
Abstract
Hemophilia B is an X-linked recessive bleeding disorder caused by abnormalities of the coagulation factor IX gene (F9). Insertion mutations in F9 ranging from a few to more than 100 base pairs account for only a few percent of all hemophilia B cases. We investigated F9 to elucidate genetic abnormalities causing severe hemophilia B in a Japanese subject. We performed PCR-mediated analysis of F9 and identified a large insertion in exon 6. Next, we carried out direct sequencing of a PCR clone of the whole insert using nested deletion by exonuclease III and S1 nuclease. We identified an approximately 2.5-kb SINE-VNTR-Alu (SVA)-F element flanked by 15-bp duplications in the antisense orientation in exon 6. Additionally, we carried out exontrap analysis to assess the effect of this retrotransposition on mRNA splicing. We observed that regular splicing at exons 5 and 6 of F9 was disturbed by the SVA retrotransposition, suggesting that abnormal FIX mRNA may be reduced by nonsense-mediated mRNA decay. In conclusion, this is the first report of SVA retrotransposition causing severe hemophilia B; only five cases of LINE-1 or Alu retrotranspositions in F9 have been reported previously.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hemophilia B is an X-linked recessive bleeding disorder occurring at a frequency of one in 25,000–30,000 male births, which is about five times less prevalent than Hemophilia A. Hemophilia B is caused by abnormalities of the coagulation factor IX gene (F9), which contains eight exons, spans 33.5-kb and is located at the distal end of the long arm of the X chromosome [1].
The coagulation factor IX (FIX) is a vitamin K-dependent serine protease and plays an important role in blood coagulation. Depending on residual FIX activity, hemophilia B is classified as severe (<1 %), moderate (1–5 %) or mild (<5–30 %) [2], and the phenotypic severity is reported to be related to the type and position of the mutations. These mutations include point mutations (missense/nonsense), deletions, insertions and splicing defects. Generally, the hemophilia B cases caused by insertions ranging from a few to more than 100 base pairs are rare and account for <5 % of all hemophilia B cases [3].
In this study, we carried out genetic analysis of F9 from a Japanese patient with hemophilia B and found a >2 kb insertion containing SINE-VNTR-Alu (SVA)-F element in exon 6. This insertion consisted of an SINE-VNTR-Alu (SVA) retrotransposon in the antisense orientation, resulting in a severe reduction of FIX activity. Furthermore, we conducted exontrap analysis to confirm the effect of this retrotransposon on an abnormal mRNA splicing. This is the first report of an SVA retrotransposition in F9 causing severe hemophilia B.
Materials and methods
Patient and DNA sample
The proband was a 1-year-old Japanese boy diagnosed with severe hemophilia B (FIX activity <1 %). He had developed a FIX inhibitor and displayed urticaria but not anaphylaxis. He had also shown proteinuria but not nephrotic syndrome. There was no hemophiliac in his family history, suggesting that he was a sporadic case of hemophilia B. The FIX activity of his mother was not checked. This study was approved by the institutional committee for research ethics and the genomic DNA sample was isolated from peripheral blood leukocytes using established methods after written informed consent from his parents was obtained [4].
F9-specific polymerase chain reaction (PCR), long-range PCR and DNA sequencing
We amplified all F9 exons and intron–exon junctions by polymerase chain reaction (PCR) using gene-specific primers as previously described [5]. PCR products were separated and the size of the amplified products was confirmed using electrophoregram with 1.5 % agarose gel. Because a band of the correct size in exon 6 was not observed, we performed long-range PCR amplification using upper and lower primers targeted to exons 5 and 6, respectively. We purified each PCR product and determined its DNA sequence as previously described [6].
Sequencing of nested deletion inserts using exonuclease III and S1 nuclease
Because we were not able to obtain the complete sequence of the large insertion via conventional sequencing methods, we cloned a long-range PCR product containing the insertion into pBluescript KS+ vector and prepared variable deletion mutants using exonuclease III (New England BioLab Japan, Inc., Tokyo, Japan) and S1 nuclease (Promega, Madison, WI) as previously described [7]. Next, we sequenced purified plasmid inserts using predesigned primers for the pBluescript KS+ vector or designed primers on newly identified regions (Table 1).
Exontrap analysis
To verify whether the large insertion in exon 6 interfered with normal mRNA splicing, we performed exontrap analysis using an exontrap cloning vector (MoBiTec, GmbH, Germany). The exontrap vector includes a 5′-exon, a 600-bp intron sequence with a multiple cloning site and a 3′-exon. Using genomic DNA samples from the patient and a normal individual, we amplified PCR fragments through exon 5 to exon 6, including intron–exon junctions, using the primer set listed in Table 1. We digested these samples with XhoI and ClaI and inserted the resulting fragments into the exontrap vector. Subsequently, we transfected COS-7 cells with these constructed vector using the calcium phosphate transfection method and cultured the cells for 16 h. After isolation of total RNA from the cells using an RNeasy mini kit (QIAGEN), we performed reverse transcription (RT) of mature mRNAs into cDNAs by PrimeScript RT Master Mix (TaKaRa, Shiga, Japan). We then carried out PCR amplification with a specific primer pair (Table 1), separated PCR products using electrophoregram, purified them and sequenced them using the process described above.
Results and discussion
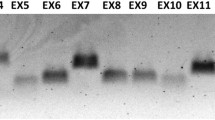
We amplified all F9 exons and intron–exon junctions of the patient’s DNA using F9-specific PCR primers. We were able to obtain accurately amplified products and confirm normal DNA sequences, except for exon 6. The results of exon 6-specific PCR (Fig. 1a) and long-range PCR through exon 5 to exon 6 (Fig. 1b) showed PCR products more than 2-kb larger than that of the normal control. We also performed multiplex ligation-dependent probe amplification (MLPA) to analyze the relative gene dosage values for all F9 exons from the patient as previously described [6], and found an absence of intragenic deletions or duplications in the F9 regions targeted by the designed probes (data not shown). These data revealed a large insertion located at the intron 5–exon 6 junction without causative mutation in any other F9 exon, suggesting that the exon 6 insertion is responsible for deficiency of the FIX activity observed in the patient.

Exon 6-specific PCR and long-range PCR covering exons 5 and 6. a Exon 6-specific PCR. PCR of the patient’s DNA results in a larger product (over 2-kb larger) than that of normal DNA. N normal; P patient. b Long-range PCR covering exons 5 and 6. Again, the PCR product from the patient’s DNA appears over 2-kb larger than that of the normal individual. N normal; P patient. c Locations and sizes of PCR amplicons from the normal F9 gene and from a predicted construct of the F9 gene of the patient based on the obtained sequence data. Upper normal F9; lower patient F9; black short arrows exon 6-specific PCR primers; grey short arrows long-range PCR primers; dup, 15-bp duplication
To sequence the entire exon 6 insertion present in the proband, we prepared variable-length deletion inserts using Exonuclease III and S1 nuclease and determined their sequences. This allowed us to identify a 2,524-bp insertion flanked on both sides by an identical 15-bp sequence (5′-TTCTAGTGCCATTTC-3′), which was intron 5–exon 6 boundary of F9 (Fig. 2a, Supplementary data). The insertion contained a 28-bp poly-T tract at the 5′ end, a GC-rich region, 18 hexamer (AGAGGG) repeats and additional AGAGC residues at the 3′ end. These features characterize an SVA retrotransposon in the antisense orientation and classify the retrotransposon in SVA family F with 15-bp target site duplications (TSDs), according to RepeatMasker (http://www.repeatmasker.org/) (Fig. 2b, c).

Schematic diagram of the SVA insertion in the F9 gene of the patient. a A large, 2,524-bp insertion flanked by 15-bp target site duplications (TSDs) located on the intron 5–exon 6 boundary was identified. b Canonical structure of a full-length, sense-oriented SVA element flanked by TSDs. TSD target site duplication; (CCCTCT)n, hexamer repeat region; Alu-like region with high homology to Alu element; VNTR variable number of tandem repeats; SINE-R short interspersed nucleotide element (retroviral origin); A(n) poly-A tail. c Structural features of the antisense-oriented SVA element sequenced from the F9 gene of the hemophilia B patient
The human genome contains active retrotransposons such as human endonuclease retrovirus (HERV), long interspersed nucleotide elements (LINEs) and short interspersed nucleotide elements (SINEs). There is a smaller retrotransposon family known as SVA (SINE-VNTR-Alu) consisting of SINE, variable number of tandem repeats (VNTRs) and Alu-like sequences, which remains active in the human genome and capable of inducing disease-causing insertions [8].
SVA retrotransposons are increasing in the human genome, even though these elements lack independent mobilization. L1, the only autonomously active retrotransposon, encodes an internal RNA polymerase (pol II) promoter, an RNA binding protein, an endonuclease and a reverse transcriptase [9]. Experimental evidence indicates that SVA retrotransposons are mobilized by L1 elements in human cultured cells [10]. For this reason, SVA elements are considered non-autonomous retrotransposons that are mobilized by L1-encoded machinery in trans.
The copy numbers of retrotransposons in the human genome have been estimated as 516,000 for L1, 1,100,000 for Alu and 2700 for SVA [8]. Retrotransposition frequencies per birth were calculated as 1 in 21 for L1, 1 in 108 for Alu and 1 in 916 for SVA. These transposition events may affect individual phenotype like single nucleotide polymorphisms (SNPs) or copy number variations (CNVs), and cause disease in some cases. A recent report finds 96 retrotransposition events resulting in single-gene diseases, including 25 cases of L1, 60 cases of Alu, 4 cases of poly-A and 7 cases of SVA [11]. Callinan and Batzer [12] reported that retrotransposition events caused about 0.27 % of all human disease and can produce insertions, deletions, genomic rearrangements and recombination between homologous elements. The seven SVA insertions were SVA-F or SVA-E retrotranspositions arranged in sense orientation, except in one case [13]. Analysis of mRNA indicated that the six SVA insertions occurring in sense orientation, regardless of whether they were located in an exon or an intron, caused exon skipping or exonization of SVA using a cryptic splice site within the SVA element; these resulted in several diseases, such as Fukuyama-type congenital muscular dystrophy (FCMD), X-linked agammaglobulinemia and autosomal recessive hypercholesterolemia (ARH) [14–16]. However, an antisense-orientated SVA retrotransposition detected in intron 32 of the TATA box binding protein-associated factor 1 (TAF1) gene showed reduced mRNA or protein levels [17]. SVA elements are likely to be hypermethylated due to their high GC content and the large number of CpG sites they contain.
In the exontrap analysis, the cDNA sample from the vector of a normal individual showed a 578-bp amplicon consisting of exons 5 and 6 and a 449-bp amplicon consisting of exon 6; in both cases, the F9 exons were flanked by the vector exons. In contrast, the cDNA sample from the patient produced abnormal 303-bp and 246-bp amplicons. Direct sequence analysis showed that the smaller amplicon consisted of only vector exons and the larger one contained a 57-bp sequence from intron 6 (c.723 + 1_57) flanked by vector exons (Fig. 3a, b). These results suggest that this insertion could disrupt regular splicing at exons 5 and 6. According to splice site prediction tools (BDGP: Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html/), the prediction score of the splice-donor site in intron 5 (D-int5) was <0.4, suggesting that the procedure successfully trapped exon 6 in the normal cDNA sample (Table 2). The SVA insertion in exon 6 dramatically lowered the prediction score of the downstream intron 5 splice-acceptor site (A-int5R) in the rear 15-bp duplication and newly introduced multiple predicted splice sites (Table 2). It also resulted in an aberrant trapped transcript consisting of a 57-bp sequence of intron 6 (c.723 + 1_57), using A-ex6 as a splice-acceptor site and D-int6# as a splice-donor site (Fig. 3c; Table 2).

Exontrap analysis. a Results of RT-PCR. In normal lane (N), 578-bp (N1) and 449-bp (N2) amplicons were observed. In patient lane (P), 303-bp (P1) and 246-bp (P2) amplicons were observed. b Schematic diagram of amplicon composition. The 578-bp (N1) amplicon resulted from trapping exons 5 and 6, whereas the 449-bp (N2) amplicon trapped only exon 6. The 303-bp fragment (P1) resulted from exon skipping together with trapping of a 57-bp sequence of intron 6 (c.723 + 1_57), whereas the 246-bp amplicon (P2) resulted from no trapping. c Predicted splice sites in the exontrap construct, with and without SVA. Downward-pointing triangles indicate splice-donor sites and upward-pointing triangles indicate splice-acceptor sites, according to splice site prediction tools (BDGP: Splice Site Prediction by Neural Network). Dashed lines denote observed splicing events
So far, there have been five reports of retrotranspositions in F9, of which three were of Alu insertions and two were of LINE-1 (L1) insertions [11]. These cases all involve insertions in exons and are predicted to cause frameshifting of F9 mRNA and premature termination of the FIX protein. However, the causative mechanism for hemophilia B remains unknown because mRNA analysis has not been performed. It is possible that these cases involve exon skipping or exonization due to the presence of cryptic splice sites in the transposed elements. In this study, we detected a TSD at the intron 5–exon 6 boundary of F9, locating the SVA retrotransposon at the beginning of exon 6. We performed exontrap analysis to detect abnormal splicing and confirmed the disturbance of regular splicing of exons 5 and 6 due to the SVA retrotransposon located in the antisense strand. Although this analysis could not precisely replicate in vivo splicing conditions, it is suggested that exon skipping or exonization using unusual splice sites may result in reduced FIX levels via nonsense-mediated mRNA decay [18].
In conclusion, we identified an SVA-F retrotransposon associated with abnormal splicing in exon 6 of F9 from a Japanese hemophilia B patient. This is the first report of SVA retrotransposition in F9 causing severe hemophilia B.
References
Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry. 1985;24:3736–50.
Bolton-Maggs PHB, Pasi KJ. Haemophilias A and B. Lancet. 2003;361:1801–9.
Li T, Miller CH, Payne AB, Craig Hooper W. The CDC hemophilia B mutation project mutation list: a new online resource. Mol Genet Genomic Med. 2013;1:238–45.
Kojima T, Tanimoto M, Kamiya T, Obata Y, Takahashi T, Ohno R, et al. Possible absence of common polymorphisms in coagulation factor IX gene in Japanese subjects. Blood. 1987;69:349–52.
Okumura K, Fujimori Y, Takagi A, Murate T, Ozeki M, Yamamoto K, et al. Skewed X chromosome inactivation in fraternal female twins results in moderately severe and mild haemophilia B. Haemophilia. 2008;14:1088–93.
Kato I, Takagi Y, Ando Y, Nakamura Y, Murata M, Takagi A, et al. A complex genomic abnormality found in a patient with antithrombin deficiency and autoimmune disease-like symptoms. Int J Hematol. 2014;100:200–5.
Takagi A, Kojima T, Tsuzuki S, Katsumi A, Yamazaki T, Sugiura I, et al. Structural organization and promoter activity of the human ryudocan gene. J Biochem. 1996;119:979–84.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, et al. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–70.
Raiz J, Damert A, Chira S, Held U, Klawitter S, Hamdorf M, et al. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012;40:1666–83.
Hancks DC, Kazazian HH. Active human retrotransposons: variation and disease. Curr Opin Genet Dev. 2012;22:191–203.
Callinan PA, Batzer MA. Retrotransposable elements and human disease. Genome Dyn. 2006;1:104–15.
Kaer K, Speek M. Retroelements in human disease. Gene. 2013;518:231–41.
Taniguchi-Ikeda M, Kobayashi K, Kanagawa M, Yu CC, Mori K, Oda T, et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature. 2011;478:127–31.
Conley ME, Partain JD, Norland SM, Shurtleff SA, Kazazian HH. Two independent retrotransposon insertions at the same site within the coding region of BTK. Hum Mutat. 2005;25:324–5.
Wilund KR, Yi M, Campagna F, Arca M, Zuliani G, Fellin R, et al. Molecular mechanisms of autosomal recessive hypercholesterolemia. Hum Mol Genet. 2002;11:3019–30.
Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80:393–406.
Hancks DC, Ewing AD, Chen JE, Tokunaga K, Kazazian HH. Exon-trapping mediated by the human retrotransposon SVA. Genome Res. 2009;19:1983–91.
Acknowledgments
The authors thank Ms. C. Wakamatsu for her expert technical assistance and Ms. M. Goto for data collection. This study was financially supported in part by the Japanese Ministry of Education, Culture, Sports, Science, and Technology Subsidy Program (25293129: T. Kojima) and the Japanese Ministry of Health, Labour and Welfare Research Subsidy Program (T. Kojima and M. S.). The authors would like to thank Enago (www.enago.jp) for the English language review.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Nakamura, Y., Murata, M., Takagi, Y. et al. SVA retrotransposition in exon 6 of the coagulation factor IX gene causing severe hemophilia B. Int J Hematol 102, 134–139 (2015). https://doi.org/10.1007/s12185-015-1765-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-015-1765-5