight (normal: upto 50). In the absence of other tests suggestive for abnormal copper metabolism, diagnosis of WD cannot be made based on an increased hepatic copper content alone (Ferenci et al. 2005a). Diagnosis requires a combination of a variety of clinical and biochemical tests. A diagnostic scoring system (Table 1) was developed at the eighth international meeting on WD, Leipzig/Germany, 2001 (Ferenci et al. 2003). Thus, if series contain just typical neurological cases frequency distribution may be quite different than in hepatic WD.
Frequency of WD in the population
On a population based approach, the incidence of WD was estimated to be at least 1:30,000–50,000 (Ireland: 17/106 live births, former East-Germany: 29 [Reilly et al. 1993; Bachmann et al. 1991]) with a gene frequency of 1:90 to 1:150. These estimations were mostly based on adolescent or adults presenting with neurologic symptoms. By a mutation based approach Wilson disease frequency in the US was about one in 55,000 births (95% confidence interval: 1:18,000 to 1:700,000) (Olivarez et al. 2001). Consequently, the detection rate in a certain population is quite variable among the various studies. For example, 150 Chinese patients represent far less than 1% of the expected number of cases in that large country, whereas 125 Austrian patients equal to about 55% of all cases (see Table 2).
Database
This review is based on two sources:
-
1.
A medline search was conducted to identify all studies published since the detection of the WD gene in 1993. Only studies done in well-defined and described populations were analyzed. Only data for index cases were used to avoid overrepresentation of a particular mutation. This approach was possible in most studies (which are marked by “a” in Tables 2, 3 and 4)
-
2.
In an ongoing genotype–phenotype study I am collecting clinical data and DNA samples from patients with WD worldwide. Currently this database contains the results from 1,050 patients mostly of European origin (Ferenci et al. 2005b). Mutation analysis in these patients was done by a stepwise procedure. First, a rapid, semi-nested PCR technique was used to detect the H1069Q mutation as described previously (Maier-Dobersberger et al. 1997). Patients not being homozygous for this mutation were further analyzed exon by exon by denaturating HPLC (WAVE mutation detection system model 4000, Transgenomics, Crewe, UK). So far, the analysis was completed for exons 3–20. Exons were amplified with published primers (Thomas et al. 1995). Samples with potential mutations identified by this approach were sequenced by the ABI Prism 310 Genetic Analyzer (Perkin Elmer; Norwalk, CT, USA). The results of the interim analysis are shown in Fig. 2.

Results of mutation analysis in 1,028 patients (887 index cases) of mostly European origin (Ferenci et al. 2005b). Exons 3–20 were examined in all patients, analysis of exons 2 and 21 is ongoing. A total of 134 mutations were identified (known mutations: 87; novel mutations: 47)
Europe
Most of the available European data are derived from Central and Eastern Europe, as well as from the Mediterranean countries. Unfortunately, there is not a single report from France. Given for the size of the country there are very limited data from the United Kingdom with just 42 index patients of British origin (Curtis et al. 1999). In general, the most common mutation is the point mutation H1069Q in exon 14 of the WD gene. Its frequency is highest in Poland and Eastern Germany and decreases to the west and to the south. About 50–80% of WD patients from these countries carry at least one allele with this mutation with an allele frequency ranging between 30 and 70% (see Table 2 and Fig. 3). South of the Alps this mutation becomes infrequent and is totally absent in Sardinia. Other common mutations in Central and Eastern Europe are located on exon 8 (2299insC, G710S), exon 15 (3400delC) and exon 13 (R969Q). The allele frequency of these mutations is substantially less than 10%.

Allele frequency of the H1069Q mutation across Europe (for details and references see Table 2)
In Mediterranean countries there is a wide range of mutations, the frequency of each of them varies considerably from country to country (see Table 3). In Sardinia, a unique deletion in the 5′ UTR (−441/−421 del) is very common (Loudianos et al. 1999). This deletion was not reported from anywhere else. Interestingly, this mutation was observed also in an isolated valley in Costa Rica (F. Hevia, personal communication) reflecting the genetic imprint of a (then) Spanish soldier from Sardinia. In mainland Spain a missense mutation in exon 6 M645R is particularly common (Margarit et al. 2005). This mutation was observed at low frequency in many other European countries. In Turkey, there is no very common mutation. Mutations are frequently located in exons 8 and 13 (Fig. 4).

Proposed algorithm for the diagnosis of WD. Score refers to the WD diagnostic score shown in Table 1
Asia
Most data from Asia are from Far Eastern areas (China, South Korea and Japan). In the Far East (see Table 4), the R778L missense mutation in exon 8 is found with an allele frequency of 14–49%. The data from India are quite divergent reflecting the ethnic diversity of this large country. The most common mutations in patients mostly from North-West India are located on chromosome 13 (Kumar et al. 2005; Gupta et al. 2005), while in patients from Eastern India (Kolkata) these mutations were not found (Gupta et al. 2005). There are no comprehensive reports from Pakistan, Bangladesh, Indonesia, the Philippines or the Arab Countries (except for Saudi Arabia).
Other continents
The study of the frequency WD mutations in the USA, Canada, and Australia is difficult because of the ethnic inhomogeneity of these immigration countries. No studies have been reported focusing on these countries. In a study involving many patients from various countries, data of 118 mostly Caucasian US patients were given. In this cohort, H1069Q was the most common mutation with an allele frequency of 35.2%, which is similar to the frequency of this mutation in most European countries (Shah et al. 1997). Except for Brazil, no information is available from Latin America. In Brazil, surprisingly the most common mutation is 3400delC in exon 15 (Deguti et al. 2004). In a limited number of cases from Chile we found also this mutation. Since this mutation is common in Europe, this finding may only apply for people of European origin. No data are available from Africa.
Mutation analysis for diagnosis of WD
Direct molecular-genetic diagnosis is difficult because of the occurrence of many mutations, each of which is rare. Furthermore, most patients are compound heterozygotes (i.e. carry two different mutations). For the H1069Q mutation we developed a semi-nested PCR technique (Maier-Dobersberger et al. 1997). Screening for mutations if it occurs with a reasonable frequency in a population can be done by denaturing HPLC analysis followed by direct sequencing or by allele-specific probes (Weirich et al. 2002). In patients from Central or Eastern Europe limited mutation analysis of exons 8, 13, 14 and 15 can be carried out within a week. A multiplex PCR for the most frequent mutations makes direct mutation analysis for diagnosis feasible (Huster et al. 2004; Lovicu et al. 2003). In other populations limited mutation testing has to be directed to other mutations (i.e. exon 8 mutations in patients originating from Far East or 3400delC in Brazil). If mutations are found on both chromosomes further diagnostic tests are not needed at all. The role of molecular genetic testing for diagnosis of WD is not yet established. In a recent study, in 126 of 145 patients from Austria mutations in exons 8, 13, 14 and 15 were detected at least on one chromosome, respectively (Ferenci et al. 2005a). Only in 34 of the 90 patients presenting with liver disease, diagnosis was made by standard clinical criteria alone. Forty-nine of the remaining 56 patients had detectable mutations. Thus, limited mutation analysis is extremely useful in the diagnostic work-up of this patient group. If both mutations are found further diagnostic tests are not needed at all. An algorithm of the diagnostic workup for patients with WD which includes genetic testing is shown in Fig. 3.
Today, mutation analysis is the only reliable tool for screening the family of an index case with known mutations; otherwise haplotype analysis can be used (Ferenci. 2005a). Haplotype analysis does not require the identification of a certain mutation. Microsatellite markers are also useful to study the segregation of the WD gene in most families. Where the markers are different at each locus in a patient, testing of at least one parent/or child of the patient is necessary to obtain the haplotype. By this approach diagnostic dilemmas in differentiating heterozygote gene carriers and affected asymptomatic siblings can be solved. For such analysis, at least one first-degree relative and the index patient is required (Maier-Dobersberger et al. 1995).
In summary, given the constant improvement of analytic tools genetic testing will become an integral part for the diagnosis of WD. Knowledge in the differences in the worldwide distribution of particular mutations will help to design shortcuts for genetic diagnosis of WD.
References
Al Jumah M, Majumdar R, Al Rajeh S, Awada A, Al Zaben A, Al Traif I et al (2004) clinical and genetic study of 56 Saudi Wilson disease patients: identification of Saudi-specific mutations. Eur J Neurol 11:121–124
Bachmann H, Lössner J, Kühn HJ, Siegemund R (1991) Occurrence, genetics and epidemiology of Wilson’s disease in east Germany. In: Czlonkowska A, van der Hamer CJA (eds) Proceedings of the 5th international symposium on Wilson's disease. Technical University Delft, pp 121–128
Caca K, Ferenci P, Kuhn HJ, Polli C, Willgerodt H, Kunath B et al (2001) High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol 35:575–581
Cater MA, Forbes J, La Fontaine S, Cox D, Mercer JFB (2004) Intracellular trafficking of the human Wilson protein: the role of the six N-terminal metal-binding sites. Biochem J 380:805–813
Curtis D, Durkie M, Balac P, Sheard D, Goodeve A. Peake I, Quarrell O, Tanner S. (1999) A study of Wilson disease mutations in Britain. Hum Mutat 14:304–311
DiDonato M, Hsu HF, Narindrasorasak S, Que L Jr, Sarkar B (2000) Copper-induced conformational changes in the N-terminal domain of the Wilson disease copper-transporting ATPase. Biochemistry 39:1890–1896
Deguti MM, Genschel J, Cancado EL, Barbosa ER, Bochow B, Mucenic M, Porta G, Lochs H, Carrilho FJ, Schmidt HH (2004) Wilson disease: novel mutations in the ATP7B gene and clinical correlation in Brazilian patients. Hum Mutat 398:1–8
Ferenci P (2005a) Wilson’s disease (clinical genomics). Clin Gastroenterol Hepatol 3:726–733
Ferenci P (2005b) Wilson’s disease. In: Bacon B, O’Grady JG, DiBisceglie A, Lake JR (eds) Comprehensive clinical hepatology, chap 24. Elsevier, Mosby, pp 351–367
Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F (2003). Diagnosis and phenotypic classification of Wilson disease. Final report of the proceedings of the working party at the 8th international meeting on Wilson disease and Menkes disease, Leipzig/Germany, 2001. Liver Int 23:139–142
Ferenci P, Steindl-Munda P, Vogel W, Jessner W, Gschwantler M, Stauber R et al (2005a). Diagnostic value of quantitative hepatic copper determination in patients with Wilson disease. Clin Gastroenterol Hepatol 3:811–818
Ferenci P, Merle U, Folhoffer A, Evstatiev R, Yurdaydin C, Bruha R et al (2005b) Phenotype–genotype correlations in Wilson Disease (WD)—results of a multinational study. Hepatology 42(Suppl 1):258A
Figus A, Angius A, Loudianos O, Bertini C; Dessi V, Loi A et al (1995) Molecular pathology and haplotype analysis of Wilson disease in Mediterranean populations. Am J Hum Genet 57:1318–1324
Firneisz G, Lakatos PL, Szalay F, Polli C, Glant TT, Ferenci P (2002) Common mutations of ATP7B in Wilson disease patients from Hungary. Am J Med Genet 108:23–28
Garcia-Villareal L, Daniels S, Shaw SH, Cotton D, Galvin M, Geskes J et al (2000) High prevalence of the very rare Wilson disease gene mutation Leu708Pro in the Island of Gran Canaria (Canary Islands. Spain): a genetic and clinical study. Hepatology 32:1329–1336
Gromadzka G, Schmidt HH, Genschel J, Bochow B, Rodo M, Tarnacka B, Litwin T, Chabik G, Clzonkowska A (2005) Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin Genet 68:524–532
Gu YH, Kodama H, Du SL, Gu QJ, Sun HJ, Ushijima H. (2003) Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson’s disease. Clin Genet 64:479–484
Gupta A, Aikath D, Neogi R, Datta S, Basu K, Mity B et al (2005) Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Hum Genet 118:49–57
Huster D, Weizenegger M, Kress S et al (2004) Rapid detection of mutations in Wilson disease gene ATP7B by DNA strip technology. Clin Chem Lab Med 42:507–510
Ivanova-Smolenskaya IA, Ovchinnikov IV, Karabanov AV, Deineko NL, Poleshchuk VV, Markova ED, Illarioshkin SN (1999) The His1069Gln mutation in the ATP7B gene in Russian patients with Wilson disease. J Med Genet 36:174
Kim EK, Yoo OJ, Song KY, Yoo HW, Choi SY, Cho SW et al (1998) Identification of three novel mutations and a high frequency of the Arg778Leu mutation in Korean patients with Wilson disease. Hum Mutat 11:275–278
Kumar S, Thapa BR, Kaur G, Prasad R (2005) Identification and molecular characterization of 18 novel mutations in the ATP7B gene from Indian Wilson disease patients: genotype. Clin Genet 67:443–445
Liu XQ, Zhang YF, Liu TT, Hsiao KJ, Zhang JM, Gu XF et al (2004) Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J Gastroenterol 10:590–593
Loudianos G, Dessi V, Lovicu M, Angius A, Nurchi A, Sturniolo GC et al (1998) Further delineation of the molecular pathology of Wilson disease in the Mediterranean population. Hum Mutat 12:89–94
Loudianos G, Dessi V, Lovicu M, Angius A, Altuntas B, Giacchino R et al (1999a) Mutation analysis in patients of Mediterranean descent with Wilson disease: identification of 19 novel mutations. J Med Genet 36:833–836
Loudianos G, Dessi V, Lovicu M, Angius A, Figus AL, Lilliu F et al (1999b) Molecular characterization of Wilson disease in the Sardinian population—evidence of a founder effect. Hum Mutat 14:294–303
Loudianos G, Kostic V, Solinas P, Lovicu M, Dessi V, Svetel M, Major T, Cao A (2003) Characterization of the molecular defect in the ATP7B gene in Wilson disease patients from Yugoslavia. Genet Test 7:107–112
Lovicu M, Dessi V, Zappu A et al (2003) Efficient strategy for molecular diagnosis of Wilson disease in the Sardinian population. Clin Chem 49:496–498
Margarit E, Bach V, Gomez D, Bruguera M, Jara P, Queralt R, Ballesta F (2005) Mutation analysis of Wilson disease in the Spanish population—identification of a prevalent substitution and eight novel mutations in the ATP7B gene. Clin Genet 68:61–68
Maier-Dobersberger Th, Rack S, Granditsch G, Korninger L, Steindl P, Mannhalter Ch, Ferenci P (1995) Diagnosis of Wilson’s disease in an asymptomatic sibling by DNA linkage analysis. Gastroenterology 109:2015–2018
Maier-Dobersberger T, Ferenci P, Polli C, Balac P, Dienes HP, Kaserer K et al (1997) Detection of the His1069Gln mutation in WD by rapid polymerase chain reaction. Ann Intern Med 127:21–26
Morgan CT, Tsivkovskii R, Kosinsky YA, Efremov RG, Lutsenko S (2004) The distinct functional properties of the nucleotide-binding domain of ATP7B, the human copper-transporting ATPase. Analysis of the Wilson disease mutations E1064A, H1069Q, R1151H, and C1104F. J Biol Chem 279:36363–36371
Okada T, Shiono Y, Hayashi H, Satoh H, Sawada T, Suzuki A et al (2000) Mutational analysis of ATP7B and genotype–phenotype correlation in Japanese with Wilson’s disease. Hum Mutat 15:454–462
Olivarez L, Caggana M, Pass KA, Ferguson P, Brewer GJ (2001) Estimate of the frequency of Wilson’s disease in the US Caucasian population: a mutation analysis approach. Ann Hum Genet 65:459–463
Panagiotakaki E, Tzetis M, Manolaki N, Loudianos G, Papatheodorou A, Manesis E et al (2004) Genotype–phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B). Am J Med Genet 131:168–173
Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M et al (1993) Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet 5:338–343
Petrukhin KE, Lutsenko S, Chernov I, Ross BM, Kaplan JH, Gilliam TC (1994) Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing, and structure/function predictions. Hum Mol Genet 3:1647–1656
Reilly M, Daly L, Hutchinson M (1993) An epidemiological study of Wilson’s disease in the Republic of Ireland. J Neurol Neurosurg Psychiatr 56:298–300
Scheinberg IH, Sternlieb I (1984) Wilson’s disease, vol 23. Major problems in internal medicine. Saunders, Philadelphia
Shah AB, Chernov I, Zjang HT, Ross B, Das K et al (1997) Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies. Genotype–phenotype correlation, and functional analyses. Am J Hum Genet 61:317–328
Shimizu N, Kawase C, Nakazono H, Hemmi H, Shimatake H, Aoki T (1995) A novel RNA splicing mutation in Japanese patients with Wilson disease. Biochem Biophys Res Commun 217:16–20
Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, Madl Ch et al (1997) Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 113:212–218
Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B et al (1993) The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 5:344–350
Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW (1995) The WD gene: spectrum of mutations and their consequences. Nat Genet 9:210–217
Todorov T, Savov A, Jelev H, Panteleeva E, Konstantinova D, Krustev Z et al (2005) Spectrum of mutations in the Wilson disease gene (ATP7B) in the Bulgarian population. Clin Genet 68:474–476
Vanderwerf SM, Cooper MJ, Stetsenko IV, Lutsenko S (2001) Copper specifically regulates intracellular phosphorylation of the Wilson’s disease protein, a human copper-transporting ATPase. J Biol Chem 276:36289–36294
Vrabelova S, Letocha O, Borsky M, Kozak L (2005) Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Genet Metab 86:277–285
Weirich G, Cabras AD, Serra S, Coni P, Nurcho AM, Faa G, Höfler H. (2002) Rapid identification of Wilson’s disease carriers by denaturing High-performance liquid chromatography. Prev Med 35:278–284
Wu Z, Wang N, Murong S, Lin M (2000) Identification and analysis of mutations of the Wilson disease gene in Chinese population. Chin Med J (Engl) 113:40–43
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferenci, P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet 120, 151–159 (2006). https://doi.org/10.1007/s00439-006-0202-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-006-0202-5