
Abstract
In a number of recent studies, we summarized the obvious errors and shortcomings that can be spotted in many (if not most) mitochondrial DNA (mtDNA) data sets published in medical genetics. We have reanalyzed here the complete mtDNA genome data published in various recent reports of East Asian families with hearing impairment, using a phylogenetic approach, in order to demonstrate the persistence of lab-specific mistakes in mtDNA genome sequencing in cases where those caveats were (deliberately) neglected. A phylogenetic reappraisal of complete mtDNAs with mutation A1555G (or G11778A) indeed supports the suggested lack of association between haplogroup background and phenotypic presentation of these mutations in East Asians. In contrast, the claimed pathogenicity of mutation T1095C in Chinese families with hearing impairment seems unsupported, basically because this mutation is rather basal in the mtDNA phylogeny, being specific to haplogroup M11 in East Asia. The roles of other haplogroup specific or associated variants, such as A827G, T961C, T1005C, in East Asian subjects with aminoglycoside-induced and non-syndromic hearing loss are also unclear in view of the known mtDNA phylogeny.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
An error doesn’t become a mistake until you refuse to correct it (Orlando A. Battista).
Introduction
Because of its unique genetic characteristics, such as maternal inheritance and absence of recombination, mutations that accumulated in the course of mtDNA evolution can be reconstructed phylogenetically from contemporary mtDNA variation. Then, based on a well-reconstructed mtDNA phylogeny, most potential errors in mtDNA data can easily be identified by cross-comparisons. Such an approach of an a posteriori quality check has successfully been applied to many data sets in human population genetics and ancient DNA studies (Bandelt 2004, 2005; Bandelt et al. 2002, 2003; Bandelt and Kivisild 2006; Forster 2003; Yao et al. 2003b; Yao and Zhang 2003), forensic genetics (Bandelt et al. 2001, 2004a, b; Röhl et al. 2001; Yao et al. 2004a), and medical genetics (Bandelt et al. 2005a, b; Salas et al. 2005a, b). A series of suggestions, caveats, as well as guides for a posteriori data checking have been put forward in these studies. In particular, we recently pointed out the problems of low ‘penetrance’ of mtDNA phylogenetic knowledge in mitochondrial disease studies, by way of reanalyzing three complete mtDNA sequencing attempts, two from Europe and one from East Asia (Bandelt et al. 2005a). It would, therefore, be interesting to see whether there is already any resonance to this broad spectrum of publications on data quality control in the medical field, especially by laboratories that had issued problematic data before; one such case (Da Pozzo et al. 2004) has most recently been clarified and settled (Da Pozzo and Federico 2005). It is understandable that the fanfare of errata (from which we, however, do not shy away ourselves) is not greeted with enthusiasm by clinical research groups, so that obviously flawed data stay as they are, without correction by the authors themselves. At least, one should expect that future lab activities would avoid committing exactly the same errors again and again.
In addition, some variants in mtDNA 12S rRNA gene, such as T1095C, A827G, T961C, and T1005C, which were recently claimed to be pathogenic in Chinese subjects with aminoglycoside-induced and non-syndromic hearing loss (Li et al. 2005b; Wang et al. 2005; Zhao et al. 2004b), need to be re-evaluated on the basis of the hitherto known mtDNA phylogeny of East Asians. A phylogenetic reappraisal of related East Asian mtDNAs with A1555G (or G11778A) and hearing impairment from the literature could provide further insights on the potential association between haplogroup background and phenotypic presentation of these mutations in East Asians (Abe et al. 1998; Hutchin and Cortopassi 1997; Qian et al. 2005; Young et al. 2005).
Materials and methods
To accentuate the need for a phylogenetic approach, we took the recently reported complete mtDNAs in East Asian families with hearing impairment (Li et al. 2004a, 2005a; Young et al. 2005; Yuan et al. 2005; Zhao et al. 2004a, b, 2005). The mtDNA complete sequences of four Chinese families with Leber hereditary optic neuropathy (LHON) (Qian et al. 2005; Qu et al. 2005) and one Chinese patient with auditory neuropathy (Wang et al. 2005) were also considered here. Since most of these patients were of Chinese origin, we focused on the available mtDNA data from East Asia for comparison. We followed the phylogenetic system for East Asian mtDNAs described in our previous work (Kivisild et al. 2002; Kong et al. 2003b; Yao et al. 2002a) with some updates (Tanaka et al. 2004). Each reported mtDNA sequence was assigned a haplogroup status according to the presence of basal haplogroup-specific mutations, as well as by matching it closely with available mtDNA complete sequences. Then, with the reported mtDNAs allocated to its respective place in the global mtDNA tree, a site-by-site audit allowed us to detect the absence of otherwise expected mutations given the phylogenetic affinities and/or clade membership of the sequence under scrutiny. The frequency of such omissions can then be contrasted with the natural spectrum of mutations at the respective positions as manifest in the published body of mtDNA genomes. This strategy has been described before in other cases of population genetics (Yao et al. 2003b), mitochondrial diseases (Bandelt et al. 2005a), and tumor studies (Salas et al. 2005b).
In total, 18 complete mtDNA sequences (Li et al. 2004a, 2005a; Qian et al. 2005; Qu et al. 2005; Wang et al. 2005; Young et al. 2005; Yuan et al. 2005; Zhao et al. 2004a, b, 2005) reported by the same laboratory came under scrutiny here. Among them, four mtDNAs reported by Young et al. (2005) have been reanalyzed in our recent paper (Bandelt et al. 2005a) but were considered again to further demonstrate the persistence of lab-specific mistakes. Some further comments about the quality of the Qian et al. (2005) data were also advanced in Salas et al. (2005a). For comparison, we incorporated some of our previously reported complete mtDNAs (Kong et al. 2003b) into the tree construction, which nearly matched some of those 18 problematic mtDNA sequences. In addition, published mtDNA data from Japanese and Chinese families with A1555G mutation and hearing impairment (Abe et al. 1998; Hutchin and Cortopassi 1997) were also reanalyzed to evaluate the potential association between this mutation and specific haplogroups in East Asia.
Results and discussion
Features of lab-specific mistakes
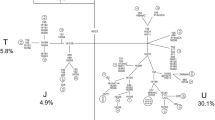
There are five major and common types of errors, namely, base shifts, reference bias, phantom mutations, base misscoring, and artificial recombination, observable in published mtDNA data in population genetics, forensic sciences, and the medical field (Bandelt et al. 2001, 2002, 2004b; Bandelt and Parson 2004; Salas et al. 2005b; Yao et al. 2004a). Batches of problematic mtDNA sequence data generated from one lab would typically show some imprint of one or two major errors throughout the data set, inasmuch as the same method was used to perform sequencing and editing. As shown in Fig. 1, several features of the lab-specific mistakes can be well discerned in the 18 reported complete mtDNAs from the same group: (1) Site 14766 was scored wrongly in 16 out of 18 complete sequences. It is most likely that a modified version of the revised Cambridge Reference Sequence (rCRS: Andrews et al. 1999) was used in scoring the mutations that still bore the erroneous T nucleotide at position 14766 of the original CRS (Anderson et al. 1981). We do not know whether the correct scoring for this position in two complete sequences most recently reported by Qian et al. (2005; sequence 14) and Yuan et al. (2005; sequence 3) represent the beginning of a correction of the data from this group, or if was accidental. This last explanation seems supported by the erratic reporting of variation at 14766 in Qian et al. (2005), where only one out of three sequences was correctly scored. (2) Private mutations of the rCRS sequence (which should be present in any sequence not closely related to it) and the basal mutations that separate rCRS from the root of haplogroup R were frequently missed. Thus, two complete mtDNAs reported by Li et al. (2004a; sequence 7) and Zhao et al. (2004a; sequence 8) were notorious in this respect: both sequences listed only some of the private mutations of the rCRS (A263G, 315+C, A8860G, and A15326G) and missed the bulk of mutations separating the ancestral sequence of haplogroup R from the ancestral sequence of haplogroup H2b to which the rCRS belongs (Loogväli et al. 2004). (3) Mutations at sites 489, 4248, 5178, 9950, 10400, and 10873 turned out to be “hotspots” of oversight. Other basal mutations, such as T9824A, C12705T, and T14318C, were also missed along the respective branches. For instance, in the D4b2b sequence (sequence 13; Qian et al. 2005), two haplogroup D4b2 specific mutations (T9824A and C5178A), one N specific mutation (T10873C), and the HV defining mutation (C14766T) were all omitted. (4) Potential phantom mutations could be observed in the five complete mtDNAs (sequences 8, 15–18) generated by Young et al. (2005) and Zhao et al. (2004a). With the exception of sequence 18, the other four mtDNAs harbored at least one rare transversion per sequence. In addition, the F3b sequence (sequence 15) ‘borrowed’ mutations T489C and T10873C from some mtDNA belonging to haplogroup M, most likely from sequence 17 or sequence 18 described in the same study (Young et al. 2005). (5) The three M11a sequences (sequences 9–11) reported by Zhao et al. (2004b) probably suffered from artificial recombination in region 16311–1719. (6) Typos for the base pair and the site position in rCRS were sporadically found in the original tables. For instance, position 3423 was wrongly marked as G instead of T, position 12358 as T instead of A, and position 10640 as G instead of T in the CRS column of Table 1 in Young et al. (2005). The nucleotides at position 14340 in the column for sequence #81 (sequence 11 in Fig. 1) and the CRS column of their Table 1 were switched (Zhao et al. 2004b). Positions 8584 and 12957 were listed as A instead of G and C instead of T, respectively, in the CRS column of Table 1 in Zhao et al. (2005). In sample WZ4 (Qian et al. 2005; sequence 12), the expected transversion G13928C was scored as a transition (G13928A) while the nucleotides at site 15784 in rCRS (T) and its expected transitional variant (C) were exchanged. This sequence also harbored a transversion T16304G instead of the transition T16304C expected in the corresponding haplogroup. Recall that the confusion between G and C constitutes a frequent clerical error (Bandelt et al. 2001, 2004a). Moreover, the basal mutation G15043A shared by all the lineages belonging to super-haplogroup M was listed as a G to C transversion in sample #WZ5 (Qian et al. 2005; sequence 13). More errors in these complete genomes are indicated in Fig. 1.

Classification tree of 27 complete mtDNAs of East Asian ancestries (plus the rCRS). Haplogroup names are inserted along the branches that determine the locations of the corresponding ancestral haplotypes. Phantom mutations or reading errors are highlighted in italics. Suffixes A, C, G, and T indicate transversions, “d” indicates a deletion, and a plus sign (+) indicates an insertion; recurrent mutations are underlined. Mutation oversights are marked along the tree by encircled numbers (1–18) that refer to sequences from the following publications: sequences 1 and 2 are from Zhao et al. (2005), sequence 3 from Yuan et al. (2005), sequence 4 from Qu et al. (2005), sequence 5 from Li et al. (2005a), sequence 6 from Wang et al. (2005), sequence 7 from Li et al. (2004a), sequence 8 from Zhao et al. (2004a), sequences 9–11 from Zhao et al. (2004b), sequences 12–14 from Qian et al. (2005), sequences 15–18 from Young et al. (2005), sequence AY972053 from Bandelt et al. (2005a), sequences AY255142, AY255145, AY255148, AY255156, AY255166, and AY255176 from Kong et al. (2003b), sequences AP008526 and AP008639 from Tanaka et al. (2004). The order of mutations on each uninterrupted branch section is arbitrary. Variation at the hypervariable sites 16182 and 16183 due to the transition at position 16189 and around position 309 are not indicated in the figure
A careful check before manuscript submission or even at the proof-reading stage could have eliminated many of these inadvertent errors. Intriguingly, the haplogroup A sequence reported by Zhao et al. (2004a; sequence 8) has an unusual mutation at 4247, which could be alternatively explained as a reading shift of the expected mutation at 4248 characteristic of haplogroup A. This sequence further shares the transition (claimed to be pathogenic) at 1494 with one haplogroup A sequence (accession no. AY255166) from Kong et al. (2003b) that contained a seeming private transition at 4257 (Fig. 1). Mutation A15326G (from the pathway between rCRS and the ancestral H2b haplotype) is missing in Li et al. (2005a; sequence 5) but quite suspiciously, a seeming private transition A15236G appears instead in this Japanese pedigree. Based on the above features, it seems that errors were introduced in these complete mtDNAs at all stages of data generation, predominantly through mutation oversight and misdocumentation. It is thus possible that more private mutations and even some potential novel pathogenic mutation(s) in some families could have been missed.
Error repetitions
After the (online) publication of our paper (Bandelt et al. 2005a), which might have alerted the group of Guan and co-workers, three new papers about mtDNA mutations in Chinese families with hearing impairment were submitted by the same group (Qian et al. 2005; Yuan et al. 2005; Zhao et al. 2005). It is instructive to see to what extent they considered the constructive suggestions for improving data quality. We now take the sequence data in the most recent paper from this group (Zhao et al. 2005) as an example of how to perform the necessary site by site audit.
Zhao et al. (2005) analyzed complete mtDNA sequences from two probands with aminoglycoside-induced and non-syndromic hearing loss. They claimed that sequences 1 (BJ105 in the original Table 1) and 2 (BJ106 in the original Table 1) belonged to haplogroups F3 and M7b, respectively, according to the haplogroup system of Tanaka et al. (2004). However, the majority of mutations observed in these two sequences do not support the claimed haplogroup status. It looks like that these authors used partial control region information to establish the haplogroup affiliation of each mtDNA (see below). An incorrect classification of mtDNA could also be found in their recent study (Qu et al. 2005), in which the B5a mtDNA was wrongly assigned to haplogroup B5b.
Figure 1 depicts the relationship of the two mtDNAs in Zhao et al. (2005) together with other near-matches from the literature. Sequence 1 harbors a string of mutations specific to haplogroup C, where, however, T14318C is missing. This sequence shared seven of the 13 apparently private substitutions (at sites 146, 4047, 5821, 6338, 7853, 12957, and 14978) of a complete sequence (accession no. AY255176) reported by Kong et al. (2003b). Note that sequence AY255176 also has a mutation at position 5987, whereas sequence 1 bears a ‘similar’ mutation at 5897 instead. It is most likely that Zhao et al. (2005) made an initial haplogroup assignment of this mtDNA to haplogroup F based on the observed variants (A73G, T146C, 249delA, A263G, 309+C or 309+CC, 315+C, and 522-523delCA) in the second hypervariable segment of the control region (HVS-II) and the apparent lack of the mutations A10398G and C10400T characteristic for haplogroup M (in which haplogroup C is embedded). An extensive research into published mtDNA data reveals two F1 mtDNAs (accession no.s AP008328 and AP008701) in the large data set of Tanaka et al. (2004) and five F1 mtDNAs in the SWGDAM database (Monson et al. 2002) with the same HVS-II mutations as sequence 1 determined by Zhao et al. (2005), although this HVS-II type was also present in 11 Chinese haplogroup C mtDNAs from our recent studies (Yao et al. 2002a, 2003a, 2004b; Kong et al. 2003a). However, it still remains unclear why Zhao et al. (2005) assigned this sequence to haplogroup F3.
Sequence 2 suffers from a number of mistakes similar to the ones observed in sequence 1. This mtDNA has all nine specific coding-region mutations of haplogroup D4b2 (relative to the root of haplogroup M) except the most prominent one, C5178A (known to be very stable), which was apparently omitted (Fig. 1). Obviously, Zhao et al. (2005) assigned this mtDNA to haplogroup M7b because the control region indeed suggests haplogroup M7b2 status in view of the mutations A73G, T199C, T204C, A263G, 309+C, 315+C, T489C, C495T, T16189C, C16223T, and T16298C (compare with sample XJ8450 [accession no. AY255173] from Kong et al. 2003b). Therefore this case most likely constitutes a sample mix between haplogroup D5b2 and M7b2 mtDNAs.
MtDNA haplogroups and phenotypic presentation of mutations A1555G and G11778A in East Asian
When examining the potential association of an mtDNA variant or mutation with a disease, it is crucial to get information about the haplogroup background of the apparent pathogenic mutation. Then, there are two main possibilities. Either, the variant analyzed belongs to a particular haplogroup; then, this means that any other variant belonging to the same haplogroup or a combination of variants of this lineage could equally be causal for the disease. Or, the lack of a particular haplogroup background could indicate a more direct implication of the mutation, although epistatic effects cannot be ignored.
Recently, we have reassessed the potential pathogenicity of mtDNA mutations in the light of an updated mtDNA phylogeny (Bandelt et al. 2005c; Kong et al. 2004; Yao et al. 2002b). This phylogenetic approach, although not in principle novel to the medical field (e.g. Brown et al. 1995, 1997; Howell et al. 1995; Hutchin and Cortopassi 1997; Torroni et al. 1997, 1999), has proved to be very helpful in assessing the origin of an mtDNA mutation. Phylogenetic analysis of mtDNAs from Spanish and Cuban families with A1555G and nonsyndromic sensorineural deafness revealed that they were members of a wide range of mtDNA haplogroups in Africans and West Eurasians, thus suggesting a multiple and random occurrence of this mutation in these lineages and undermining the association of mtDNA haplogroup background in maternally transmitted sensorineural deafness (Estivill et al. 1998; Torroni et al. 1999). In contrast, phylogenetic analyses of LHON families in Europe and populations of European ancestry indicated that mtDNA haplogroup J played a role in the expression of LHON by increasing the penetrance of the primary mutations G11778A and T14484C (Brown et al. 1997; Howell et al. 1995; Torroni et al. 1997; Carelli et al. 2006). Because the mtDNAs of East Asians and Europeans occupy different branches of the world mtDNA phylogeny showing a marked difference (Kong et al. 2003b; Palanichamy et al. 2004; Richards et al. 2002; Tanaka et al. 2004), the above phylogenetic results in Europeans might not automatically apply to East Asians. It is, therefore, necessary and worthwhile to re-evaluate mutations A1555G and G11778A in East Asians according to an updated mtDNA tree.
Previous phylogenetic analyses of LHON pedigrees have demonstrated that among the three primary mtDNA mutations, G11778A and T14484C showed a preferential association with haplogroup J, in particular with subhaplogroups J1c and J2b (Carelli et al. 2006), whereas mutation G3460A was distributed randomly along the phylogenetic trees and had no association with particular West Eurasian specific haplogroups (Brown et al. 1997; Torroni et al. 1997; Man et al. 2004). In the current compiled data sets, mutation G11778A was present in four of the 19 mtDNAs that could be assigned to haplogroups B5a2, F1, D4b2b, and M10a, respectively (Fig. 1). Hence, the occurrence of G11778A in Chinese pedigrees is of multiple origin. There does not seem to be haplogroup-specific mutation(s) that would increase the penetrance of the primary mutation G11778A as well as the risk of disease expression in East Asians (Qian et al. 2005).
In Fig. 1, nine of the 19 mtDNAs from Chinese families (including two Japanese mtDNAs) with hearing impairment harbored mutation A1555G, and they are scattered over a wide range of haplogroups (F3b, A, N9a1, D4b2, D4b2b, D4a, and C). Although haplogroup D4 has a high prevalence (5/9) for mutation A1555G, it seems unlikely that these D4 mtDNAs with A1555G all stemmed from a recent common ancestor inasmuch as they harbor many private mutations, which suggests a considerably earlier divergence among them. It should be mentioned that two previous studies that performed a phylogenetic analysis of mtDNAs in Chinese and Japanese pedigrees with A1555G and hearing loss also revealed a multiple origin pattern for this mutation (Abe et al. 1998; Hutchin and Cortopassi 1997), although their data contained some obvious errors. Table 1 presents the tentative haplogroup classification of these pedigrees. In the 13 Japanese families analyzed by Abe et al. (1998), the mtDNAs thriving in five matrilines could be assigned to haplogroup B5b. In particular, families 1, 2, and 6 shared the same B5b2 haplotype (probably due to a recent founder event), whereas family 11 differed from these families at sites 207 and 16291 (disregarding A16182C). The mtDNAs in two matrilines (3 and 4) belonged to haplogroup A. Four families (7, 8, 9, and 10) had mtDNAs from haplogroup D4. Finally, the mtDNAs of families 5 and 13 could be classified into haplogroups F1b1 and M7b2, respectively. The data reported by Hutchin and Cortopassi (1997) contained many phantom mutations, among which is the notorious C16085T transition that signposted the presence of phantom mutations in many other cases (Brandstätter et al. 2005). RFLP or HVS-I data in general contain not enough information for reliable haplogroup assignment, but several haplogroups could still be safely inferred by motif recognition and near-matching with available data (Yao et al. 2002a). Based on the above phylogenetic classification results (Fig. 1; Table 1), it is evident that mutation A1555G is recurrent in East Asian pedigrees and that mtDNA background may not play a decisive role in the phenotypic presentation of the A1555G mutation. In Japan, there seems to have been a founder effect for the B5b type with mutation A1555G, which may parallel the situation with the Spanish pedigrees carrying haplogroup H3 mtDNAs (Achilli et al. 2004; Torroni et al. 1999).
T1095C and aminoglycoside-induced and non-syndromic hearing impairment
The association of mutation T1095C with hearing impairment was first reported in two Italian families (Tessa et al. 2001; Thyagarajan et al. 2000), but no information was provided as to which haplogroup the affected mtDNAs belonged. Moreover, Thyagarajan et al. (2000) did not detect this mutation in 270 samples (including 22 Japanese, 26 African-Americans, 54 individuals of Italian maternal origin, 20 Parkinson disease patients, and 148 Americans of presumed West Eurasian maternal origin). Guan and colleagues identified the presence of this mutation in three Chinese subjects with a variable phenotype of aminoglycoside-induced and non-syndromic hearing impairment and one female subject with auditory neuropathy (Wang et al. 2005; Zhao et al. 2004b). Based on two more lines of evidence, absence of this mutation in 364 Chinese control samples and the report of the Italian cases, they claimed that this mutation was involved in the pathogenesis of hearing impairment. Unfortunately, these authors failed to notice that mutation T1095C, together with ten other mutations, actually defines a basal branch of the East Asian mtDNA phylogeny, namely haplogroup M11 (Fig. 1). Thus, in Wang et al.’s study dealing with the implications of T1095C in the auditory neuropathy, their assertion that “the following mutations are novel missense polymorphisms in the Chinese population: A8108G (I175V) mutation in the CO2, G11969A (A406T) in the ND4, and C14340T (V112M) in the ND6” (p. 29) is incorrect and their subsequent discussion on the high degree of evolutionary conservation of I175V and V112M, as well as the potential association with the auditory neuropathy phenotype, is thus ill-based.
Hitherto, there were eight complete mtDNA sequences of haplogroup M11 available, including four complete sequences from Zhao et al. (2004b) and Wang et al. (2005). We have summarized the phylogenetic relationship among these eight mtDNAs in Fig. 1. One can discern two subhaplogroups, M11a (characterized by C14340T) and M11b (characterized by G10685A, C13890T, and A14790G), within haplogroup M11. This haplogroup is generally rather rare in East Asians, but it has been spotted sporadically in Japanese (1/162, Imaizumi et al. 2002; 1/572, Tanaka et al. 2004), Korean (4/593, Lee et al. 2006; 1/523, Jin et al. 2005 and references therein), and Chinese (3/263, Yao et al. 2002a; 0/232, Kong et al. 2003a; 2/76, Yao et al. 2003a; 1/105, Rao et al. 2003; 4/252, Yao et al. 2004b; 1/55, Zhang et al. 2005). It is then not surprising that Li et al. (2005b) found only one sample with this mutation in 128 pediatric Chinese subjects suffering from sensorineural hearing loss. The fact that the T1095C mutation was absent in 364 Chinese controls in Zhao et al. (2004a) does not significantly conflict with the frequency expected for this population, and cannot lend support to the clinical association—contrary to the claims in Li et al. (2005b). The absence of this mutation in a large cohort of “Caucasians” and “African-Americans” (Thyagarajan et al. 2000) is concordant with the absence of M11 in matrilines that are not of East Asian origin. In short, the claimed pathogenicity of the haplogroup M11 specific mutation T1095C in aminoglycoside-induced and non-syndromic hearing impairment in Chinese does not receive support from the phylogenetic perspective, and therefore would still await further confirmation by functional assay.
Roles of A827G, T961C, T1005C, and other related mutations in hearing loss
The hitherto known phylogeny of East Asian mtDNAs based on complete sequences could provide further information for us to reappraise the results of a recent study of mitochondrial 12S rRNA gene in 128 Chinese paediatric subjects with aminoglycoside-induced and non-syndromic hearing loss. In that study, Li et al. (2005b) marked variants A827G, T961C, and T1005C as “known and putative pathogenic mutations” in their Tables 1 and 2 and aimed at arguing in favor of pathogenic roles of these variants in hearing loss. But these authors failed to notice that mutation A827G, together with C15535T, defines haplogroup B4b’d that includes the entire Native American haplogroup B2 (Bandelt et al. 2003; Kong et al. 2003b). Further, T1005C is one of the characteristic mutations of haplogroup F2 (Kong et al. 2003b, 2004), whereas mutation T961C is specific for two subhaplogroups (labeled “A1b” and “N9a2a” by Tanaka et al. 2004) of haplogroups A5 and N9a, respectively. The C insertion at the cytosine homopolymeric track that runs from position 956 to 960, which was scored as 956iC by Tanaka et al. (2004), 960+C by Kong et al. (2003b), and 961-C insertion by Li et al. (2005), is in the list of haplogroup characteristic mutations for B5b1 (Tanaka et al. 2004). The statement by Li et al. (2005) that the mitochondrial 12S rRNA gene is a hotspot for deafness-associated mutations in the Chinese populations may therefore no longer be sustainable on the basis of the phylogenetic information.
A suggested association of particular variants appearing e.g. in T1095C carriers, such as A2238G or T2885C (Zhao et al. 2004b), without any formal (statistical and functional) support whatsoever, is an inadequate praxis. There is a general tendency to suspect any mtDNA variant found in patients of positive association as long as the variant is non-synonymous or modifies a highly evolutionarily conserved position (McFarland et al. 2004). The risk with this praxis is that it creates a false expectation and may mislead subsequent research efforts towards erroneous topics (thus creating an ascertainment bias), as is the case of mtDNA studies in tumorigenesis (Salas et al. 2005b). This caveat is particularly appropriate for human mtDNA association studies, where we have the privileged knowledge of a robust phylogeny based on >2,500 complete sequences and phylogeographic distribution information for >40,000 individual HVS-I sequences. For instance, T2885C is a well-known polymorphism, which is among the defining variants for sub-Saharan African branch L2’3 (Herrnstadt et al. 2002); note that this mutation is even highly conserved from an evolutionary perspective, which effectively constitutes the only argument in favor of its positive association with the disease by Zhao et al. (2004b). Hence, from the phylogenetic perspective, transition T2885C is unlikely to have anything to do with hearing impairment. Li et al. (2004b) claimed (through biochemical assays) to have demonstrated the implications of T3308C and T5655C in deafness—at least when these two variants were in combination with T7511C. These authors failed to notice that both T3308C and T5655C are among the string of mutations characteristic of haplogroup L1b (Rocha et al. 1999; Herrnstadt et al. 2002) which is highly prevalent in sub-Saharan Africans (Salas et al. 2002, 2004). Therefore, Li et al.’s statement that “...the presence of the ND1 T3308C and tRNA Ala T5655C mutations in the African family but the absence of these mtDNA mutations in the French and Japanese families seem to account for different penetrance between two pedigrees” (p. 875) cannot be justified. It would actually be very surprising to see these two L1b mutations in a Japanese or East Asian lineage. The title of the article “Cosegregation of the G7444A mutation in the mitochondrial...with aminoglycoside-induced and nonsyndromic hearing loss” by Yuan et al. (2005) clashes with the fact that G7444A is a common variant within the West European prevalent haplogroup V (Ingman et al. 2000; Finnilä et al. 2001; Kivisild et al. 2006). Moreover, G7444A has already been found in two H sequences (Herrnstadt et al. 2002), and one L1b sequence (Kivisild et al. 2006), suggesting that this mutation rather constitutes a normal polymorphism.
A call for the phylogenetic error surveillance
As has been exemplified many times before, a phylogenetic method employing the full body of published mtDNA sequences is very useful to pinpoint potential errors in mtDNA findings. The kind of problems we have seen in the ongoing work of Guan and colleagues would be very well solvable, and inadvertent misreports of mtDNA variation could be prevented from seeing the light of publication if a posteriori quality check would be carried out. If, however, clinical research would stay immune against criticism and advice and ignore good scientific practice, then the burden solely rests on the editors of a scientific journal to have proper refereeing and editing carried out. An unfiltered, very rapid dissemination of clinical research results (lacking any in-depth reviewing process), however, would enhance the construction of a ‘parallel world’ of ‘exciting’ results without solid basis—just as it was and is the case with mtDNA investigations in cancer research (Salas et al. 2005b).
A phylogenetic approach is also indispensable to provide insight into the role that a particular mutation could possibly play in regard to pathogenicity. The reappraisal of mtDNAs showing mutation A1555G or G11778A indicates multiple random occurrences of these mutations in East Asian lineages. Therefore, the mtDNA haplogroup background does not seem to play any decisive role in aminoglycoside-induced and nonsyndromic hearing loss, at least, given the information provided so far. The association between haplogroup M11 (T1095C) and hearing loss receives no support from phylogenetic analysis and awaits further confirmation with focused experimental designs. Despite the—as we believe—premature labeling of the T1095C mutation as being a “confirmed” pathogenic mutation by MITOMAP (www.mitomap.org), there is no compelling evidence yet for this status. There are further instances where MITOMAP status of “confirmed” pathogenic came into reconsideration when more stringent rules were applied (Mitchell et al. 2006). This does not preclude the possibility of some indirect influence to the manifestation of hearing impairment, but the previous approaches fell short of the goal by ignoring the fact that T1095C is a basal polymorphism of the East Asian mtDNA phylogeny. Similarly, the pathogenicity of these haplogroup specific or associated variants in the 12S rRNA gene of East Asian families with hearing impairment, such as A827G, T1005C, 960+C, and T961C, if at all, should all be labeled as provisional or unclear, at best, for the time being. When applying a pathogenicity scoring system along the lines suggested by Mitchell et al. (2006), it would be desirable to give additional negative scores to a mutation depending on its age in the worldwide mtDNA phylogeny. For example, one could subtract score k if the estimated maximum age of the targeted mutation in the worldwide mtDNA phylogeny would be rounded down to 2k times, say, 750 years. Such negative scoring would assist in avoiding an ascertainment bias in the case of mutations specific to rather rare basal haplogroups, which are not routinely found in mtDNA screenings of small control groups. We are aware that such a scoring system might not apply in some cases. For instance, under the ‘thrifty genotype hypothesis’ (Neel 1962), here reconsidered in an mtDNA context, the genetic predisposition to a disease could be the consequence of adaptation to an ancient lifestyle characterized by fluctuating and unpredictable environmental conditions; the switch to different environmental conditions would make this thrift genotype no longer advantageous, then giving rise to the disease phenotype. This could well be applied to complex diseases (which differ in prevalence across ethnic groups) where the mtDNA could play some role under this hypothesis (e.g. diabetes, obesity, and hypertension).
In summary, considering the importance of phylogenetic knowledge in mtDNA medical studies and the (inadvertent) neglect of this knowledge in the field, we feel it is indispensable to call for attention to the suggestions, caveats, and guides for a posteriori data checking based on mtDNA phylogeny, as exemplified in the current study and our previous studies (Bandelt et al. 2001, 2002, 2005a, c; Salas et al. 2005a, b; Yao et al. 2002b, 2003b, 2004a), to essentially improve the data quality and properly explain the observed mutation(s) in the clinical research.
References
Abe S, Usami S, Shinkawa H, Weston MD, Overbeck LD, Hoover DM, Kenyon JB, Horai S, Kimberling WJ (1998) Phylogenetic analysis of mitochondrial DNA in Japanese pedigrees of sensorineural hearing loss associated with the A1555G mutation. Eur J Hum Genet 6:563–569
Achilli A, Rengo C, Magri C, Battaglia V, Olivieri A, Scozzari R, Cruciani F, Zeviani M, Briem E, Carelli V, Moral P, Dugoujon JM, Roostalu U, Loogväli EL, Kivisild T, Bandelt H-J, Richards M, Villems R, Santachiara-Benerecetti AS, Semino O, Torroni A (2004) The molecular dissection of mtDNA haplogroup H confirms that the Franco-Cantabrian glacial refuge was a major source for the European gene pool. Am J Hum Genet 75:910–918
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147
Bandelt H-J (2004) Etruscan artifacts. Am J Hum Genet 75:919–920
Bandelt H-J (2005) Mosaics of ancient mitochondrial DNA: positive indicators of nonauthenticity. Eur J Hum Genet 13:1106–1112
Bandelt H-J, Achilli A, Kong Q-P, Salas A, Lutz-Bonengel S, Sun C, Zhang Y-P, Torroni A, Yao Y-G (2005a) Low “penetrance” of phylogenetic knowledge in mitochondrial disease studies. Biochem Biophys Res Commun 333:122–130
Bandelt H-J, Herrnstadt C, Yao Y-G, Kong Q-P, Kivisild T, Rengo C, Scozzari R, Richards M, Villems R, Macaulay V, Howell N, Torroni A, Zhang Y-P (2003) Identification of Native American founder mtDNAs through the analysis of complete mtDNA sequences: some caveats. Ann Hum Genet 67:512–524
Bandelt H-J, Kivisild T (2006) Quality assessment of DNA sequence data: autopsy of a mis-sequenced mtDNA population sample. Ann Hum Genet (Jan 17, 2006, Epub ahead of print; doi: 10.1111/j.1529-8817.2005.00234.x)
Bandelt H-J, Kong Q-P, Parson W, Salas A (2005b) More evidence for non-maternal inheritance of mitochondrial DNA? J Med Genet 42:957–960
Bandelt H-J, Lahermo P, Richards M, Macaulay V (2001) Detecting errors in mtDNA data by phylogenetic analysis. Int J Legal Med 115:64–69
Bandelt H-J, Parson W (2004) Fehlerquellen mitochondrialer DNS-Datensätze und Evalution der mtDNS-Datenbank “D-Loop-BASE. Rechtsmedizin 14:251–257
Bandelt H-J, Quintana-Murci L, Salas A, Macaulay V (2002) The fingerprint of phantom mutations in mitochondrial DNA data. Am J Hum Genet 71:1150–1160
Bandelt H-J, Salas A, Bravi C (2004a) Problems in FBI mtDNA database. Science 305:1402–1404
Bandelt H-J, Salas A, Lutz-Bonengel S (2004b) Artificial recombination in forensic mtDNA population databases. Int J Legal Med 118:267–273
Bandelt H-J, Yao Y-G, Kivisild T (2005c) Mitochondrial genes and schizophrenia. Schizophr Res 72:267–269
Brandstätter A, Sänger T, Lutz-Bonengel S, Parson W, Béraud-Colomb E, Wen B, Kong Q-P, Bravi CM, Bandelt H-J (2005) Phantom mutation hotspots in human mitochondrial DNA. Electrophoresis 26:3414–3429
Brown MD, Sun F, Wallace DC (1997) Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet 60:381–387
Brown MD, Torroni A, Reckord CL, Wallace DC (1995) Phylogenetic analysis of Leber’s hereditary optic neuropathy mitochondrial DNA’s indicates multiple independent occurrences of the common mutations. Hum Mutat 6:311–325
Carelli V, Achilli A, Valentino ML, Rengo C, Semino O, Pala M, Olivieri A, Mattiazzi M, Pallotti F, Carrara F, Zeviani M, Leuzzi V, Carducci C, Valle G, Simionati B, Mendieta L, Salomao S, Belfort R, Sadun AA, Torroni A (2006) Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber Hereditary Optic Neuropathy pedigrees. Am J Hum Genet 78 (Jan 27, 2006, Epub ahead of print; http://www.ajhg.org/AJHG/journal/issues/v78n4/43264/43264.html)
Chen Y-S, Olckers A, Schurr TG, Kogelnik AM, Huoponen K, Wallace DC (2000) MtDNA variation in the South African Kung and Khwe-and their genetic relationships to other African populations. Am J Hum Genet 66:1362–1383
Da Pozzo P, Cardaioli E, Radi E, Federico A (2004) Sequence analysis of the complete mitochondrial genome in patients with mitochondrial encephaloneuromyopathies lacking the common pathogenic DNA mutations. Biochem Biophys Res Commun 324:360–364
Da Pozzo P, Federico A (2005) Commentary to mitDNA research for the pathogenesis of mitochondrial disorders. Biochem Biophys Res Commun 336:1003–1004
Estivill X, Govea N, Barcelo E, Badenas C, Romero E, Moral L, Scozzari R, D’Urbano L, Zeviani M, Torroni A (1998) Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am J Hum Genet 62:27–35
Finnilä S, Lehtonen MS, Majamaa K (2001) Phylogenetic network for European mtDNA. Am J Hum Genet 68:1475–1484
Forster P (2003) To err is human. Ann Hum Genet 67:2–4
Herrnstadt C, Elson JL, Fahy E, Preston G, Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal MF, Davis RE, Howell N (2002) Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups. Am J Hum Genet 70:1152–1171; erratum 71:448–449
Howell N, Kubacka I, Halvorson S, Howell B, McCullough DA, Mackey D (1995) Phylogenetic analysis of the mitochondrial genomes from Leber hereditary optic neuropathy pedigrees. Genetics 140:285–302
Hutchin TP, Cortopassi GA (1997) Multiple origins of a mitochondrial mutation conferring deafness. Genetics 145:771–776
Imaizumi K, Parsons TJ, Yoshino M, Holland MM (2002) A new database of mitochondrial DNA hypervariable regions I and II sequences from 162 Japanese individuals. Int J Legal Med 116:68–73; erratum 119:183
Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408:708–713
Jin HJ, Kwak KD, Hong SB, Shin DJ, Han MS, Tyler-Smith C, Kim W (2005) Forensic genetic analysis of mitochondrial DNA hypervariable region I/II sequences: an expanded Korean population database. Forensic Sci Int (2005 Jun 28, Epub ahead of print; doi:10.1016/j.forsciint.2005.03.026)
Kivisild T, Shen P, Wall DP, Do B, Sung R, Davis K, Passarino G, Underhill PA, Scharfe C, Torroni A, Scozzari R, Modiano D, Coppa A, de Knijff P, Feldman M, Cavalli-Sforza LL, Oefner PJ (2006) The role of selection in the evolution of human mitochondrial genomes. Genetics 172:373–387
Kivisild T, Tolk H-V, Parik J, Wang Y, Papiha SS, Bandelt H-J, Villems R (2002) The emerging limbs and twigs of the East Asian mtDNA tree. Mol Biol Evol 19:1737–1751; erratum 20:162
Kong Q-P, Yao Y-G, Liu M, Shen S-P, Chen C, Zhu C-L, Palanichamy MG, Zhang Y-P (2003a) Mitochondrial DNA sequence polymorphisms of five ethnic populations from northern China. Hum Genet 113:391–405
Kong Q-P, Yao Y-G, Sun C, Bandelt H-J, Zhu C-L, Zhang Y-P (2003b) Phylogeny of East Asian mitochondrial DNA lineages inferred from complete sequences. Am J Hum Genet 73:671–676; erratum 75:157
Kong Q-P, Yao Y-G, Sun C, Zhu C-L, Zhong L, Wang C-Y, Cai W-W, Xu X-M, Xu A-L, Zhang Y-P (2004) Phylogeographic analysis of mitochondrial DNA haplogroup F2 in China reveals T12338C in the initiation codon of the ND5 gene not to be pathogenic. J Hum Genet 49:414–423
Lee HY, Yoo JE, Park MJ, Chung U, Shin KJ (2006) Mitochondrial DNA control region sequences in Koreans: identification of useful variable sites and phylogenetic analysis for mtDNA data quality control. Int J Legal Med 120:5–14
Li R, Ishikawa K, Deng J-H, Heman-Ackah S, Tamagawa Y, Yang L, Bai Y, Ichimura K, Guan M-X (2005a) Maternally inherited nonsyndromic hearing loss is associated with the T7511C mutation in the mitochondrial tRNASerUCN gene in a Japanese family. Biochem Biophys Res Commun 328:32–37
Li R, Xing G, Yan M, Cao X, Liu X-Z, Bu X, Guan M-X (2004a) Cosegregation of C-insertion at position 961 with the A1555G mutation of the mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. Am J Med Genet A 124:113–117
Li X, Fischel-Ghodsian N, Schwartz F, Yan Q, Friedman RA, Guan M-X (2004b) Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res 32:867–877
Li Z, Li R, Chen J, Liao Z, Zhu Y, Qian Y, Xiong S, Heman-Ackah S, Wu J, Choo DI, Guan M-X (2005b) Mutational analysis of the mitochondrial 12S rRNA gene in Chinese pediatric subjects with aminoglycoside-induced and non-syndromic hearing loss. Hum Genet 117:9–15
Loogväli EL, Roostalu U, Malyarchuk BA, Derenko MV, Kivisild T, Metspalu E, Tambets K, Reidla M, Tolk H-V, Parik J, Pennarun E, Laos S, Lunkina A, Golubenko M, Barać L, Peričić M, Balanovsky OP, Gusar V, Khusnutdinova EK, Stepanov V, Puzyrev V, Rudan P, Balanovska EV, Grechanina E, Richard C, Moisan JP, Chaventré A, Anagnou NP, Pappa KI, Michalodimitrakis EN, Claustres M, Gölge M, Mikerezi I, Usanga E, Villems R (2004) Disuniting uniformity: a pied cladistic canvas of mtDNA haplogroup H in Eurasia. Mol Biol Evol 21:2012–2021
Man PYW, Howell N, Mackey DA, Nørby S, Rosenberg T, Turnbull DM, Chinnery PF (2004) Mitochondrial DNA haplogroup distribution within Leber hereditary optic neuropathy pedigrees. J Med Genet 41:e41
McFarland R, Taylor RW, Elson JL, Lightowlers RN, Turnbull DM, Howell N (2004) Proving pathogenicity: when evolution is not enough. Am J Med Genet A 131:107–108
Mitchell AL, Elson JL, Howell N, Taylor RW, Turnbull DM (2006) Sequence variation in mitochondrial complex I genes: mutation or polymorphism? J Med Genet 43:175–179
Monson KL, Miller KWP, Wilson MR, DiZinno JA, Budowle B (2002) The mtDNA Population Database: an integrated software and database resource for forensic comparison. Forensic Sci Commun 4, no. 2. (http://www.fbi.gov/hq/lab/fsc/backissu/april2002/miller1.htm)
Neel JV (1962) Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14:353–362
Palanichamy Mg, Sun C, Agrawal S, Bandelt H-J, Kong Q-P, Khan F, Wang C-Y, Chaudhuri TK, Palla V, Zhang Y-P (2004) Phylogeny of mitochondrial DNA macrohaplogroup N in India, based on complete sequencing: implications for the peopling of South Asia. Am J Hum Genet 75:966–978
Qian Y, Zhou X, Hu Y, Tong Y, Li R, Lu F, Yang H, Mo JQ, Qu J, Guan M-X (2005) Clinical evaluation and mitochondrial DNA sequence analysis in three Chinese families with Leber’s hereditary optic neuropathy. Biochem Biophys Res Commun 332:614–621
Qu J, Li R, Tong Y, Hu Y, Zhou X, Qian Y, Lu F, Guan M-X (2005) Only male matrilineal relatives with Leber’s hereditary optic neuropathy in a large Chinese family carrying the mitochondrial DNA G11778A mutation. Biochem Biophys Res Commun 328:1139–1145
Rao L, Wu MY, Liang WB, Zhang L (2003) Sequence polymorphisms of the mitochondrial DNA control region in 105 Chinese Han population. J Forensic Sci 48:891–895
Richards M, Macaulay V, Torroni A, Bandelt H-J (2002) In search of geographical patterns in European mitochondrial DNA. Am J Hum Genet 71:1168–1174
Rocha H, Flores C, Campos Y, Arenas J, Vilarinho L, Santorelli FM, Torroni A (1999) About the “pathological” role of the mtDNA T3308C mutation.... Am J Hum Genet 65:1457–1459
Röhl A, Brinkmann B, Forster L, Forster P (2001) An annotated mtDNA database. Int J Legal Med 115:29–39
Salas A, Carracedo Á, Macaulay V, Richards M, Bandelt H-J (2005a) A practical guide to mitochondrial DNA error prevention in clinical, forensic, and population genetics. Biochem Biophys Res Commun 335:891–899
Salas A, Richards M, De la Fé T, Lareu MV, Sobrino B, Sánchez-Diz P, Macaulay V, Carracedo Á (2002) The making of the African mtDNA landscape. Am J Hum Genet 71:1082–1111; erratum (http://www.stats.gla.ac.uk/~vincent/bantu/errata.html)
Salas A, Richards M, Lareu MV, Scozzari R, Coppa A, Torroni A, Macaulay V, Carracedo Á (2004) The African diaspora: mitochondrial DNA and the Atlantic slave trade. Am J Hum Genet 74:454–465
Salas A, Yao Y-G, Macaulay V, Vega A, Carracedo Á, Bandelt H-J (2005b) A critical reassessment of the role of mitochondria in tumorigenesis. PloS Med 2:e296
Tanaka M, Cabrera VM, González AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M, Shinoda K, Umetsu K, Yamada Y, Oshida Y, Sato Y, Hattori N, Mizuno Y, Arai Y, Hirose N, Ohta S, Ogawa O, Tanaka Y, Kawamori R, Shamoto-Nagai M, Maruyama W, Shimokata H, Suzuki R, Shimodaira H (2004) Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res 14:1832–1850
Tessa A, Giannotti A, Tieri L, Vilarinho L, Marotta G, Santorelli FM (2001) Maternally inherited deafness associated with a T1095C mutation in the mDNA. Eur J Hum Genet 9:147–149
Thyagarajan D, Bressman S, Bruno C, Przedborski S, Shanske S, Lynch T, Fahn S, DiMauro S (2000) A novel mitochondrial 12SrRNA point mutation in Parkinsonism, deafness, and neuropathy. Ann Neurol 48:730–736
Torroni A, Cruciani F, Rengo C, Sellitto D, López-Bigas N, Rabionet R, Govea N, López De Munain A, Sarduy M, Romero L, Villamar M, del Castillo I, Moreno F, Estivill X, Scozzari R (1999) The A1555G mutation in the 12S rRNA gene of human mtDNA: recurrent origins and founder events in families affected by sensorineural deafness. Am J Hum Genet 65:1349–1358
Torroni A, Petrozzi M, D’Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, De Negri A, Scozzari R (1997) Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet 60:1107–1121
Wang Q, Li R, Zhao H, Peters JL, Liu Q, Yang L, Han D, Greinwald JH Jr., Young W-Y, Guan M-X (2005) Clinical and molecular characterization of a Chinese patient with auditory neuropathy associated with mitochondrial 12S rRNA T1095C mutation. Am J Med Genet A 133:27–30
Yao Y-G, Bravi CM, Bandelt H-J (2004a) A call for mtDNA data quality control in forensic science. Forensic Sci Int 141:1–6
Yao Y-G, Kong Q-P, Bandelt H-J, Kivisild T, Zhang Y-P (2002a) Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am J Hum Genet 70:635–651
Yao Y-G, Kong Q-P, Man X-Y, Bandelt H-J, Zhang Y-P (2003a) Reconstructing the evolutionary history of China: a caveat about inferences drawn from ancient DNA. Mol Biol Evol 20:214–219
Yao Y-G, Kong Q-P, Wang C-Y, Zhu C-L, Zhang Y-P (2004b) Different matrilineal contributions to genetic structure of ethnic groups in the silk road region in china. Mol Biol Evol 21:2265–2280
Yao Y-G, Kong Q-P, Zhang Y-P (2002b) Mitochondrial DNA 5178A polymorphism and longevity. Hum Genet 111:462–463
Yao Y-G, Macaulay V, Kivisild T, Zhang Y-P, Bandelt H-J (2003b) To trust or not to trust an idiosyncratic mitochondrial data set. Am J Hum Genet 72:1341–1346; 1348–1349
Yao Y-G, Zhang Y-P (2003) Pitfalls in the analysis of ancient human mtDNA. Chinese Sci Bull 48:826–830
Young W-Y, Zhao L, Qian Y, Wang Q, Li N, Greinwald JH Jr, Guan M-X (2005) Extremely low penetrance of hearing loss in four Chinese families with the mitochondrial 12S rRNA A1555G mutation. Biochem Biophys Res Commun 328:1244–1251
Yuan H, Qian Y, Xu Y, Cao J, Bai L, Shen W, Ji F, Zhang X, Kang D, Mo JQ, Greinwald JH, Han D, Zhai S, Young W-Y, Guan M-X (2005) Cosegregation of the G7444A mutation in the mitochondrial COI/tRNA(Ser(UCN)) genes with the 12S rRNA A1555G mutation in a Chinese family with aminoglycoside-induced and nonsyndromic hearing loss. Am J Med Genet A 138A:133–140
Zhang Y, Xu Q, Cui H, Cui Y, Lin H, Kim K, Lee J (2005) Haplotype diversity in mitochondrial DNA hypervariable region I, II and III in a Korean ethnic group from northeast China. Forensic Sci Int 151:299–301
Zhao H, Li R, Wang Q, Yan Q, Deng J-H, Han D, Bai Y, Young W-Y, Guan M-X (2004a) Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am J Hum Genet 74:139–152
Zhao L, Wang Q, Qian Y, Li R, Cao J, Hart LC, Zhai S, Han D, Young W-Y, Guan M-X (2005) Clinical evaluation and mitochondrial DNA sequence analysis in two Chinese families with aminoglycoside-induced and non-syndromic hearing loss. Biochem Biophys Res Commun 336:967–973
Zhao L, Young W-Y, Li R, Wang Q, Qian Y, Guan M-X (2004b) Clinical evaluation and sequence analysis of the complete mitochondrial genome of three Chinese patients with hearing impairment associated with the 12S rRNA T1095C mutation. Biochem Biophys Res Commun 325:1503–1508
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yao, YG., Salas, A., Bravi, C.M. et al. A reappraisal of complete mtDNA variation in East Asian families with hearing impairment. Hum Genet 119, 505–515 (2006). https://doi.org/10.1007/s00439-006-0154-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-006-0154-9