
Abstract
The Asiatic black bear (Ursus thibetanus) is a protected species from eastern Asia. In China, the Asiatic black bear occurs in 17 provinces from northeast to southwest regions. To date, information on microbial diversity in the gut of the Asiatic black bears from different populations remains limited. To determine the species composition and community structure of the gut microbiota in the Asiatic black bear, we characterized 36 fecal samples from Sichuan, Yunnan, and Heilongjiang provinces, China, by pyrosequencing the 16S V3–V4 hypervariable regions using the Illumina Miseq platform. Results showed that Firmicutes and Proteobacteria were the predominant phyla in the samples, which were largely comprised Escherichia-Shigella, Peptostreptococcaceae_incertae_sedis, Turicibacter, Streptococcus, and Clostridium. By analyzing the community structure from these 36 samples, we found that there were significant differences in the species diversity and richness between Sichuan, Yunnan, and Heilongjiang populations. In conclusion, our results reveal the species composition and structure of the gut microbiota in captive black bears in China, and suggest that biogeography could affect the black bear’ gut microbiota.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gut microbiota share a special relationship with their hosts, and advances in genomics are updating these relationships, indicating a long history of co-evolution. (Ley et al. 2008; Sun et al. 2016). Many factors, such as genetics, diets, and geography, affect the community structure of gut microbiota (Muegge et al. 2011; Godoy-Vitorino et al. 2012; Phillips et al. 2012; Yatsunenko et al. 2012; Kong et al. 2014). It has been well established that gut microbiota play an essential role in nutritional uptake, energy harvest, food digestion, vitamin synthesis, and metabolism in humans and other animals (Hooper et al. 2002; Bäckhed et al. 2005).
The Asiatic black bear (Ursus thibetanus), also known as moon bear and white-chested bear, is a protected animal from eastern of Asia. In China, the Asiatic black bear occurs in 17 provinces from northeast to southwest regions. Previous studies on bacterial diversity in feces from bear species were carried out based on culture-(in)dependent microflora analysis (Schwab et al. 2009), and mini-clone libraries were built to determine the number of clones (Glad et al. 2010). A recent study compared the gut microbiota of giant pandas, red pandas, and black bears and suggested a divergent evolution of the gut microbiota in the three species (Li et al. 2015). Another study analyzed the gut microbiota of brown bears and found that the species composition differed seasonally between hibernation and active phases (Sommer et al. 2016). Therefore, information on the microbial diversity in the gut of the Asiatic black bears is ecologically relevant.
As a large omnivore, the bacterial community of Asiatic black bears is interesting. To understand whether this species has distinctive gut microbiota in different regions, a comprehensive analysis of the bacterial community is required. The development of high-throughput next-generation sequencing has resulted in the characterization of complex microbiota populations. Thus, the aim of the present study was to investigate the bacterial community in the feces of black bears from different populations.
In the present study, we used high-throughput sequencing to investigate the microbial community in the feces of captive Asiatic black bears from three different captive populations, and a total of 36 fecal samples were collected from Sichuan, Yunnan, and Heilongjiang provinces in China. We aimed to reveal differences in the community structure and species composition of the gut microbiota in captive Asiatic black bears in the three populations and to identify the shared and unique bacterial species of gut microbiota in these black bears.
Materials and methods
Ethic statement
Before sample collection, all animal works were approved by the Institutional Animal Care and Use Committee of the Chongqing University under permit number CBE-A20140702.
All experiments were performed in accordance with the approved guidelines and regulations.
Sample collection
Fecal samples from captive black bears were collected immediately after their natural defecation, frozen using liquid nitrogen and sent to the laboratory in foam boxes with dry ice. All samples were obtained from the inside of the feces using sterilized equipment, in which there was no contact with soil or other pollution sources. Twelve fecal samples were collected from the Conservation and Research Center for Bear Species in Sichuan Province, eight samples from the Conservation and Research Center for Bear Species in Yunnan Province, and sixteen samples from the Conservation and Research Center for Bear Species in Heilongjiang Province (for details, see Table S1). All samples were collected from healthy animals.
DNA isolation, PCR amplification, and Miseq sequencing
The total genomic DNA was isolated from fecal samples using the Qiagen QIAamp Fast DNA Stool Mini Kit according to the manufacturer’s instructions. The concentration of isolated DNA was determined using a Nano-Drop 2000 spectrophotometer. PCR amplification of the 16S rRNA gene was performed based on the literature (Dennis et al. 2013) using PCR primers specific for the 515–806 (V3–V4) regions. The PCR assays were carried out in triplicate as follows: 20 ml reaction solutions with 10 ng template DNA, 4 μl PCR reaction buffer, 0.4 mM of each primer, 2.5 mM dNTPs, and 0.5 U TransStart-FastPfu DNA polymerase (TransGen Biotech). The PCR conditions were as follows: 95 °C for 4 min, followed by 27 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s, and a final extension of 72 °C for 10 min. All PCR products were visualized on agarose gels (2% in TAE buffer) containing ethidium bromide, and purified with a DNA gel extraction kit (Axygen, China). Prior to sequencing, the DNA concentration of each PCR product was determined using a QuantiFluor™-ST fluorescent quantitative system (Promega, USA), and mixed with the appropriate proportion based on sequencing requirements. PE amplicon libraries were constructed, and sequencing was performed using the Illumina Miseq platform at Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China.
Sequence analysis
Before analysis, sequences were demultiplexed and quality-filtered using QIIME (version 1.9.1) with the following criteria: (1) the 300 bp reads were truncated at any site receiving an average quality score <20 over a 50 bp sliding window, discarding the truncated reads that were shorter than 50 bp; (2) exact barcode matching, two nucleotide mismatch in primer matching, reads containing ambiguous characters were removed; and (3) only sequences that overlapped longer than 10 bp were assembled according to their overlap sequence. Reads that could not be assembled were discarded.
Sets of sequences with at least 97% identified were defined as an OTU (operational taxonomic unit), and chimeric sequences were identified and removed using UCHIME (Edgar et al. 2011). The taxonomy of each 16S rRNA gene sequence was analyzed using RDP Classifier (http://rdp.cme.msu.edu/) against the SILVA ribosomal RNA gene database using a confidence threshold of 70% (Cole et al. 2009; Quast et al. 2013).
Ecological and statistical analyses
Rarefaction curves were plotted for each sample to determine the abundance of communities and sequencing data of each sample (Amato et al. 2013). Alpha-diversity analyses, including community richness parameters (Chao1, ACE, Rank-Abundance curves), community diversity parameters (Shannon, Simpson, Shannon–Wiener curves), and a sequencing depth index (Good’s coverage), were calculated using the mothur software (Schloss et al. 2011). Beta diversity measurements, including gut microbiota trees, were calculated as described (Jiang et al. 2013), and principal coordinate analyses (PCoA) based on OTU compositions were determined. Bacterial taxonomic distributions of sample communities were visualized using the R package software. A Venn diagram was implemented using the R package to show unique and shared OTUs. Linear discriminant analysis effect size (LEfSe) (Segata et al. 2011), which takes into account both statistical significance and biological relevance, was conducted to identify OTUs differentially represented between the black bears from the three different regions. Differences between populations were analyzed using a one-way ANOVA. P < 0.05 was considered statistically significant.
Results
Taxonomic composition
Across the 36 samples, an average of 16,307 high-quality reads were obtained after denoising. The number of reads, coverage, the number of operational taxonomic units (OTUs), and statistical estimates of species richness from each sample at a genetic distance of 3% are presented in Table S2. The rarefaction, Shannon–Wiener, and rank-abundance curves generated using the R software, as shown in Figs. S1–S9, were clustered at the 97% phylotype similarity level. These sequences were assigned to 357 OTUs in total, and the content ranged from 28 to 173 in each sample.
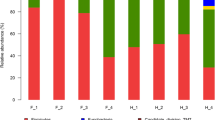
A total of 12 prokaryotic phyla were identified from the 16S rRNA gene sequences (Fig. 1a). In the feces of the 36 captive black bears, Firmicutes was predominant, represented by 43.6–96.4% of the 16S rRNA gene sequences. The second most abundant phylum was Proteobacteria, with 3.58–56.3% in each sample. Firmicutes and Proteobacteria were the only two prokaryotic phyla in all the samples. At the genus level, detected OTUs were distributed among 192 different bacterial genera in total (Fig. 1b). In the three distribution groups, the top five abundant genera have been listed in Table 1. In the Sichuan group, 90 genera were detected, and in the Yunnan group, 76 genera were detected. In contrast, a more diverse community of 184 genera was detected in the Heilongjiang group. No significant differences were observed in the relative abundance of gut microbiota at the phylum level among three populations of bears (Fig. 2a), but at the genus level, Streptococcus, Escherichia-Shigella, and Turicibacter presented a significant differences in the three populations (Fig. 2b). Bacterial community richness (measured by the total number of observed OTUs) in the H group was significantly higher than that in the S group and Y group (Fig. 2c, parametric test, P < 0.001). The bacterial community diversity was measured by both the Shannon index and the inverse Simpson index. Consistently, both indices were significantly higher in H group than in S group and Y groups (Fig. 2d, e, parametric test, P < 0.001).

Fecal bacterial community at the phylum (a) and genus (b) levels. a, b Relative abundance of bacterial groups in the feces of 36 black bears. Less than 1% abundance of the phyla or genus was merged into others

Statistical comparison of the relative abundance (a, b) and alpha diversities (c–e) of gut microbiota among the three bear populations. a Comparison of dominant phyla in the S, Y, and H groups. b Comparison of dominant genera in the S, Y, and H groups. c Bacterial community richness (measured by the total number of observed OTUs) in the S, Y, and H groups. d Bacterial community diversity (measured by Shannon index) in the S, Y, and H groups. e Bacterial community diversity (measured by inverse Simpson index) in the S, Y, and H groups. A P < 0.05 was considered statistically significant
Community structures
We examined the relationship between the gut microbiota of the 36 feces samples from three different regions using Bray–Curtis distances, which were represented by a dendrogram. Each branch on the tree represented one sample of gut microbiota (Fig. 3a). Interestingly, the gut microbiota of the black bears in Sichuan and Yunnan clustered together, but those of the black bears in Heilongjiang located on different branches.

Relationship of the gut microbiota of the black bears from three populations. a Clustering analysis of the evolution of the gut microbiotas in the black bears from Sichuan (S group), Yunnan (Y group), and Heilongjiang (H group) provinces. Gut microbiota trees were generated using the UPGMA (unweighted pair group method with arithmetic mean) algorithm based on the Bray–Curtis distances generated by mothur. b Principal coordinate analysis (PCoA) of microbial communities. Squares, diamonds, and circulars represent the gut microbiotas of black bears from Sichuan (S group), Yunnan (Y group), and Heilongjiang (H group) provinces, respectively. Distances between symbols on the ordination plot reflect relative dissimilarities in community structures
We used principal coordinate analysis (PCoA) to examine the community structures of the gut microbiotas of the black bears in the three regions (Fig. 3b). On the PCoA plot, each symbol represents one gut microbiota. Similar to the cluster analysis, bacterial communities from the black bears in Sichuan and Yunnan clustered tightly and were separated from those from the black bears in Heilongjiang along principal coordinate axis 1 (PC1), which explained the largest amount of variation (35.25%). This result is consistent with the bacterial community analysis, where the black bears in Heilongjiang were found to possess a significantly higher number of OTUs than those in Sichuan and Yunnan.
Unique and shared bacterial taxa
Next, we examined the shared and unique bacterial taxa between the gut microbiotas of the captive black bears from the three groups (Sichuan, Yunnan, and Heilongjiang) using our sequencing data. The shared seven OTUs from the 36 samples are shown in Fig. 4a. We used linear discriminant analysis effect size (LEfSe) to identify OTUs differentially represented between the captive black bears from the three regions (Fig. 4b). Although OTU153 and OTU191 were shared by all the samples, their relative abundance was differentially represented in the three groups. OTU153 was affiliated with the genus Escherichia-Shigella and higher in the Sichuan and Yunnan than Heilongjiang groups (Fig. 4c). OTU191 was affiliated with the genus Streptococcus and higher in the Yunnan and Sichuan than Heilongjiang groups (Fig. 4d). OTU324, which was affiliated with the genus Pseudomonas vranovensis, was significantly higher in Heilongjiang than in the Sichuan and Yunnan groups (Fig. 4e), and this genus could not be detected in most of fecal samples from the Sichuan and Yunnan groups.

Unique and shared OTUs presented in the black bears from three populations. a OTUs differentially represented between black bears from Sichuan (S group), Yunnan (Y group), and Heilongjiang (H group) identified by linear discriminant analysis coupled with effect size (LEfSe). b Venn diagram showing the unique and shared OTUs (3% distance level) from Sichuan (S group), Yunnan (Y group), and Heilongjiang (H group). The distribution of the most differentially distributed OTUs: OUT153 (more abundant in S group), OUT191 (more abundant in Y group), and OTU324 (more abundant in H group) were illustrated in c, d, and e, respectively, identified by linear discriminant analysis coupled with effect size (LEfSe)
Discussion
Some previous studies on bacterial diversity in feces from bear species were carried out based on culture-(in)dependent microflora analysis (Schwab et al. 2009), or mini-clone libraries were developed to determine the number of clones (Glad et al. 2010). These reports uncovered some (un)culturable gut microbiota species and dominant gut microbiota flora, but the results did not indicate the real gut microbiota structure of the bears. The development of high-throughput next-generation sequencing has allowed us to resolve the gut microbiota community composition at an unprecedented level compared with the traditional cultural-based and PCR-DGGE way. In the present study, we extensively analyzed the gut microbiota community in captive black bears in China, and revealed the species composition of microbes existed in the intestinal tract of captive Asiatic black bears. For example, we found that Turicibacter, a non-culturable bacterium (Bosshard et al. 2002; Cuív et al. 2011), was a major component of the gut microbiota community in the black bears, and widely distributed in all samples (Fig. S10). Clostridium is widely detected in the feces of other bear species (Schwab and Ganzle 2011). In our study, Clostridium_sensu_stricto_1 constituted a core microbiota in Asiatic black bear fecal samples, and was relatively abundant in all samples (Fig. S11). Our results revealed a comprehensive bacterial abundance and diversity in the gut flora of captive Asiatic black bears. Consistent with some previous studies (Zhu et al. 2011; Li et al. 2015), we found the gut microbiota of Asiatic black bears are similar to those of giant pandas at the phylum level. Similar to that in other omnivores, such as humans (Ley et al. 2006), pigs (Lu et al. 2014), and mouse (Gu et al. 2013), Firmicutes was the most dominant phylum in the gut microbiota of black bears. The results of comparison of the relative abundance of gut microbiota among the three bear populations also suggested that black bears presented relatively stable community structure and abundance of gut microbiota at the phylum level. However, at the genus level, black bears exhibited a variable and complex community structure and abundance of gut microbiota.
Interestingly, we observed that the bacterial communities between the samples from the northeast (H group) and southwest (S and Y groups) in China are significantly different, and a total of 52 genera were differentially represented in the three groups using LEfSe (Fig. S12). Sichuan (30°55′N 103°43′E) and Yunnan (23°22′N 103°24′E) share similar geography and climate conditions, whereas Heilongjiang (44°35′N 129°34′E) is located far from Sichuan and Yunnan. The three groups shared similar host genetic background and diets (zoo breeding mode), but their bacterial community structure was different. In addition, the samples from Heilongjiang had significantly higher community diversity and species richness than the samples from Sichuan and Yunnan. Although we found different fecal microbiota compositions among Sichuan and Yunnan groups, there were no significant differences in the richness and diversity. The results from the taxonomic composition and community structures suggested the geographical population pattern of the gut microbiota of Asiatic black bears in the three regions. Many factors contribute to the community structure of host intestinal microbiota, such as diet (Turnbaugh et al. 2009; Muegge et al. 2011), geography (Yatsunenko et al. 2012), and seasonal changes (Sommer et al. 2016; Sun et al. 2016).
Heilongjiang is the highest latitude region in China, and has special environmental conditions, such as a cold climate. Black bears inhabiting this region have high-energy requirements, and greater abundance of gut microbiota would result in higher active metabolism and energy harvest. Our results suggest that biogeographical and climatic factors contributed to the community structure of the gut microbiota of the black bears, and future studies with larger sample sizes and refined diets are necessary to determine the major cause of this geographical variation.
In conclusion, our study characterized the gut microbiota in captive black bear populations using the next-generation sequencing techniques, and found that, based on the 16S rRNA sequencing analysis, the gut microbiota of these black bears in Sichuan and Yunnan were clustered closer than those of black bears in Heilongjiang. We also identified shared and unique bacterial taxa represented in the black bears from the three different regions. Our study provides insights into the composition and structure of the gut microbiota in black bears and enhances our understanding of the effect of biogeography on the species diversity of the intestinal microbiota of their host. Further studies are still required to determine whether black bears in the wild have specific gut microbiota as compared to that in other large omnivore species.
References
Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, Gaskins HR, Stumpf RM, Yildirim S, Torralba M (2013) Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J 7:1344–1353
Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI (2005) Host-bacterial mutualism in the human intestine. Science 307:1915–1920
Bosshard PP, Zbinden R, Altwegg M (2002) Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int J Syst Evol Micr 52:1263–1266
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen A, McGarrell DM, Marsh T, Garrity GM (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
Cuív PÓ, Klaassens ES, Durkin AS, Harkins DM, Foster L, McCorrison J, Torralba M, Nelson KE, Morrison M (2011) Draft genome sequence of Turicibacter sanguinis PC909, isolated from human feces. J Bacteriol 193:1288–1289
Dennis KL, Wang YW, Blatner NR, Wang SY, Saadalla A, Trudeau E, Roers A, Weaver CT, Lee JJ, Gilbert JA, Chang EB, Khazaie K (2013) Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res 73:5905–5913
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Glad T, Bernhardsen P, Nielsen KM, Brusetti L, Andersen M, Aars J, Sundset MA (2010) Bacterial diversity in faeces from polar bear (Ursus maritimus) in Arctic Svalbard. BMC Microbiol 10:10
Godoy-Vitorino F, Goldfarb KC, Karaoz U, Leal S, Garcia-Amado MA, Hugenholtz P, Tringe SG, Brodie EL, Dominguez-Bello MG (2012) Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J 6:531–541
Gu SH, Chen DD, Zhang JN, Lv XM, Wang K, Duan LP, Nie Y, Wu XL (2013) Bacterial community mapping of the mouse gastrointestinal tract. PLoS One 8:e74957
Hooper LV, Midtvedt T, Gordon JI (2002) How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 22:283–307
Jiang XT, Peng X, Deng GH, Sheng HF, Wang Y, Zhou HW, Tam NFY (2013) Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb Ecol 66:96–104
Kong F, Zhao J, Han S, Zeng B, Yang J, Si X, Yang B, Yang M, Xu H, Li Y (2014) Characterization of the gut microbiota in the red panda (Ailurus fulgens). PLoS One 9:e87885
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology—human gut microbes associated with obesity. Nature 444:1022–1023
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI (2008) Evolution of mammals and their gut microbes. Science 320:1647–1651
Li Y, Guo W, Han SS, Kong FL, Wang CD, Li DS, Zhang HM, Yang MY, Xu HL, Zeng B, Zhao JC (2015) The evolution of the gut microbiota in the giant and the red pandas. Sci Rep 5:10185
Lu XM, Lu PZ, Zhang H (2014) Bacterial communities in manures of piglets and adult pigs bred with different feeds revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biot 98:2657–2665
Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, Henrissat B, Knight R, Gordon JI (2011) Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332:970–974
Phillips CD, Phelan G, Dowd SE, McDONOUGH MM, Ferguson AW, Delton Hanson J, Siles L, ORDÓÑEZ-GARZA N, San Francisco M, Baker RJ (2012) Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol Ecol 21:2617–2627
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6:e27310
Schwab C, Ganzle M (2011) Comparative analysis of fecal microbiota and intestinal microbial metabolic activity in captive polar bears. Can J Microbiol 57:177–185
Schwab C, Cristescu B, Boyce MS, Stenhouse GB, Ganzle M (2009) Bacterial populations and metabolites in the feces of free roaming and captive grizzly bears. Can J Microbiol 55:1335–1346
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12:R60
Sommer F, Ståhlman M, Ilkayeva O, Arnemo JM, Kindberg J, Josefsson J, Newgard CB, Fröbert O, Bäckhed F (2016) The gut microbiota modulates energy metabolism in the hibernating brown bear Ursus arctos. Cell Rep 14:1655–1661
Sun B, Wang X, Bernstein S, Huffman MA, Xia D-P, Gu Z, Chen R, Sheeran LK, Wagner R, Li J (2016) Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci Rep 6:26035
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP (2012) Human gut microbiome viewed across age and geography. Nature 486:222–227
Zhu LF, Wu Q, Dai JY, Zhang SN, Wei FW (2011) Evidence of cellulose metabolism by the giant panda gut microbiome. P Natl Acad Sci USA 108:17714–17719
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This work was supported by National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (2014ZX09301306-007), the Natural Science Foundation Project of CQ CSTC (cstc2015jcyjA10094), the Visiting Scholar Foundation of the Key Laboratory of Biorheological Science and Technology (Chongqing University), and the Ministry of Education (CQKLBST-2012-007).
Conflict of interest
The authors declare no competing financial interests.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Data availability
The raw 16S rRNA sequences supporting the results of this study are available in the GenBank Database under the accession No. SRX1244940.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Song, C., Wang, B., Tan, J. et al. Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol Genet Genomics 292, 407–414 (2017). https://doi.org/10.1007/s00438-016-1282-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-016-1282-0