
Abstract
Pummelo (Citrus grandis) is one of the most important gene pools for citrus breeding programmes. A high-density linkage map is a valuable tool for functional genomics and genetic breeding studies. A newly developed genome sequence-based marker technology, restriction site-associated DNA (RAD) sequencing, has proven to be powerful for the rapid discovery and genotyping of genome-wide SNP markers and for the high-density genetic map construction. We present the construction of a high-density genetic map of pummelo using RAD sequencing. An F1 population of 124 individuals and its parents (‘Pingshan’ pummelo and ‘Guanxi’ pummelo) were applied to the map construction. One thousand five hundred forty three high-quality single nucleotide polymorphism (SNP) markers were developed and mapped to nine linkage groups. In addition, 20 simple sequence repeat (SSR) markers were included and showed general consistency with the SNP markers. These 1563 markers constituted a total genetic length of 976.58 cM and an average of 0.62 cM between adjacent loci. The number of markers within each linkage group (LG) ranged from 81 (for LG4) to 285 (for LG2). A comparison of the genetic maps to the published sweet orange (Citrus sinensis) genome revealed both conservation and variations. The alignment of the LGs from this map was also shown in comparison with a previously genetic linkage map from pummelo. This study showed that the RAD sequencing allowed the rapid discovery of a large number of SNPs in the pummelo. The SNP-based high-density genetic map for pummelo was successfully generated by using these designed SNP markers. The completed genetic map is a valuable resource for further pummelo genetic studies and provides useful information for gene positional cloning, MAS breeding and C. grandis genome assembly.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Citrus (2n = 18), part of the Rutaceae family, comprise some of the most economically important fruit crops in the world, hence the importance developing new improved varieties. Conventional breeding of citrus is difficult due to the long juvenile periods, large plant size, male sterility, incompatibility and the many generations necessary to carry out selections and introgression (Soost and Cameron 1975). Molecular breeding is an efficient strategy to overcome such limitations. High-density genetic maps are necessary tools for efficient molecular breeding including quantitative trait locus (QTL) mapping and marker-assisted selection (MAS). Citrus is suitable for genetic linkage mapping studies because of the low haploid chromosome number (2n = 18) and genome size (1C = 0.62 pg) (Ollitrault et al. 1994). However, most citrus varieties are polyembryonic, and hybrids from crosses between them are usually difficult to acquire (Corazza-Nunes et al. 2002; Grosser et al. 2004). Thus, this biological characteristic has hindered the development of citrus genetic maps.
Pummelo (Citrus grandis Osbeck), one of the three basic species of citrus, has unique biological characteristics compared with other types of citrus (except for citron), such as monoembryony. It is native to tropical and subtropical regions in Asia and widely cultivated in China and other Southeast Asian countries (Corazza-Nunes et al. 2002). The long history and diversified climates enable these areas to harbour high genetic diversity of pummelo (Ye 1997; Liu and Deng 2007; Gmitter et al. 2009). This genetic diversity provides valuable genetic resources (resistance to Citrus tristeza virus (CTV) and diversity of colour, sugar and sourness) for the citrus breeding programmes (Garnsey et al. 1996; Ramón-Laca 2003). In the past, the pummelo gene pool contributed to the genesis of many important species and cultivars including sweet and sour oranges (Wu et al. 2014a), grapefruit, tangelos and other hybrids (Nicolosi et al. 2000; Barkley et al. 2006; García-Lor et al. 2012). For these reasons, the construction of pummelo genetic map is essential for further genetic studies. Over the last 25 years, several genetic maps have been constructed in pummelo using many types of markers, including isozyme, restriction fragment length polymorphism (RFLP), randomly amplified polymorphic DNA (RAPD), amplified fragment length polymorphisms (AFLP), simple sequence repeats (SSRs) and cleaved amplified polymorphic sequences (CAPSs) (Cai et al. 1994; Luro et al. 1996; Bernet et al. 2010; Ollitrault et al. 2012). There is a growing need for generating a high-density genetic map for pummelo. Construction of a high-density genetic map requires a large number of markers. Until now, the genetic maps of pummelo have been constructed mainly by hybridization-based DNA markers. The number of these markers was limited because the traditional molecular marker technologies are highly time-, labour- and cost-intensive (Ahmad et al. 2003).
In recent years, SNP markers have gained popularity in genetic mapping due to their stability, heritability, low cost, high-throughput efficiency and abundant genetic variation. With the development of next-generation sequencing (NGS) technologies, whole genome sequencing (WGS) (Shulaev et al. 2010; Argout et al. 2011; Wu et al. 2013) and restriction site-associated DNA (RAD) sequencing approaches have been used successfully for numerous SNP discoveries in many crops (Baird et al. 2008; Barchi et al. 2011; Chutimanitsakun et al. 2011; Gaur et al. 2012; Scaglione et al. 2012; Hirakawa et al. 2013). In these 2 years, the WGS strategy was also successfully used for the SNP detection in the pummelos (Xu et al. 2013; Wu et al. 2014a). Over hundreds of thousands of SNPs were identified by the re-sequencing data of several pummelos. Compared to the WGS strategy, the RAD sequencing approach can reduce the genome complexity by restricting sequencing to the flanks of DNA adjacent to recognition sites of a chosen restriction endonuclease (Varshney et al. 2009). The overall sequencing costs are reduced while still identifying a large number of SNP markers. In the last 2 years, RAD sequencing has been used successfully for high-density genetic map construction in many fruit crops including pear, grape and pineapple (Wang et al. 2012a; Wai 2013; Wu et al. 2014b). Wang et al. (2012a) successfully constructed a high-density genetic map with 1814 high-quality RAD markers for grape. By using the RAD sequencing approach, Wai (2013) successfully mapped 1240 high-quality SNP markers into 30 linkage groups (LGs) with a combined length of 2227 cM in pineapple. Recently, the high-density genetic maps of pear comprising 2005 SNP markers were also successfully constructed (Wu et al. 2014b). These results showed that RAD sequencing is a powerful high-throughput approach for the construction of high-density genetic maps in many fruit crops. We speculated that the RAD sequencing approach could also be used successfully to construct a genetic linkage map in citrus.
In this study, we generated an F1 segregating population derived from two pummelo cultivars: ‘Guanxi’ pummelo and ‘Pingshan’ pummelo. A high-density genetic linkage map was constructed using the RAD sequencing approach. Not only does this genetic map make it possible to facilitate future QTL analysis and early marker-assisted selection within pummelo breeding programmes, but it also provides a genetic framework for the C. grandis reference genome assembly.
Materials and methods
Development of a mapping population and DNA extraction
An F1 population of 124 progeny derived from the Guanxi pummelo and Pingshan pummelo cross was used to generate the linkage map. Pingshan pummelo was employed as the male parent. The seedlings of the F1 progeny were planted in the National Center for Citrus Breeding at Huazhong Agricultural University (HZAU), Wuhan.
Young leaf samples were harvested from each individual F1 plant and the two parents. These samples were immediately frozen in liquid nitrogen and kept at −80 °C. Genomic DNA was extracted by a modified CTAB method (Cheng et al. 2003).
RAD library preparation and Illumina sequencing
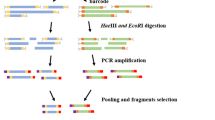
Genomic DNA from each sample (both parents and 124 F1 progeny) was digested with the restriction endonuclease EcoRI (recognition site 5′G^AATTC3′) and processed into RAD libraries according to a previously described protocol (Baird et al. 2008). In brief, 300 ng of genomic DNA was digested for 60 min at 37 °C with 20 units (U) of EcoRI (New England Biolabs [NEB]) in a 50 μL reaction. Samples were heat inactivated for 20 min at 65 °C followed by addition of 2.0 μL of 100 nM P1 Adapter(s), a modified Solexa© adapter (2006 Illumina, Inc.). The EcoRI P1 adapters each contained a unique multiplex sequence index (barcode), which is found in the first four nucleotides of the Illumina sequence read. P1 adaptors (100 nM) were added to each sample along with 1 μL of 10 mM rATP (Promega), 1 μL 10× NEB Buffer 4, 1.0 μL (1000 U) T4 DNA Ligase (high concentration, Enzymatics, Inc.) and 5 μL H2O. The reaction was incubated at room temperature for 20 min and then heat inactivated for 20 min at 65 °C. The samples were pooled and randomly sheared with a Bioruptor (Diagenode) to an average size of 500 bp. DNA fragments ranging in size from 300 to 700 bp were separated on a 1.5 % agarose gel (Sigma) and isolated using a MinElute Gel Extraction Kit (Qiagen). The dsDNA ends were polished by end blunting enzymes (Enzymatics, Inc.), and the samples were then purified using a MinElute column (Qiagen). Fifteen units of Klenow exo- (Enzymatics) was used to add adenine (Fermentas) overhangs to the 3′end of the DNA by incubating at 37 °C for 10 min. After subsequent purification, 1 μL of 10 μM P2 adapter, a divergent modified Solexa© adapter (Illumina, Inc.), was ligated to the obtained DNA fragments at 18 °C. The samples were then purified as above and eluted in a volume of 50 μL. Following quantification using a Qubit fluorimeter, 20 ng was used in a PCR amplification reaction containing 2 μL Phusion Master Mix (NEB), 5 μL 10 μM modified Solexa© Amplification primer mix (Illumina, Inc.) and sterile water to 100 μL. Phusion PCR settings followed product guidelines for a total of 18 cycles. Samples were gel purified, excising DNA from a size range of 300 to 700 bp and diluted to 1 nM. The constructed RAD libraries were sequenced on the NGS Illumina platform HiSeq2000 at the BGI, China, following the manufacturer’s protocol.
Sequence analysis and SNP discovery
Raw Illumina reads with more than five bases showing a quality score below 30 were discarded. Sequences with ambiguous barcode sequences were also eliminated. Reads were separated by individuals using the barcodes. These filtered reads were aligned to the sweet orange genome (http://citrus.hzau.edu.cn/orange/) by SOAP2 software (Li et al. 2009b). Then, the SNP markers were determined by SOAPsnp (http://soap.genomics.org.cn/soapsnp.html) in all the F1 population (Li et al. 2009a). Four strict criteria were used for filtering individuals and mapping SNP markers: (1) individuals with reads number more than 3.5 M; (2) minimum depth of 5 for descendants and minimum depth of 7 for parents; (3) expected segregation ratios (1:2:1 or 1:1) using a chi-square test (P value ≤0.01) and (4) markers with ≤20 % missing value. SNPs with missing genotype calls in one parent were also excluded from further analysis.
SSR genotyping
Sequence information of SSR markers were obtained from previous published paper (Ollitrault et al. 2012). The segregation data for SSR markers in the same population were detected, and 20 of them were used for constructing an integrated map. PCR amplification was carried out in 10 μL reactions, each containing 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, 1.0 U Taq DNA polymerase (Fermentas), corresponding 1× reaction buffer, 0.1 μmol/L of each primer pair and 50 ng of genomic DNA. PCR amplification was conducted using the following program: 94 °C for 5 min, 32 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s, followed by a final step at 72 °C for 10 min. The success of amplification was monitored by analysis of the reaction product with 2 % agarose gel electrophoresis. The successful amplification products were separated by 6 % polyacrylamide gel electrophoresis (PAGE) and visualized by a simplified silver staining method previously described by Ruiz et al. (2000).
Genetic linkage map construction
The ‘two-way pseudo-testcross’ approach (Grattapaglia and Sederoff 1994) was applied to construct the genetic linkage map. The ratios of marker segregation were calculated by chi-square test, and only markers that satisfied with the expected Mendelian segregation ratio were included for mapping. The filtered markers contained different segregation types, including <hk×hk>, <lm×ll> and <nn×np>. Linkage analysis was performed by JoinMap® 4.0 software (Van 2006). After the SNP marker genetyping data had been imported, the cross-pollination (CP) population option was used for data mining. On the basis of a LOD threshold of 8.0, 1563 SNP markers were grouped into nine LGs, and the maps were constructed using the default maximum likelihood parameters. The Kosambi mapping function was used to calculate the genetic distance between markers.
Results
RAD sequencing
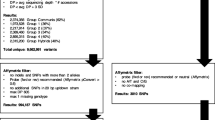
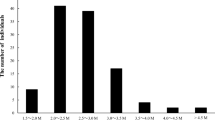
Based on the stringent filtering criteria described in M&M, ~56.8 G of high-quality sequencing data was filtered. The average high-quality data in parents and individuals were ~0.82 and ~0.45 G, respectively. The Q20 ratio was 95.6 %, and the guanine-cytosine (GC) content was 36.8 %. Of these high-quality data, ~886 Mb were from Pingshan with 10,108,354 reads and ~793 Mb from Guanxi with 9,678,764 reads. Obtained from the libraries for the 124 individuals were ~628.48 million reads. The number of reads for the individuals ranged from 2.53 to 8.00 M, with an average of 5.07 M reads (Fig. 1). Removing individuals with poor reads number (reads number fewer than 3.5 M) resulted in the removal of six individuals.

Distribution of reads number for F1 individuals. The x-axis indicates the interval of reads number; the y-axis indicates the number of individuals
SNP marker discovery and genotyping
The high-quality Illumina sequences were used for identification of SNPs. A total of 194,231 putative SNPs were determined by SOAP2 and SOAPsnp in all the 120 F1 population level (two parents and 118 individuals). After filtering (minimum read depth of 7, no missing), 63,760 (32.83 %) SNP loci were retained between the parents. Most of the SNPs were transition-type SNPs, with the C/T and A/G types accounting for 27.9 and 27.7 % of the SNPs, respectively. The other four SNP types were transversions and included A/C, G/T, A/T and C/G, ranging in percentages from 7.6 to 13.6 % (Table 1). Five hundred twenty homozygous SNPs have been removed because of no segregation in progeny. Stringent filtering criteria (see ‘Methods’) were applied to identify and remove SNPs with a high level of missing data and/or Mendelian errors. With the miss rate 20 % in all the 118 individuals, 7992 (12.53 %) SNP markers have been retained. After the chi-square test, 1543 (19.53 %) SNP markers were used to construct the genetic map in the final. The SNP location information is listed in Supplementary material 1. The marker codes ‘lm×ll’ and ‘nn×np’ represent markers with one parent heterozygous (providing 1:1 segregation ratios), and ‘hk×hk’ represents markers with both parents heterozygous (providing 1:2:1 segregation ratios). Of these 1543 SNP markers, 498 were present in the parental configuration lm×ll (i.e. heterozygous only in Pingshan), 520 in the configuration nn×np (i.e. heterozygous only in Guanxi) and the remaining 525 in the configuration hk×hk (i.e. heterozygous in both parents).
Linkage map construction
To anchor and align the current map with previously published maps for pummelo, 30 previously published SSR markers distributed among the nine linkage groups of the genetic map, which came from Ollitrault et al. (2012), were screened on the parental genotypes. As a result, 20 polymorphic markers were identified. A total of 20 markers were mapped to the eight LGs of the current map (Fig. 2; Supplementary material 2). Using JoinMap 4.0 to construct the genetic map, 1543 SNP markers and 20 SSRs were assigned to nine linkage groups (LG1–LG9) for the integrated map with a grouping LOD value of 8 (Fig. 2). Moreover, 500 (31.99 %) markers fell into nine LGs for Pingshan (male) and 511 (32.69 %) markers for Guanxi (female), with a grouping LOD value of 7 and 8, respectively (Supplementary material 3). A complete list of all marker information used in this study is provided in Supplementary material 1. Taking into account the size of all LGs, marker coverage amounted to 641.01 cM for Pingshan (male), 602.07 cM for Guanxi (female) and 976.58 cM for the integrated map (Table 2). On average, the intervals between two adjacent mapped markers were 1.28, 1.18 and 0.62 cM for the Pingshan, Guanxi and integrated maps, respectively. The pummelo genome size is approximately 383 Mb (Ollitrault et al. 2012), making the average marker site density of physical intervals in the integrated map at 245.04 Kbp per marker.

Integrated linkage group for ‘Guanxi’ pummelo × ‘Pingshan’ pummelo
The maximum number of markers was 151 for the female (LG5), 148 for the male (LG2) and 285 for the integrated map (LG2). The minimum number of markers was 6 for the female (LG6), 5 for the male (LG8) and 81 for the integrated map (LG4). The length per LG also varied widely. The longest LGs were 120.65 cM for the female (LG9), 145.77 cM for the male (LG9) and 139.26 cM for the integrated map (LG9). The shortest LGs were 12.05 cM for the female (LG8), 8.22 cM for the male (LG8) and 75.42 cM for the integrated map (LG6). The average distance between adjacent markers ranged from 0.43 cM (LG2 and LG3) to 1.25 cM (LG4) for the integrated map (Table 3). A high-quality linkage map should have short distances between adjacent markers and evenly distributed markers (Wang et al. 2012b). Our results showed that most of the SNP markers were evenly mapped in nine linkage groups (Table 3). Among the 1572 intervals in 9 different linkage groups, 1301 intervals were smaller than 1 cM (83.84 %), and only 15 intervals (0.95 %) were larger than 5 cM. Thirteen (0.83 %) gaps with a distance >5 cM between adjacent markers occurred in nine of the linkage groups: LG1 (1), LG2 (1), LG3 (1), LG4 (2), LG5 (2), LG6 (1), LG7 (1), LG8 (3) and LG9 (1). Only two (0.13 %) gaps with a distance >10 cM between adjacent markers occurred in LG4 and LG8 (Table 3, Fig. 3). These results indicated that this genetic map is of high quality and high resolution.

The x-axis indicates the position in each linkage group in 1 cM intervals, and the y-axis indicates the number of markers within 1 cM bin
The sweet orange is an inter-specific hybrid between pummelo and mandarin. The genome of pummelo had a high similarity with the sweet orange genome. In order to assess the quality of this genetic map, a dot-plot diagram was used to evaluate the physical distances of the mapped SNP markers on the sweet orange genome sequence (Xu et al. 2013) plotted against their genetic positions on the pummelo genetic map (Fig. 4). According to our results, most of the markers showed good linear agreement between the physical and genetic maps. However, there were also chromosomes showing discrepancy of some regions. Among the nine LGs, signifying high and low discrepancy could be observed in the some regions of LG2 and LG7.

Collinear analysis of the consensus between integrated pummelo genetic maps and sweet orange genome. The x-axis indicates the genetic position of each SNP marker; the y-axis indicates the physical position of each SNP marker
Comparison with published maps of pummelo
The linkage map constructed in this study was also aligned to the two pummelo genetic maps developed by Ollitrault et al. (2012). In 20 single-locus SSR loci of the SNP-based linkage map, 20 were identified as having corresponding loci in the 8 chromosomes of the ‘Chandler’ map. All LGs except for LG7 consisted of at least one SSR, which was used for locating the corresponding chromosome number for each LG and is convenient for map comparison. The comparison showed that the corresponding LGs were collinear, except LG2 (Supplementary material 2). For collinear LGs, all the markers were comparative, with the same order and similar genetic distance. For example, CX2019, JI-GA01 and MEST080 located at 0, 80.554 and 96.76 cM of LG4 in our map, respectively, and were located at 0, 83.6 and 101.8 cM, respectively, on LG01 in Chandler. For LG2, some of the conserved loci had a reversed order. As we can see, mCrCIR04B06, CF-TTC02, mCrCIR05A05, mCrCIR06B07 and MEST525 located at 8.9, 77.5, 104, 105.3 and 123.2 cM of LG2 in our map, respectively, but were located at 7.8, 52.2, 79.1, 109.4 and 94.2 cM, respectively, on LG02 in Chandler. The order of two markers (mCrCIR06B07 and MEST525) in our map was reverse to the Chandler map. But in the ‘pink’ map, these two markers were showed the same order with our maps, with the location of 94.7 and 99.5 cM, respectively. In addition, the marker numbers between these SSRs were significantly increased in our map. For example, there were five markers between CX0118 and CF-CA14 and one marker between CF-CA14 and CX0029 in ‘pummelo’ map, respectively, but there were 155 SNP markers between CX0118 and CF-CA14 and 27 SNPs between CF-CA14 and CX0029 in our map. These markers provided us additional information about these loci for future research.
Discussion
Constructing a high-density linkage map is a vital prerequisite for both sophisticated genetic analysis such as QTL mapping and efficient molecular breeding such as MAS aiming at important agronomic traits. One of the challenges of high-density genetic map construction was to identify enough molecular markers. In previous research of citrus genetic maps, limited mapping markers were identified using traditional marker technologies such as RFLP, RAPD, SSRs and EST-based markers (Chen et al. 2008). SNP markers have been recognized as important candidate markers due to their high abundance and relatively even distribution across the genome (Brookes 1999; Chagné et al. 2007). SNP discovery in citrus using traditional methods has been shown to be a difficult and tedious process. The RAD sequencing approach used in this study enabled the identification and mapping of an unprecedented number of SNP markers. Moreover, the methods employed here are more cost-effective than the array-based methods of SNP detection and scoring (Ward et al. 2013). These results also demonstrate the value of RAD sequencing for the development of large numbers of mapping SNP markers and for the production of the most saturated linkage maps.
Before RAD sequencing technique was developed, a number of other platforms were available for the SNP identification, such as high-resolution melt (HRM) analysis and high-throughput SNP arrays. The HRM analysis is a closed-tube method for rapid identification of SNP within PCR amplicons. The advantage of HRM analysis is the dosage sensitivity and ability to detect high levels of heterozygosity (Han et al. 2012). But, the throughput of the SNP identification is much lower than the NGS-based techniques. The cost of SNP genotyping using HRM analysis was much higher than NGS-based techniques in a large population. The high-throughput SNP arrays were effective and cost-efficient to discover and genotype the predetermined set of SNPs (Yan et al. 2010). By using the high-throughput SNP arrays, a large numbers of SNPs were successful called in a hybrid population of 88 progeny and 103 citrus accessions (Fujii et al. 2013). Therefore, the production of a high-quality SNP array requires a substantial investment of resources. Additionally, these predetermined markers are specific to the population in which they were developed; hence, the heterozygous SNPs between species will be hardly transferred. The sequencing-based techniques has been proved to be an efficient method for the simultaneous discovery and genotyping of large numbers of SNPs in large amount of individuals (Davey et al. 2011). Genotyping of the same markers in new populations can be achieved with further sequencing runs. Terol et al. (2008) successfully identified 6617 putative SNPs through the BAC end sequencing in citrus. With the developing of the NGS-based techniques, the WGS became an important strategy for the SNP mining. By using the WGS strategy, hundreds of thousands of SNPs have been successfully identified in several important pummelos (Xu et al. 2013; Wu et al. 2014a). The identified SNPs in these two studies provided us important molecular marker information for the future research in pummelo. Despite the attractiveness of this approach, the NGS-based techniques still have a very serious disadvantage in its high probability of sequence error. To overcome this problem, high coverage of a specific sequence must be obtained. By using the restriction enzymes, the RAD sequencing approach can reduce the genome size and complexity, resulting in the requirement of very little data to achieve high coverage. Moreover, the diversity of restriction enzymes available makes them an extremely versatile assay tool. This method can greatly improve the throughput of SNP identification and genotyping.
The distribution of these 1543 SNP markers over the physical sequence was provided in the Supplementary material 1. Ollitrault et al. (2012) claimed that pummelo harbours a sequence that is ~387 Mb. By using the RAD seq, we manipulated ~95,439 restriction sites and each contained an ~82-bp sequence in parents and 124 individuals. We totally obtained ~15.65 Mb genome sequence, which resulted in a 4.04 % coverage of the pummelo genome. The number of SNP markers identified in the two parents was crucial for construction of the high-density genetic map. To maximize the chances of detecting segregating SNPs in the parents, the parents were sequenced at twice the depth of the individuals. The greater sequencing depth in the parents made it possible to maximize the determination of all polymorphic loci and also highly improved the marker numbers in the genetic linkage map. By using the RAD seq, a comprehensive integrated genetic linkage map with 1543 SNP markers was constructed covering 9 linkage groups. To date, a total of seven genetic linkage maps of pummelo have been developed over the past decade (Table 4). The earliest pummelo linkage map consisted of 52 markers, encompassing 553 cM and distributed on seven linkage groups with an average distance of 10.6 cM between markers (Durham et al. 1992). Since then, many efforts have been made to construct genetic linkage maps of pummelo. However, these genetic maps were mainly constructed with dominant, non-transferable RAPD or AFLP. With the development of molecular markers, SSRs have been widely used for genetic map construction due to their transferability, ease of assay and co-dominance. Recently, two inter-specific F1 segregating populations between Citrus clementina and C. grandis (Chandler pummelo× ‘Nules’ Clementine and Nules Clementine× ‘Pink’ pummelo) were used to establish the pummelo genetic map mainly based on SSRs (Ollitrault et al. 2012). The Chandler pummelo genetic map consisted of 151 markers (141 SSRs, 5 SNPs and 5 Indels) covering 828.6 cM and spread among nine linkage groups with a marker density of 5.5 cM/marker. The Pink pummelo genetic map consisted of 81 markers (67 SSRs, 7 SNPs and 7 Indels) covering 633.9 cM and spread among nine linkage groups with a marker density of 7.8 cM/marker. To our knowledge, our genetic linkage map has the shortest average genetic intervals (0.62 cM/marker), and the number of transferable markers on the map is more than on the previously published genetic maps of pummelo (Table 4). Furthermore, all 1563 markers could be attributed unequivocally to the nine linkage groups, indicating that the map should cover the entire pummelo genome. The size of the population (118 individuals) used in this study was a little smaller for the high saturated map construction. The total size of this F1 population is more than 500; we can improve the density of this genetic map by genotyping more individuals further. We added 20 SSR loci from previously published maps of pummelo to our genetic map. Then, we can compare these markers’ orders and genetic distances to the previously published reported SSR pummelo maps. Most of the markers showed the good collinear with previous published pummelo maps. We also observed that the order of two markers (mCrCIR06B07 and MEST525) in our map was reverse to the Chandler map. The reason why these two markers are located differently might be due to genome structure differences between the different pummelo cultivars.
High-density genetic linkage maps are valuable and important resources that provide a framework for many genomic analyses (Yagi et al. 2013): (1) This high-density genetic map can be used in genomic platforms to anchor and orient scaffolds onto pseudo-chromosomes for the comparative analysis of mandarin, pummelo and orange. The high-density genetic map constructed in Clementine (Ollitrault et al. 2012) with almost 1000 markers had been successfully used for the Clementine reference genome assemble (Wu et al. 2014a). (2) The genetic map can enhance the efficiency of citrus molecular breeding programmes with MAS. The female parent of the F1 population Guanxi pummelo is an important cultivar in China. There are high demands for the improvement of this cultivar. Efficiency of MAS breeding depends on the degree of saturation of genetic maps. This map represents a significant improvement over existing marker resources. It will be beneficial for the molecular breeding programmes of pummelo. (3) The genetic map can be used for QTL analysis of key agronomic traits in pummelo. A principal problem with many QTL mapping efforts is the limited size of the mapping population (Li et al. 2006; Zhao et al. 2007). The total size of this F1 population is more than 500. We can further extend these markers to a larger population, providing an important resource for QTL analysis in future work.
In conclusion, we have constructed a high-density linkage map of a cross between Guanxi pummelo and Pingshan pummelo cultivars using SNP markers derived from the next-generation RAD sequencing. The genetic linkage map is comprised of 1543 SNP loci and 20 SSR loci covering 976.58 cM with an average distance of 0.62 cM between loci. It significantly improves the marker density compared with previously published pummelo genetic maps. Our study provides a valuable genetic resource for QTL analysis, MAS, map-based gene cloning and the sequence assembly of the pummelo reference genome.
References
Ahmad R, Struss D, Southwick SM (2003) Development and characterization of microsatellite markers in citrus. J Am Soc Hortic Sci 128:584–590
Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN, Abrouk M, Murat F, Fouet O, Poulain J, Ruiz M, Roguet Y, Rodier-Goud M, Barbosa-Neto JF, Sabot F, Kudrna D, Ammiraju JS, Schuster SC, Carlson JE, Sallet E, Schiex T, Dievart A, Kramer M, Gelley L, Shi Z, Bérard A, Viot C, Boccara M, Risterucci AM, Guignon V, Sabau X, Axtell MJ, Ma ZR, Zhang YF, Brown S, Bourge M, Golser W, Song X, Clement D, Rivallan R, Tahi M, Akaza JM, Pitollat B, Gramacho K, D’Hont A, Brunel D, Infante D, Kebe I, Costet P, Wing R, McCombie WR, Guiderdoni E, Quetier F, Panaud O, Wincker P, Bocs S, Lanaud C (2011) The genome of Theobroma cacao. Nat Genet 43:101–108
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376
Barchi L, Lanteri S, Portis E, Acquadro A, Vale G, Toppino L, Rotino G (2011) Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genomics 12:304
Barkley N, Roose M, Krueger R, Federici C (2006) Assessing genetic diversity and population structure in a citrus germplasm collection utilizing simple sequence repeat markers (SSRs). Theor Appl Genet 112:1519–1531
Bernet GP, Fernandez-Ribacoba J, Carbonell EA, Asins MJ (2010) Comparative genome-wide segregation analysis and map construction using a reciprocal cross design to facilitate citrus germplasm utilization. Mol Breed 25:659–673
Brookes AJ (1999) The essence of SNPs. Gene 234:177–186
Cai Q, Guy CL, Moore GA (1994) Extension of the linkage map in Citrus using random amplified polymorphic DNA (RAPD) markers and RFLP mapping of cold-acclimation-responsive loci. Theor Appl Genet 89:606–614
Chagné D, Batley J, Edwards D, Forster JW (2007) Single nucleotide polymorphism genotyping in plants. In: Association mapping in plants. Springer, pp 77–94
Chen CX, Bowman KD, Chio YA, Dang PM, Rao MN, Huang S, Soneji JR, McCollum TG, Gmitter FG (2008) EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliata. Tree Genet Genome 4:1–10
Cheng YJ, Guo WW, Yi HL, Pang XM, Deng XX (2003) An efficient protocol for genomic DNA extraction from Citrus species. Plant Mol Biol Rep 21:177–178
Chutimanitsakun Y, Nipper RW, Cuesta-Marcos A, Cistué L, Corey A, Filichkina T, Johnson EA, Hayes PM (2011) Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley. BMC Genomics 12:4
Corazza-Nunes M, Machado M, Nunes W, Cristofani M, Targon M (2002) Assessment of genetic variability in grapefruits (Citrus paradisi Macf.) and pummelos (C. maxima (Burm.) Merr.) using RAPD and SSR markers. Euphytica 126:169–176
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
Durham RE, Liou PC, Gmitter FG, Moore GA (1992) Linkage of restriction fragment length polymorphisms and isozymes in Citrus. Theor Appl Genet 84:39–48
Fujii H, Shimada T, Nonaka K, Kita M, Kuniga T, Endo T, Ikoma Y, Omura M (2013) High-throughput genotyping in citrus accessions using an SNP genotyping array. Tree Genet Genome 9:145–153
García-Lor A, Luro F, Navarro L, Ollitrault P (2012) Comparative use of InDel and SSR markers in deciphering the interspecific structure of cultivated citrus genetic diversity: a perspective for genetic association studies. Mol Genet Genomics 287:77–94
Garnsey S, Su H, Tsai M (1996) Differential susceptibility of pummelo and Swingle citrumelo to isolates of citrus tristeza virus. In: Proceedings of the 13th Conference of the International Organization of Citrus Virologists, Riverside, CA: IOCV, pp 138–146
Gaur R, Azam S, Jeena G, Khan AW, Choudhary S, Jain M, Yadav G, Tyagi AK, Chattopadhyay D, Bhatia S (2012) High-throughput SNP discovery and genotyping for constructing a saturated linkage map of chickpea (Cicer arietinum L.). DNA Res 19:357–373
Gmitter FG, Soneji JR, Rao MN (2009) Citrus breeding. In: Breeding plantation tree crops: temperate species. Springer, pp 105–134
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Grosser JW, Medina-Urrutia V, Ananthakrishnan G, Serrano P (2004) Building a replacement sour orange rootstock: somatic hybridization of selected mandarin + pummelo combinations. J Am Soc Hortic Sci 129:530–534
Han YH, Khu DM, Monteros M (2012) High-resolution melting analysis for SNP genotyping and mapping in tetraploid alfalfa (Medicago sativa L.). Mol Breed 29:489–501
Hirakawa H, Shirasawal K, Ohyama A, Fukuoka H, Aoki K, Rothan C, Sato S, Isobe S, Tabata S (2013) Genome-wide SNP genotyping to infer the effects on gene functions in tomato. DNA Res 20:221–233
Li XM, Quigg RJ, Zhou J, Xu SZ, Masinde G, Mohan S, Baylink DJ (2006) A critical evaluation of the effect of population size and phenotypic measurement on QTL detection and localization using a large F2 murine mapping population. Genet Mol Biol 29:166–173
Li RQ, Li YR, Fang XD, Yang HM, Wang J, Kristiansen K, Wang J (2009a) SNP detection for massively parallel whole-genome resequencing. Genome Res 19:1124–1132
Li RQ, Yu C, Li YR, Lam TW, Yiu SM, Kristiansen K, Wang J (2009b) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Liu YZ, Deng XX (2007) Citrus breeding and genetics in China. Asian Australas J Plant Sci Biotechnol 1:23–28
Luro F, Laigret F, Lorieux M, Ollitrault P (1996) Citrus genome mapping with molecular markers: two maps obtained by segregation analysis of progeny of one intergeneric cross. In: Proc Intl Soc Citricult, pp 862–866
Nicolosi E, Deng Z, Gentile A, La Malfa S, Continella G, Tribulato E (2000) Citrus phylogeny and genetic origin of important species as investigated by molecular markers. Theor Appl Genet 100:1155–1166
Ollitrault P, Dambier D, Luro F, Duperray C (1994) Nuclear genome size variations in citrus. Fruits 49:475–476
Ollitrault P, Terol J, Chen CX, Federici CT, Lotfy S, Hippolyte I, Ollitrault F, Bérard A, Chauveau A, Cuenca J, Costantino G, Kacar Y, Mu L, Garcia-Lor A, Froelicher Y, Aleza P, Boland A, Billot C, Navarro L, Luro F, Roose ML, Gmitter FG, Talon M, Brunel D (2012) A reference genetic map of C. clementina hort. ex Tan.; citrus evolution inferences from comparative mapping. BMC Genomics 13:593
Ramón-Laca L (2003) The introduction of cultivated citrus to Europe via Northern Africa and the Iberian Peninsula. Econ Bot 57:502–514
Ruiz C, Paz Breto M, Asíns MJ (2000) A quick methodology to identify sexual seedlings in citrus breeding programs using SSR markers. Euphytica 112:89–94
Sankar AA, Moore GA (2001) Evaluation of inter-simple sequence repeat analysis for mapping in Citrus and extension of the genetic linkage map. Theor Appl Genet 102:206–214
Scaglione D, Acquadro A, Portis E, Tirone M, Knapp S, Lanteri S (2012) RAD tag sequencing as a source of SNP markers in Cynara cardunculus L. BMC Genomics 13:3
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP, Burns P, Davis TM, Slovin JP, Bassil N, Hellens RP, Evans C, Harkins T, Kodira C, Desany B, Crasta OR, Jensen RV, Allan AC, Michael TP, Setubal JC, Celton JM, Rees DJG, Williams KP, Holt SH, Rojas JJR, Chatterjee M, Liu B, Silva H, Meisel L, Adato A, Filichkin SA, Troggio M, Viola R, Ashman TL, Wang H, Dharmawardhana P, Elser J, Raja R, Priest HD, Bryant DW Jr, Fox SE, Givan SA, Wilhelm LJ, Naithani S, Christoffels A, Salama DY, Carter J, Girona EL, Zdepski A, Wang WQ, Kerstetter RA, Schwab W, Korban SS, Davik J, Monfort A, Denoyes-Rothan B, Arus P, Mittler R, Flinn B, Aharoni A, Bennetzen JL, Salzberg SL, Dickerman AW, Velasco R, Borodovsky M, Veilleux RE, Folta KM (2010) The genome of woodland strawberry (Fragaria vesca). Nat Genet 43:109–116
Soost R, Cameron J (1975) Citrus. Advances in fruit breeding, pp 507–540
Terol J, Naranjo MA, Ollitrault P, Talon M (2008) Development of genomic resources for Citrus clementina: characterization of three deep-coverage BAC libraries and analysis of 46,000 BAC end sequences. BMC Genomics 9:423
Van OJ (2006) JoinMap 4 Software for the calculation of genetic linkage maps in experimental populations Kyazma BV, Wageningen, Netherlands
Varshney RK, Nayak SN, May GD, Jackson SA (2009) Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 27:522–530
Wai CM (2013) Development of Pineapple (Ananas comosus L.) Genetic map with restriction site associated DNA (RAD) markers. In: Plant and Animal Genome XXII Conference, Plant and Animal Genome, San Diego, CA, pp 452
Wang N, Fang LC, Xin HP, Wang LJ, Li SH (2012a) Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. BMC Plant Bio 12:148
Wang WX, Huang S, Liu YM, Fang ZY, Yang LM, Hua W, Yuan SX, Liu SY, Sun JF, Zhuang M, Zhang YY, Zeng AS (2012b) Construction and analysis of a high-density genetic linkage map in cabbage (Brassica oleracea L. var. capitata). BMC Genomics 13:523
Ward JA, Bhangoo J, Fernández-Fernández F, Moore P, Swanson JD, Viola R, Velasco R, Bassil N, Weber CA, Sargent DJ (2013) Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genomics 14:2
Weber CA, Moore GA (2003) Mapping freeze tolerance quantitative trait loci in a Citrus grandis×Poncirus trifoliata F1 pseudo-testcross using molecular markers. J Am Soc Hortic Sci 128:508–514
Wu J, Wang ZW, Shi ZB, Zhang S, Ming R, Zhu SL, Khan MA, Tao ST, Korban SS, Wang H, Chen NJ, Nishio T, Xu X, Cong L, Qi KJ, Huang XS, Wang YT, Zhao X, Wu JY, Deng C, Gou CY, Zhou WL, Yin H, Qin GH, Sha YH, Tao Y, Chen H, Yang YN, Song Y, Zhan DL, Wang J, Li LT, Dai MS, Gu C, Wang YZ, Shi DH, Wang XW, Zhang HP, Zeng L, Zheng DM, Wang CL, Chen MS, Wang GB, Xie L, Sovero V, Sha SF, Huang WJ, Zhang SJ, Zhang MY, Sun JM, Xu LL, Li Y, Liu X, Li QS, Shen JH, Wang JY, Paull RE, Bennetzen JL, Wang J, Zhang SL (2013) The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res 23:396–408
Wu GA, Prochnik S, Jenkins J, Salse J, Hellsten U, Murat F, Perrier X, Ruiz M, Scalabrin S, Terol J, Takita MA, Labadie K, Poulain J, Couloux A, Jabbari K, Cattonaro F, Fabbro CD, Pinosio S, Zuccolo A, Chapman J, Grimwood J, Tadeo FR, Estornell LH, Muñoz-Sanz JV, Ibanez V, Herrero-Ortega A, Aleza P, Pérez-Pérez J, Ramón D, Brunel D, Luro F, Chen CX, Farmerie WG, Desany B, Kodira C, Mohiuddin M, Harkins T, Fredrikson K, Burns P, Lomsadze A, Borodovsky M, Reforgiato G, Freitas-Astúa J, Quetier F, Navarro L, Roose M, Wincker P, Schmutz J, Morgante M, Machado MA, Talon M, Jaillon O, Ollitrault P, Gmitter F, Rokhsar D (2014a) Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat Biotechnol 32:656–662
Wu J, Li LT, Li M, Khan MA, Li XG, Chen H, Yin H, Zhang SL (2014b) High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J Exp Bot: eru311
Xu Q, Chen LL, Ruan XA, Chen DJ, Zhu AD, Chen CL, Bertrand D, Jiao WB, Hao BH, Lyon MP, Chen JJ, Gao S, Xing F, Lan H, Chang JW, Ge XH, Lei Y, Hu Q, Miao Y, Wang L, Xiao SX, Biswas MK, Zeng WF, Guo F, Cao HB, Yang XM, Xu XW, Cheng YJ, Xu J, Liu JH, Luo JH, Tang ZH, Guo WW, Kuang HH, Zhang HY, Roose ML, Nagarajan N, Deng XX, Ruan YJ (2013) The draft genome of sweet orange (Citrus sinensis). Nat Genet 45:59–66
Yagi M, Yamamoto T, Isobe S, Hirakawa H, Tabata S, Tanase K, Yamaguchi H, Onozaki T (2013) Construction of a reference genetic linkage map for carnation (Dianthus caryophyllus L.). BMC Genomics 14:734
Yan JB, Yang XH, Shah T, Sánchez-Villeda H, Li JS, Warburton M, Zhou Y, Crouch JH, Xu YB (2010) High-throughput SNP genotyping with the GoldenGate assay in maize. Mol Breed 25:441–451
Ye YM (1997) The diversity centre of pummelo germplasm. South Chin Fruit (China) 26:3–5
Zhao HH, Fernando RL, Dekkers JC (2007) Power and precision of alternate methods for linkage disequilibrium mapping of quantitative trait loci. Genetics 175:1975–1986
Acknowledgments
This work was funded by the Ministry of Science and Technology of China (2011CB100600), the National Natural Science Foundation of China (31221062) and the Huazhong Agricultural University Scientific &Technological Self-innovation Foundation (2012YB11).
Data archiving statement
Genetic map is now under process of submission to Citrus Genome Database and will be completed during the review.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W.-W. Guo
Supplementary materials
Below is the link to the electronic supplementary material.
Supplementary material 1
List of markers, linkage groups, genetic distances, and genome (sweet orange) location of SNPs and SSRs. (XLS 339 kb)
Supplementary material 2
Comparison of SSR markers with previously published linkage maps. (XLS 15 kb)
Supplementary material 3
Genetic maps for Guanxi pummelo (female) and Pingshan pummelo (male). (GIF 200 kb)
Rights and permissions
About this article

Cite this article
Guo, F., Yu, H., Tang, Z. et al. Construction of a SNP-based high-density genetic map for pummelo using RAD sequencing. Tree Genetics & Genomes 11, 2 (2015). https://doi.org/10.1007/s11295-014-0831-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-014-0831-0