
Abstract
Charcot-Marie-Tooth disease (CMT) is one of the most common inherited neurological disorders with a prevalence estimated at 1/2500. The axonal form of this disorder is referred to as Charcot-Marie-Tooth type 2 disease (CMT2). Recently, a large Chinese family with CMT2 was found in the Hunan and Hubei provinces of China. The known loci for CMT1A, CMT2D, CMT1B (the same locus is also responsible for CMT2I and CMT2J), CMT2A, CMT2E, and CMT2F were excluded in this family by linkage analysis. A genome-wide screening was then carried out, and the results revealed linkage of CMT2 to a locus at chromosome 12q24. Haplotype construction and analyses localized this novel locus to a 6.8-cM interval between microsatellite markers D12S366 and D12S1611. The maximal two-point LOD score of 6.35 and multipoint LOD score of 8.08 for marker D12S76 at a recombination fraction (θ) of 0 strongly supported linkage to this locus. Thus, CMT2 neuropathy in this family represents a novel genetic entity that we have designated as CMT2L.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Charcot-Marie-Tooth disease (CMT) is one of the most common inherited neurological disorders with a prevalence estimated at 1/2500 (Skre 1974) and is clinically characterized by distal weakness and atrophy of the limb muscles, mild sensory loss, and absence of tendon reflexes. On the basis of electrophysiological properties and histopathology, CMT has been divided into two main types: one is the demyelinating type consisting of CMT1 (autosomal dominant form) and CMT4 (autosomal recessive form), and the other is the axonal type CMT2 (both autosomal dominant and autosomal recessive forms; Dyck and Lambert 1968; Buchthal and Behse 1977; Harding and Thomas 1980). CMT1 and CMT4 are distinguished from axonal forms by reduced nerve-conduction velocities (NCVs) with values of less than 38m/s for the median motor nerve, segmental de- and remyelination, and onion bulb formation. CMT2 shows normal or only slightly reduced NCVs. Nerve pathology reveals axonal loss and regenerative sprouting.
At present, four loci responsible for autosomal dominant CMT1 have been mapped, and all corresponding causative genes have been identified. CMT1A is mainly associated with a 1.5-Mb duplication on chromosome 17p11.2–12 (Lupski et al. 1991; Raeymaekers et al. 1991) with a gene-dosage effect for the peripheral myelin protein 22 (PMP22) gene (Matsunami et al. 1992; Patel et al. 1992; Timmerman et al. 1992; Valentijin et al. 1992b; Warner et al. 1996). PMP22 point mutations have been found in rare CMT1A patients lacking the duplication (Valentijn et al. 1992a; Fabrizi et al. 1999). CMT1B is caused by point mutations in the myelin protein zero (MPZ) gene at 1q21.3-q23 (for a review, see Lupski 1998). CMT1C is linked to chromosome 16p13.1-p12.3 and is associated with mutations in the lipopolysaccharide-induced tumor necrosis factor (LITAF) gene, also known as the “small integral membrane protein of lysosome/late endosome (SIMPLE) gene” (Street et al. 2003). CMT1D, which has been mapped to chromosome 10, is associated with mutations in the early growth response 2 (EGR2) gene, also known as “Krox-20” (Warner et al. 1998).
Four CMT2 entities have been confirmed to be caused by mutations in different genes; interestingly, these genes are also responsible for demyelinating CMT. Autosomal dominant CMT2I and CMT2J result from mutations in the MPZ gene (Boerkoel et al. 2002; Misu et al. 2000), which is also responsible for CMT1B. The S194X mutation in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene is associated with both autosomal recessive CMT2G and CMT2K (Cuesta et al. 2002; Birouk et al. 2003), and the same gene mutation also results in the autosomal recessive demyelinating CMT phenotype CMT4A (Baxter et al. 2002). To date, among seven loci specific for axonal forms only, four genes have been identified: the kinesin superfamily (KIF1B) gene for CMT2A (Zhao et al. 2001), the RAS-related GTP-binding protein 7 (RAB7) gene for CMT2B (Verhoeven et al. 2003), the glysyl tRNA synthetase (GARS) gene for CMT2D (Antonellis et al. 2003), and the neurofilament light chain (NEFL) gene for CMT2E (Mersiyanova et al. 2000; De Jonghe et al. 2001). The other three loci include CMT2C at chromosome 12q23-q24 (Klein et al. 2003), CMT2F on 7q11-q21 (Ismailov et al. 2001), and a form of hereditary motor and sensory neuropathy with dominant proximal involvement at 3q13.1 (Takashima et al. 1999).
In CMT1, the demyelination is not the direct cause of muscle atrophy and weakness but, instead, appears to initiate cytoskeleton modification and other changes in axons that finally result in axonal loss (Sahenk 1999; Scherer 1999). In view of this, the identification of genes directly involved in axonal damage in CMT2 may supply us with a more direct path toward understanding the pathogenesis of CMT. In the study presented here, we report a Chinese family with autosomal dominant CMT2 that shows linkage to a new locus that lies on 12q24 and that represents a novel genetic entity that we have designated as CMT2L.
Materials and methods
A large six-generation family from the Hunan and Hubei provinces of China with autosomal-dominant CMT2 were studied. Twenty six members of the family were clinically investigated, and six affected individuals were electrophysiologically diagnosed.
Genomic DNA of all 26 family members was isolated from peripheral blood samples by standard techniques. A genome-wide screening was performed by using 382 microsatellite markers from the ABI Prism Linkage Mapping Set Version 2 (PE Applied Biosystems, Foster City, Calif., USA). Information concerning additional markers to confirm and refine the interval was accessed through the Genome Database. Multiplexed polymerase chain reaction products were separated by capillary electrophoresis in an ABI PRISM 3100 Automated sequencer (PE Applied Biosystems). Microsatellite marker-allele data were analyzed by GENESCAN version 3.0 and GENOTYPER version 2.1 (PE Applied Biosystems).
Two-point and multipoint linkage analyses were performed with the MLINK and LINKMAP programs of the LINKAGE package (version 5.1; Lathrop et al. 1984). CMT2L was assumed to be an autosomal dominant trait with full penetrance. The disease frequency was set at 0.0001. Equal male and female recombination rates were assumed. Marker allele frequencies were set at 1/n, where n is the number of alleles observed. For multipoint analysis, the genetic distances between loci were obtained from the Marshfield sex-averaged linkage map (http://research.marshfieldclinic.org/genetics/Map_Markers/maps/IndexMapFrames.html) and converted to recombination fractions by using the Kosambi map function. Haplotypes were assigned on the basis of the minimization of number of recombinations.
The study was approved by the Expert Committee (equivalent to an Institutional Review Board) of Xiangya Hospital of the Central South University in China, and informed consent was obtained from all family members.
Results
Clinical information
Eighteen individuals were diagnosed as CMT2. Onset of the disease was between 15 and 33 years of age with weakness of the lower limbs. Two patients had weakness and atrophy in both proximal and distal muscles of the lower limbs; all other patients presented a typical CMT2 phenotype, i.e., symmetrical muscle wasting and a predominating weakness of the distal parts of the lower limbs, decreased or absent deep tendon reflexes, and mild to moderate sensory impairment including pain and touch. No evidence of painless injury or ulceration was seen in any of the patients. Six patients who initially presented with lower limb involvement demonstrated variable distal weakness and wasting in their upper extremities. Pes cavus presented in 14 patients. Three affected members of the family also had scoliosis. We noted that all the patients became severely affected only in advanced age, but even the elderly patients never became wheelchair-dependent.
In six patients who were subjected to electrophysiological examinations, the NCV of the median nerve was normal, ranging from 56.7 to 69.2 m/s (mean: 64.8 m/s). Sensory nerve action potentials were decreased (not more than 23.2 μV in the median nerve) or absent in all the six patients. Electromyograms revealed signs of denervation with large motor unit potentials, fibrillation potentials, and positive sharp waves. A superficial peroneal nerve biopsy for patient IV:13 confirmed the presence of axonal neuropathy with an important loss of large myelinating fibers and a large number of clusters with mostly thinly myelinated axons. These clinical and electrophysiological findings supported the diagnosis of CMT2.
Linkage analysis
Prior to performing a genome-wide screening of the family, the known loci of CMT1A, CMT2D, CMT1B, CMT2A, CMT2E, and CMT2F were excluded by linkage analysis (data not shown). A genome screen was subsequently undertaken by using markers spaced, on average, at 10-cM intervals. Linkage was established to chromosome 12q24 when a significant LOD score was obtained with the consecutive marker D12S86 (Table 1). No other markers analyzed from the genome screen gave a LOD score ≥3.0. Ten additional markers around D12S86 were tested in the family. The results of the two-point analysis between the disease phenotype and the additional marker loci are shown in the Table 1. Multipoint linkage analysis with markers D12S366–D12S76–D12S1611 resulted in a maximum LOD score of 8.08 at D12S76 (Fig. 1).

Multipoint linkage analysis at markers D12S366-D12S76-D12S1611. The maximal LOD score of 8.08 is reached at D12S76. The genetic distances between the markers are in centimorgans
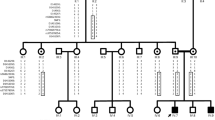
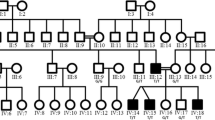
The most probable disease haplotype was constructed according to the order of the Marshfield genetic map and the National Center for Biotechnology Information physical map, build 34 (Fig. 2). A particular haplotype associated with the disease was found in all affected members of the family. Three key recombinants were identified. The recombined haplotype of patients V:1, V:2, V:4, and V:5 suggested recombination between markers D12S366 and D12S1619 during meiosis in III:2, their grandmother. This localized the CMT2L gene telomeric to marker D12S366. This site was also supported by another recombination found in healthy individual V:7 (currently 41 years old) who inherited alleles of three centromeric markers D12S2082, D12S1720, and D12S366 from the linked haplotype of the affected mother IV:5, also thereby localizing the disease-causing gene telomeric to D12S366. Alleles of the following three markers D12S1619, D12S86, and D12S1666 may also have come from the same haplotype, but their origin could not be traced with certainty. The third key recombinantion was deduced from patient IV:1. Although the exact haplotype of her affected mother III:2 was unknown, it could be deduced that crossing-over had occurred centromeric to D12S1611. Combining the information of these three recombinantions located the CMT2L gene to a 6.8-cM region between markers D12S366 and D12S1611 (Fig. 3).

Halpotype analysis of the Chinese CMT2 family (filled symbols affected individuals, open symbols unaffected individuals). The haplotype segregating with the disease is boxed, the haplotypes in the dotted boxes (individuals IV:1 and V:7) are discussed in the text, and the haplotypes in brackets are deduced

Genetic map (sex-averaged) of the chromosome 12 markers used in this study. The genetic distances were obtained from the Marshfield map. Loci that appear on the same line map to the same genetic location. The order of these markers was obtained from the chromosome 12 q-arm metric physical map. Markers defining the CMT2L genetic interval are in bold
In the Human Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway), more than 70 genes have been mapped between D12S366 and D12S1611, four of them functionally similar or related to known CMT genes.
Discussion
The family reported here shows autosomal dominant axonal CMT syndrome. Except for the finding that two patients had weakness and atrophy in both proximal and distal muscles of the lower limbs, all other patients presented a typical CMT2 phenotype. The involvement of proximal muscles is rare in hereditary neuropathies, except in motor neuronopathies. Furthermore, this is considered as an exclusion criterion when it is restricted to these muscles (De Jonghe et al. 1998). However, some cases have been reported with autosomal dominant or autosomal recessive CMT1 and CMT2 and proximal muscles involvement (Bird et al. 1997; Bolino et al. 2000; Bouhouche et al. 1999; Gabreels-Festen et al. 1991). The way in which the same mutation can result in the involvement of proximal muscles in some patients, while leaving these muscles intact in others, is unclear.
Loci for several neuromuscular diseases have been linked to chromosome 12 in the vicinity of CMT2L, including scapuloperoneal spinal muscular atrophy (SPSMA; 12q24.1-q24.31; Isozumi et al. 1996), congenital distal spinal muscular atrophy (SMAL; 12q23-q24; van der Vleuten et al. 1998), CMT2C (12q23-q24; Klein et al. 2003), and distal hereditary motor neuropathy type II (distal HMN II; 12q24; Timmerman et al. 1996). The first three loci are at least 10 cM centromeric to CMT2L, whereas the minimal genetic region for distal HMN II overlaps the region of CMT2L by 5 cM. Because patients with CMT2C have special clinical features, i.e., diaphragmatic, vocal cord, and intercostal paralysis, whereas no diaphragm or vocal fold weakness were seen in our reported family, we consider CMT2C to be a distinct entity, both clinically and genetically. SPSMA, SMAL, and distal HMN II are characterized as spinal amyotrophies. Clinically, they are distinguished by several features, most notably by the absence of sensory involvement. However, patients with CMT2L show sensory involvement as revealed by physical examination, electrophysiology, and neural pathology. Therefore, we consider that SPSMA, SMAL, and distal HMN II are distinct diseases. Nonetheless, that the genes responsible for these diseases, especially for distal HMN II, are allelic to CMT2L remains a possibility. A large Mongolian family with a conserved haplotype and variable phenotype characterized by both spinal amyotrophy (dSMAV) and CMT2D has been reported (Antonellis et al. 2003). Therefore, it is still possible that these diseases can be explained by a common genetic abnormality.
Further studies are needed to demonstrate the specific gene alteration in this new genetic form of autosomal dominant CMT2 in order to bring possible new insights to the understanding of the pathogenic mechanism(s) involved in these degenerative diseases.
References
Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED (2003) Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72:1293–1299
Baxter RV, Ben Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, Hentati F, Ben Hamida M, Bel S, Stenger JE, Gilbert JR, Pericak-Vance MA, Vance JM (2002) Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet 30:21–22
Bird TD, Kraft GH, Lipe HP, Kenney KL, Sumi SM (1997) Clinical and pathological phenotype of the original family with Charcot-Marie-Tooth type 1B: a 20-year study. Ann Neurol 41:463–469
Birouk N, Azzedine H, Dubourg O, Muriel MP, Benomar A, Hamadouche T, Maisonobe T, Ouazzani R, Brice A, Yahyaoui M, Chkili T, Le Guern E (2003) Phenotypical features of a Moroccan family with autosomal recessive Charcot-Marie-Tooth disease associated with the S194X mutation in the GDAP1 gene. Arch Neurol 60:598–604
Boerkoel CF, Takashima H, Garcia CA, Olney RK, Johnson J, Berry K, Russo P, Kennedy S, Teebi AS, Scavina M, Williams LL, Mancias P, Butler IJ, Krajewski K, Shy M, Lupski JR (2002) Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann Neurol 51:190–201
Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MA, Georgiou DM, Christodoulou K, Hausmanowa-Petrusewicz I, Mandich P, Schenone A, Gambardella A, Bono F, Quattrone A, Devoto M, Monaco AP (2000) Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet 25:17–19
Bouhouche A, Benomar A, Birouk N, Mularoni A, Meggouh F, Tassin J, Grid D, Vandenberghe A, Yahyaoui M, Chkili T, Brice A, LeGuern E (1999) A locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 1q21.2-q21.3. Am J Hum Genet 65:722–727
Buchthal F, Behse F (1977) Peroneal muscular atrophy (PMA) and related disorders. I. Clinical manifestations as related to biopsy findings, nerve conduction and electromyography. Brain 100:41–66
Cuesta A, Pedrola L, Sevilla T, Garcia-Planells J, Chumillas MJ, Mayordomo F, LeGuern E, Marin I, Vilchez JJ, Palau F (2002) The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30:22–25
De Jonghe P, Timmerman V, Van Broeckhoven C (1998) 2nd Workshop of the European CMT Consortium: 53rd ENMC International Workshop on Classification and Diagnostic Guidelines for Charcot-Marie-Tooth Type 2 (CMT2-HMSN II) and Distal Hereditary Motor Neuropathy (distal HMN-Spinal CMT), 26–28 September 1997, Naarden, The Netherlands. Neuromuscul Disord 8:426–431
De Jonghe P, Mersivanova I, Nelis E, Del Favero J, Martin JJ, Van Broeckhoven C, Evgrafov O, Timmerman V (2001) Further evidence that neurofilament light chain gene mutations can cause Charcot-Marie-Tooth disease type 2E. Ann Neurol 49:245–249
Dyck PJ, Lambert EH (1968) Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 18:603–618
Fabrizi GM, Cavallaro T, Taioli F, Orrico D, Morbin M, Simonati A, Rizzuto N (1999) Myelin uncompaction in Charcot-Marie-Tooth neuropathy type 1A with a point mutation of peripheral myelin protein-22. Neurology 53:846–851
Gabreels-Festen AA, Joosten EM, Gabreels FJ, Jennekens FG, Gooskens RH, Stegeman DF (1991) Hereditary motor and sensory neuropathy of neuronal type with onset in early childhood. Brain 114:1855–1870
Harding AE, Thomas PK (1980) The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 103:259–280
Ismailov SM, Fedotov VP, Dadali EL, Polyakov AV, Van Broeckhoven C, Ivanov VI, De Jonghe P, Timmerman V, Evgrafov OV (2001) A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2F) maps to chromosome 7q11-q21. Eur J Hum Genet 9:646–650
Isozumi K, DeLong R, Kaplan J, Deng HX, Iqbal Z, Hung WY, Wilhelmsen KC, Hentati A, Pericak-Vance MA, Siddique T (1996) Linkage of scapuloperoneal spinal muscular atrophy to chromosome 12q24.1-q24.31. Hum Mol Genet 5:1377–1382
Klein CJ, Cunningham JM, Atkinson EJ, Schaid DJ, Hebbring SJ, Anderson SA, Klein DM, Dyck PJ, Litchy WJ, Thibodeau SN, Dyck PJ (2003) The gene for HMSN2C maps to 12q23–24: a region of neuromuscular disorders. Neurology 60:1151–1156
Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446
Lupski JR (1998) Charcot-Marie-Tooth disease: lessons in genetic mechanisms. Mol Med 4:3–11
Lupski JR, Montes de Oca-Luna R, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI (1991) DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66:219–232
Matsunami N, Smith B, Ballard L, William Lensch M, Robertson M, Albertsen H, Oliver Hanemann C, Muller HW, Bird TD, White R, Chance PF (1992) Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot-Marie-Tooth 1A. Nat Genet 1:176–179
Mersiyanova IV, Perepelov AV, Polyakov AV, Sitnikov VF, Dadali EL, Oparin RB, Petrin AN, Evgrafov OV (2000) A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am J Hum Genet 67:37–46
Misu K, Yoshihara T, Shikama Y, Awaki E, Yamamoto M, Hattori N, Hirayama M, Takegami T, Nakashima K, Sobue G (2000) An axonal form of Charcot-Marie-Tooth disease showing distinctive features in association with mutations in the peripheral myelin protein zero gene (Thr124Met or Asp75Val). J Neurol Neurosurg Psychiatry 69:806–811
Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask BJ, Pentao L, Jackson Snipes G, Garcia CA, Francke U, Shooter EM, Lupski JR, Suter U (1992) The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat Genet 1:159–165
Raeymaekers P, Timmerman V, Nelis E, DeJonghe P, Hoogendijk JE, Baas F, Barker DF, Martin JJ, De Visser M, Bolhuis PA, Van Broeckhoven C (1991) Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). Neuromuscul Disord 1:93–97
Sahenk Z (1999) Abnormal Schwann cell-axon interactions in CMT neuropathies. The effects of mutant Schwann cells on the axonal cytoskeleton and regeneration-associated myelination. Ann N Y Acad Sci 883:415–426
Scherer S (1999) Axonal pathology in demyelinating disease. Ann Neurol 41:771–780
Skre H (1974) Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet 6:98–118
Street VA, Bennett CL, Goldy JD, Shirk AJ, Kleopa KA, Tempel BL, Lipe HP, Scherer SS, Bird TD, Chance PF (2003) Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology 60:22–26
Takashima H, Nakagawa M, Suehara M, Saito M, Saito A, Kanzato N, Matsuzaki T, Hirata K, Terwilliger JD, Osame M (1999) Gene for hereditary motor and sensory neuropathy (proximal dominant form) mapped to 3q13.1. Neuromuscul Disord 9:368–371
Timmerman V, Nelis E, Van Hul W, Nieuwenhuijsen BW, Chen KL, Wang S, Ben Othman K, Cullen B, Leach RJ, Hanemann CO, De Jonghe P, Raeymaekers P, Ommen G-JB van, Martin J-J, Muller HW, Vance JM, Fischbeck KH, Van Broeckhoven C (1992) The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat Genet 1:171–175
Timmerman V, De Jonghe P, Simokovic S, Lofgren A, Beuten J, Nelis E, Ceuterick C, Martin JJ, Van Broeckhoven C (1996) Distal hereditary motor neuropathy type II (distal HMN II): mapping of a locus to chromosome 12q24. Hum Mol Genet 5:1065–1069
Valentijn LJ, Baas F, Wolterman RA, Hoogendijk JE, Bosch NHA van den, Zorn I, Gabreels-Festen AAWM, Visser M de, Bolhuis PA (1992a) Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1A. Nat Genet 2:288–291
Valentijn LJ, Bolhuis PA, Zorn I, Hoogendijk JE, Bosch N van den, Hensels GW, Stanton VPJ, Housman DE, Fischbeck KH, Ross DA, Nicholson GA, Meershoek EJ, Dauwerse HG, Ommen G-JB van, Baas F (1992b) The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nat Genet 1:166–170
Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, FitzPatrick D, Schmedding E, De Vriendt E, Jacobs A, Van Gerwen V, Wagner K, Hartung H-P, Timmerman V (2003) Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 72:722–727
Vleuten AJ van der, Ravenswaaij-Arts CM van, Frijns CJ, Smits AP, Hageman G, Padberg GW, Kremer H (1998) Localisation of the gene for a dominant congenital spinal muscular atrophy predominantly affecting the lower limbs to chromosome 12q23-q24. Eur J Hum Genet 6:376–382
Warner LE, Roa BB, Lupski JR (1996) Absence of PMP22 coding region mutations in CMT1A duplication patients: further evidence supporting gene dosage as a mechanism for Charcot-Marie-Tooth disease type 1A. Hum Mutat 8:362–365
Warner LE, Mancias P, Butler IJ, McDonald CM, Keppen L, Gene Koob K, Lupski JR (1998) Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet 18:382–384
Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N (2001) Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 105:587–597
Acknowledgements
We thank the participating families for their cooperation throughout this study. This study was co-funded by a grant from the National 863 High-tech Project (2001AA227011, 2002AA711A07) and National Natural Science Foundation of China (grant nos. 39900047 and 30300200). We are also grateful to Vincent Timmerman (University of Antwerp, Belgium) who kindly donated some of the primers for exclusion analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tang, Bs., Luo, W., Xia, K. et al. A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2L) maps to chromosome 12q24. Hum Genet 114, 527–533 (2004). https://doi.org/10.1007/s00439-004-1102-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-004-1102-1