
Abstract
In the study, we developed new markers for phylogenetic relationships and intraspecies differentiation in Coffea. Nana and Divo, two novel Ty1-copia LTR-retrotransposon families, were isolated through C. canephora BAC clone sequencing. Nana- and Divo-based markers were used to test their: (1) ability to resolve recent phylogenetic relationships; (2) efficiency in detecting intra-species differentiation. Sequence-specific amplification polymorphism (SSAP), retrotransposon-microsatellite amplified polymorphism (REMAP) and retrotransposon-based insertion polymorphism (RBIP) approaches were applied to 182 accessions (31 Coffea species and one Psilanthus accession). Nana- and Divo-based markers revealed contrasted transpositional histories. At the BAC clone locus, RBIP results on C. canephora demonstrated that Nana insertion took place prior to C. canephora differentiation, while Divo insertion occurred after differentiation. Combined SSAP and REMAP data showed that Nana could resolve Coffea lineages, while Divo was efficient at a lower taxonomic level. The combined results indicated that the retrotransposon-based markers were useful in highlighting Coffea genetic diversity and the chronological pattern of speciation/differentiation events. Ongoing complete sequencing of the C. canephora genome will soon enable exhaustive identification of LTR-RTN families, as well as more precise in-depth analyses on contributions to genome size variation and Coffea evolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Global changes occurring as a result of rapid climate change and forest degradation (FAO 2006) are endangering biodiversity in tropical areas (Gardner et al. 2009). In this setting, wild coffee trees (Coffea genus) native to tropical forests of Africa, Madagascar, Mascarenes and Comoros are especially at risk. Indeed, out of the 103 currently described Coffea species, 72 (approximately 70%) are threatened with extinction as a result of the quantitative and qualitative decline in their habitat (Davis et al. 2006). The three botanical sections defined by Chevalier (1947), i.e., Eucoffea (West and Central African species), Mozambicoffea (East African species) and Mascarocoffea (from Madagascar, Comoros and Mascarenes) are also characterized by several morpho-physiologic traits. F1 interspecific hybrid sterility was the first evidence of genetic divergence between eastern and western-central African species (Louarn 1992). High divergence was also reported between Malagasy and African species (Charrier 1978). This divergence is generally associated with (1) genome size variation (0.50–0.88 pg/1C, Noirot et al. 2003), higher genome size differences between parental species and higher F1 hybrid sterility and (2) distinct rDNA chromosomal distributions (Hamon et al. 2009). Early phylogenetic studies (26 species/taxa) based on ITS (Lashermes et al. 1997) and cpDNA sequence divergence (Cros et al. 1998) indicated that the Coffea genus was geographically organized within four clades i.e., West and Central Africa, Central Africa, East Africa and Madagascar. A more complete molecular phylogenic analysis of the genus (86 species, Maurin et al. 2007), based on four plastid and one nuclear regions, supported the major geographical lineages but highlighted large unresolved branches. Similarly, the use of microsatellite markers did not completely resolve the relationships among Coffea species (Cubry et al. 2008). However, at a lower taxonomic level, studies using molecular markers, such as RFLPs (Dussert et al. 1999) and microsatellites (Gomez et al. 2009), detected genetic differentiation within C. canephora—five major genetic groups (A–E). Hence, to overcome the low species differentiation and insufficient marker variability and informativeness, it would be essential to develop new approaches able to detect diversity at the required taxonomic level.
Retrotransposons are classified according to the presence or not of a long terminal repeat (LTR) at their extremities. LTR-retrotransposons are generally subdivided into Ty1-copia and Ty3-gypsy groups based on their internal coding region organization (Wicker et al. 2007; Hill et al. 2005). LTR-retrotransposons are abundant in plant genomes, and numerous families have been identified and characterized (Vitte and Panaud 2005). The replicative mechanism (via an RNA intermediate) results in preservation of the original copy and its amplification throughout the genome through copy–paste mechanisms. Stress could induce transpositional activity and de novo insertions that generate rapid genetic variability (Melayah et al. 2001). In some extreme cases, intense transpositional LTR-retrotransposon activity results in a rapid increase in their copy number, leading to profound changes in the genome organization such as a rapid increase in genome size (Bennetzen 2000; Kalendar et al. 2000; Piegu et al. 2006). These structural features, as well as the activity and dispersion, of LTR-retrotransposons have prompted their widespread exploitation as effective molecular markers for genetic diversity analysis between genera (Pearce et al. 2000; Kumar and Hirochika 2001; Schulman 2007). Two popular retrotransposon-based marker techniques, based on PCR detection of anonymous insertion polymorphism at the genome scale, have been developed in plants: sequence-specific amplified polymorphism (SSAP, Waugh et al. 1997) and retrotransposon-microsatellite amplified polymorphism (REMAP, Kalendar et al. 1999). SSAP detects insertion polymorphism in LTR-retrotransposons next to a specified restriction site, while REMAP detects LTR-retrotransposons inserted close to a microsatellite. SSAP and REMAP polymorphism patterns reflect the situation encountered at the whole genome level and the history of LTR-retrotransposon transpositional activity.
In addition to SSAP and REMAP, the locus-specific retrotransposon-based insertional polymorphism technique (RBIP, Flavell et al. 1998) appears to be an interesting method for detecting polymorphism with respect to integration of an element at a particular locus. The RBIP method requires retrotransposon and flanking sequence information for primer design. RBIP is the only retrotransposon-based marker method yielding co-dominant markers. Using these methods, retrotransposon-based markers have proven to have great potential for detecting diversity at different taxonomic levels, analyzing evolutionary history and resolving complex pedigrees or species lineages (Pearce et al. 2000; Bousios et al. 2007; Natali et al. 2007; Petit et al. 2007; Sanz et al. 2007; D’Onofrio et al. 2010).
In this study, we identified the first two Coffea LTR-retrotransposons (LTR-RTN) and performed a comparative analysis of these LTR-RTNs as potential sources of polymorphic markers for Coffea genetic studies. We used RBIP, SSAP and REMAP approaches to reveal the genetic diversity and infer the evolutionary history of Coffea (182 accessions studied, corresponding to 32 species). We aimed to: (1) characterize the two new LTR-RTN families, Nana and Divo, identified in C. canephora; (2) assess both elements as markers and compare their insertional patterns; and (3) examine their relative contributions to gaining further insight into Coffea species relationships and, at a lower taxonomic level, within species differentiation.
Materials and methods
Plant materials and DNA isolation
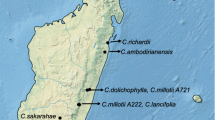
In all, 182 accessions belonging to 31 Coffea species, plus one accession of the genus most closely related to Coffea (Robbrecht and Manen 2006), i.e., Psilanthus ebracteolatus from Côte d’Ivoire, were studied. The 31 Coffea species are representatives of the natural geographic distribution of the genus, i.e., West and Central Africa, East Africa and Indian Ocean islands (Madagascar and Comoros) (Fig. 1).

Natural geographic distribution of the Coffea species studied, according to Charrier (1978), Davis et al. (2006) and Noirot et al. (2003). Because of overlap in some species distributions, four Africa maps denoted in a–d were used. Africa a: 1, C. liberica var liberica; 2, C. liberica var dewevrei; 3, C. congensis; 4, C. kapakata; 5, C. pseudozanguebariae; 6, C. racemosa. Africa b: 1, C. stenophylla; 2, C. heterocalyx; 3, C. anthonyi; 4, C. sessiliflora; 5, C. salvatrix. Africa c: 1, C. humilis; 2, C. brevipes; 3, C. sp Congo, C. sp Ngongo2, C. sp Mayombe; 4, C. costatifructa. Africa d: 1, C. canephora; 2, C. arabica; 3, C. eugenioides. Comoros: 1, C. humblotiana. Madagascar: 2, C. tsirananae; 3, C. heimii; 4, C. perrieri; 5, C. millotii; 6, C. lancifolia; 7, C. sakarahae; 8, C. resinosa; 9, C. dolichophylla; 10, C. bertrandii. Dotted line shows geographic distribution of the Coffea genus
Except for C. canephora, fresh leaves corresponding to 20 African Coffea species (total: 81 accessions) and P. ebracteolatus (1 accession) were harvested on plants maintained in IRD tropical greenhouses (Montpellier, France), and 10 Mascarocoffea accessions (belonging to 10 species) were provided by the National Research Centre for Applied Rural Development (FOFIFA), Madagascar. DNA was purified using the Qiagen DNeasy Plant Maxi Kit® or the DNeasy Plant Mini Kit® (Quiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Quantification was done using a NanoDrop TM 1000 Spectrophotometer (LabTech, France).
Regarding C. canephora sampling, DNAs from 78 wild and cultivated accessions from Côte d’Ivoire, Cameroon, Congo and Central African Republic were kindly provided by S. Dussert. Those from the 12 wild Ugandan accessions were kindly supplied by P. Musoli from NARO/COREC (Uganda). These accessions were representative of the genetic diversity range within this species (five genetic groups, namely A–E, Gomez et al. (2009) plus the Ugandan group, Cubry et al. 2008).
The species and number of accessions used according to the technique applied, the geographic origin of accessions and the code species used on the genetic trees obtained in this study are given in Table 1.
LTR-retrotransposon characterization and annotation
Two different C. canephora BAC clone libraries were used for this study. One (Leroy et al. 2005) gave us access to the 49N24 BAC clone containing a caffeic acid O-methyltransferase (CcCOMT) gene. The second library constructed from the C. canephora doubled haploid genotype DH200-94 (de Kochko et al. 2010) provided high-density filters for the DNA–DNA hybridization step to extrapolate the LTR-RTNs minimum copy number.
The 49N24 BAC clone sequence was analyzed using BLASTN algorithms (Altschul et al. 1997) against public and private databases. LTR-RTNs were identified and annotated by similarity searches against plant sequences. Detailed analyses of LTR-RTNs were performed with the EMBOSS package (Rice et al. 2000) and by dot-plot (Sonnhammer and Durbin 1995). Protein domains were identified using Pfam (http://pfam.sanger.ac.uk). Final annotation was performed with the Artemis tool (Rutherford et al. 2000). Two LTR-RTN sequences, named Nana and Divo, were deposited in GenBank (HM755953 and HM755952). The specific Nana and Divo primers used in this study were designed using Primer 3 (http://frodo.wi.mit.edu/); the primer sequences are given in Table 2.
Nana and Divo minimum copy number estimation
The DH200-94 HindIII BAC library consists of 36,864 clones with an average insert size of 135 kb (de Kochko et al. 2010). This BAC library represents 7.9 equivalent haploid C. canephora genomes (704 Mb, Noirot et al. 2003). LTR and gag probes for each LTR-RTN were amplified with the specific primers listed in Table 2. After purification with the QIAquick PCR purification kit (Qiagen), the probes were quantified before labeling with [α32P]-dATP random priming using the Prime-a-Gene® labeling kit (Promega) according to the manufacturer’s instructions. DNA–DNA hybridizations were performed using the protocol described in Bustamante-Porras et al. (2007). Membranes were then placed for 3–4 h under a screen sensitive to 32P rays (Phosphor Screen, Amersham®, Amersham, UK). Images were then analyzed with GIMP2.6.6 software (http://www.gimp.org). Double signals in concordance with the deposit pattern in elementary squares on the high-density filters were scored as positive as shown on one membrane hybridized with the LTR Divo probe (Fig. 2).

Southern hybridization with a 32P radiolabeled Divo LTR probe. The DH200-94 BAC high-density filter shown contains 18,432 clones (de Kochko et al. 2010). Only double signals on each elementary square are considered
Nana and Divo expression assessment in C. canephora
Specific primers were designed on the predicted gag gene of each LTR-RTN as described in Guyot et al. (2009) (Table 2). PCR amplification was performed on cDNA libraries obtained from C. canephora young leaves and fruits at different development and maturation stages (Mahesh et al. 2006).
Survey of Nana and Divo insertion at the 49N24 locus using retrotransposon-based polymorphism
For retrotransposon-based polymorphism (RBIP) experiments, three types of primers were designed as follows: one (RBIP-2) in the LTR region (oriented toward the exterior of the element) and two others (RBIP-1 and RBIP-3) in the flanking region outside the RTN (Table 2; Fig. 3). The PCR was performed in a 10-μL total volume with 25 ng template DNA, 0.1 μM primers, 0.2 mM dNTPs, 5 mM MgCl2 and 0.5 U Taq® DNA polymerase Promega buffer 1×. The following program was used: −94°C for 4 min, 27 cycles at 94°C for 45 s, 53°C for 1 min, 72°C for 1 min and 72°C for 8 min.

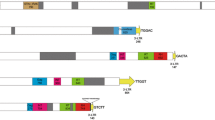
Schematic representation of the structural features of Nana and Divo LTR-retrotransposons. Hatched boxes represent LTRs while black boxes and gray boxes, respectively, represent the predicted coding regions and protein domains (Zn zinc finger, Pr protease, Int integrase and Rvt reverse transcriptase). Small triangles symbolize the positions of primers (given in Table 2) used for PCR amplifications as follows: gray triangles PCR amplifications of gag genes, white triangles PCR amplifications of LTRs and black triangles PCR amplifications of insertion sites
The PCR products were visualized after migration on 1% agarose gel and ethidium bromide staining. The presence of an LTR-RTN at the investigated locus was noted when the amplification was positive with the RBIP-1/RBIP-2 pair, but failed with the RBIP-1/RBIP-3 pair. On the contrary, the absence of an LTR-RTN at this same locus led to a PCR product with the RBIP-1–RBIP-3 pair and no amplification with the RBIP-1–RBIP-2 pair.
All C. canephora accessions (90), one accession per wild species, one C. arabica and one P. ebracteolatus accession were used for RBIP experiments (see Table 1).
Survey and analyses of Nana and Divo genome-wide insertion polymorphism using SSAP and REMAP techniques
For SSAP analyses, the primers used for the pre-amplification step were the EcoRI adapter derived from the AFLP technique, as used in Coulibaly et al. (2003), and RBIP-2. Total DNA (400 ng) was digested with EcoRI (10 U/μg) overnight at 37°C. Ligation of adapters on 200 ng of digested DNA was performed by adding 50 ng EcoRI adapter, 1 U T4 ligase and T4 ligase buffer 1× (Promega) followed by 6 h incubation at room temperature (50 μl final volume). Pre-amplification was done in a 40 μL total volume with 20 ng DNA, 0.2 μM of each primer (EcoRI adapter and RBIP-2), 0.2 mM dNTPs, 0.5 U GoTaq® DNA polymerase (Promega, Madison, WI, USA) and 1× reaction buffer. PCR conditions were similar to those used for RBIP, but 35 cycles were performed and the annealing temperature was 50°C. For selective amplification, the EcoRI adapter was extended in 3′ by -A, -T, -C or –G, while the M13 sequence was added to RBIP-2 in 5′. Selective amplification was performed in a 15 μl final volume with 0.5 μl pre-amplification and 35 cycle amplification with a touchdown annealing temperature: 60 to 55°C for 30 s.
For REMAP analyses, primers facing outward from the LTRs were combined with five anchored simple sequence repeat (SSR) primers from Joshi et al. (2000). These primers were selected for their ability to amplify Coffea genomic DNA and were used in combination with the RBIP-2-M13 primer. The PCR conditions were similar to those used for RBIP amplifications, with the following modifications: 0.2 mM primers (ISSR, RBIP-2 and dyed M13), 35 amplification cycles and 55°C annealing temperature.
PCR products obtained with REMAP or SSAP procedures were analyzed on an automated 16-capillary ABI Prism® 3130xl (Applied Biosystems, Foster City, CA, USA) sequencer. Data reads were done with GeneMapper® Software v.3.7 (Applied Biosystems).
SSAP and REMAP techniques provide dominant markers scored according to the presence (1) and absence (0) of each band. Hence, genetic parameters [mean number of insertion sites (i.e., bands) per accession, number of monomorphic sites for the presence and expected heterozygosity over all sites] and F ST values (Lynch and Milligan 1994) between species were calculated using the AFLPsurv program (http://www.ulb.ac.be/sciences/lagev/aflp-surv.html). Genetic similarities were calculated from the binary presence/absence matrices using Dice’s coefficient (Dice 1945). Neighbor-joining trees were constructed using DARwin Version 5.0.155 software (http://darwin.cirad.fr/darwin) with bootstrapping (1,000 replicates).
For these experiments, 8 Mascarocoffea accessions (each one corresponding to one species), 70 accessions from Africa (i.e., nine Coffea species represented by 1–10 accessions, depending on their availability), 17 C. canephora accessions (among the 90 available, selected to be representative of the five intraspecific genetic groups and intergroup hybrids) and 1 P. ebracteolatus accession were used (see Table 1).
Results
Identification, characterization and annotation of two novel Ty1-copia retrotransposon families—Nana and Divo
In an attempt to characterize CcCOMT encoding genes, the BAC clone 49N24 was previously isolated from a C. canephora BAC library (Leroy et al. 2005); 190 kb of the BAC clone was sequenced using the Roche FLX 454 pyrosequencing technique. Detailed annotations of the contigs revealed the presence of two full-length LTR-RTNs belonging to the Ty1-copia group—they were named Nana and Divo (accession no. HM755953 and HM755952, respectively).
Nana and Divo were found to be 5,749 and 4,396 bp in size, respectively. Each LTR-RTN had similar sized 5′ and 3′ LTRs, and high nucleotide identity was noted for each LTR pair, but it was slightly higher for Divo (94.5%) than Nana (90.5%). The two LTR-RTNs differed in terms of LTR pair size (Nana 5′ LTR, 677 bp, and 3′ LTR, 641 bp; Divo 5′ LTR, 407 bp, and 3′ LTR, 385 bp) and sequence. Moreover, Nana and Divo did not share any significant sequence similarity and hence belonged to two different families. The reconstructed putative coding sequences for Nana and Divo showed strong similarities to the plant LTR- RTN Ty1-copia group. For Nana, the protease (PR), integrase (INT) and reverse transcriptase (RT) motifs were found in this order, while only the RT motif was detected for Divo (Fig. 3).
For both Nana and Divo, similarity searches (BLASTn) of the EST division of GenBank (dbEST, August 2010) did not detect any similarity with known sequences (imperfect match in the predicted gag domain of Nana, DV666936, E value 10e−105, 78% identity). To test Nana and Divo expression, primers were designed within the Nana and Divo predicted gag domains (Fig. 3), and PCR amplifications were performed in C. canephora fruit and leaf cDNA libraries (Mahesh et al. 2006). No PCR amplification was observed for the two LTR-RTNs.
Minimum copy Nana and Divo insertion number in C. canephora
A minimum Nana and Divo copy number evaluation was performed after hybridization of the DH200-94 C. canephora Hind III BAC clones spotted on two filters (de Kochko et al. 2010) with two LTR-RTN specific probes (LTR and gag). Despite probable underestimation of the copy number (inability to distinguish unique or multiple insertions within a single BAC clone, incomplete genome cloning within the BAC library, lack of the probe corresponding sequence in a degenerated LTR-RTN), this experiment gave the minimum copy number of each LTR-RTN in the C. canephora genome (Table 3). Screening with LTR and gag probes, respectively, revealed 368 and 200 positive hybridization signals for Nana versus 442 and 1,317 for Divo. We used the estimated genome coverage (7.9×) for the DH200-94 HindIII BAC library and extrapolated the copy number of LTR and gag probes per genome as 46 and 25 versus 55 and 165 for Nana and Divo, respectively (Table 3). Based on the assumption that the simultaneous hybridization patterns of both LTR and gag probes represented the presence of an almost complete copy of LTR-RTN, 22 Nana and 43 Divo complete copies were detected (Table 3).
Divo and Nana 49N24 locus insertion polymorphism survey
Among C. canephora accessions, the RBIP technique revealed marked differences in 49N24 locus insertion polymorphism between the two LTR-RTNs. Nana was present at this locus in all accessions tested regardless of their nature (wild or cultivated) and genetic diversity group (Table 4). On the contrary, insertion polymorphism was noted for Divo. For instance, among wild accessions, Divo was only present in the heterozygous state and only detected in accessions from genetic groups D (9/26) and E (1/8). Among the cultivated accessions, homozygous accessions for presence (of Divo) were only detected in the genetic group A (1/8 accessions) and intergroup hybrids AE (1/5) and DE (1/10). Heterozygous accessions belonged to genetic groups A (3/8), E (2/6) and to intergroup hybrids AE (1/5) and DE (3/10).
Among the remaining Coffea species, the RBIP technique also revealed substantial differences in insertion polymorphism patterns between Nana and Divo. Divo was absent from all analyzed Coffea species and from P. ebracteolatus, while Nana exhibited insertion polymorphism (Table 5). For instance, species native to East Africa (Mozambicoffea) were homozygous for presence (of Nana) while Nana was absent from Mascarocoffea (species from Indian Ocean islands) and P. ebracteolatus. Species from West and Central Africa (Eucoffea) appeared to be homozygous for presence (4/17), for absence (8/17) or heterozygous (3/17).
Nana and Divo genome-wide polymorphism patterns in the Coffea genus
To evaluate Nana and Divo polymorphism on the genome scale, we analyzed genetic diversity in 95 accessions belonging to 18 Coffea species and in one P. ebracteolatus accession (Table 1), using both SSAP and REMAP techniques. The mean number of bands (representing LTR-RTN insertion sites) per accession ranged from 2 to 17.5 for Divo and 2.5 to 15.2 for Nana, irrespective of the technique used (Table 6). Apart from C. arabica, the highest numbers were obtained for C. canephora (7.7–15.2 bands).
Regarding each species represented by more than two accessions, few monomorphic loci for presence (1–4) were detected. All bands recorded for Psilanthus but one were also recorded for Coffea. Regarding species represented by equal sample size, high levels of polymorphic sites at 5% (57.1–100%) were generally obtained, regardless of the technique and species. The average expected heterozygosity (H E) calculated for all sites (i.e., loci) was low and ranged from 0.13 to 0.20, regardless of the LTR-RTN and technique used.
Genetic relationships were inferred using Nana and Divo genome-wide polymorphisms. With Nana, except for the C. liberica var. liberica/C. liberica var. dewevrei (Lib/Dew) pair, medium to strong differentiation between species (pairwise F ST ranged from 0.055 to 0.23) was clearly noted regardless of the technique used. Conversely, with Divo, no or weak differentiation was detected between species (average F ST: <0.08), except for pairs involving C. canephora (F ST from 0.10 to 0.29 depending on the technique).
Considering each LTR-RTN separately, REMAP and SSAP techniques generated non-redundant information (Mantel test coefficient values, r = 0.19 and 0.21 with Nana and Divo, respectively, nonsignificant at P ≤ 0.05). Hence, combining REMAP and SSAP data for each LTR-RTN to produce phylogenetic trees was fully justified and NJ trees were produced for each Nana and Divo (Fig. 4). Considerable differences were observed between tree topologies. Nana revealed eight major distinct clusters (Fig. 4a). Most of them corresponded to separate species, except for the C. heterocalyx/C. eugenioides/C. arabica (Het/Eug/Ara) grouping. The eight species from Indian Ocean islands (Mascarocoffea) were clustered and located close to C. racemosa (RAC, eastern African species). P. ebracteolatus (PSI) did not cluster with any other Coffea accession. A completely different tree topology was found for Divo (Fig. 4b). Divo-based markers: (1) distinguished C. liberica var. liberica (LIB) and C. canephora (CAN) from the remaining species, and (2) revealed marked intra-C. canephora differentiation. To a lesser extent, some of the Mascarocoffea species were found to be grouped together. For the remaining Coffea species, there was no clear separation corresponding to the species classification. The accessions were found to be highly intermixed despite the presence of long branches corresponding to high but unshared polymorphism.

Neighbor-joining tree representing genetic relationships between 95 accessions from 18 Coffea species and one accession from Psilanthus ebracteolatus. The tree was based on the Dice dissimilarity matrix calculated from both REMAP and SSAP bands obtained with Nana (a) and Divo (b). Bootstrap values were calculated with 1,000 replicates, and only values greater than or equal to 50 are shown. The main groups are outlined. Species and corresponding botanical section codes are as indicated in Table 1
Discussion
Nana and Divo LTR-RTNs: two Ty1-copia families with unrelated insertion patterns
LTR-RTNs are known to represent a major plant genome component, but no LTR-RTN had been previously identified and described in Coffea. Only transposable elements were observed in an analysis of the 46C02 BAC clone sequence (Guyot et al. 2009). In this study, the two LTR-RTNs, i.e., Nana and Divo, isolated from the 49N24 BAC clone, belong to two novel Ty1-copia group families, since no similar sequences have been deposited in public databases. The failure in their amplification from C. canephora cDNA libraries suggested that, under the tested conditions, Nana and Divo were not expressed in C. canephora and therefore that they were inactive. Given the higher nucleotide identity between LTR sequence pairs for Divo (94.5%) than for Nana (90.5%), and despite the more complete sequence of Nana as compared to Divo at the investigated site, Nana seemed to be older than Divo. Furthermore, the estimated total minimum number of Nana and Divo putative complete copies (when both LTR and gag sequences were present) in the C. canephora genome, i.e., higher for Divo than for Nana (43 and 22, respectively), indicated more recent transpositional activity for Divo than for Nana. The low expected heterozygosity calculated for all loci probably indicates ancient transpositional activities of the two LTR-RTNs, even though several rounds could have occurred during evolution of the Coffea genus, which could explain the high levels of polymorphic sites per species.
The evolutionary relationship patterns revealed by retrotransposon-based markers are often dependent on the underlying retrotransposon activity over time. In this study, we performed a comparative analysis of the structure and information associated with each retrotransposon.
Nana and Divo had different insertion timings: consequences for understanding evolution of the Coffea genus
Combining RTNs with different evolutionary histories provides higher resolution for studying complex species relationships (Pearce et al. 2000; Konovalov et al. 2010; Tam et al. 2007). Hence, recently active RTN families should provide polymorphic markers that are informative at the intraspecific level but uninformative at higher taxonomic levels (i.e., genus or interspecific level). Conversely, RTN families that have been inactive over a long evolutionary period will give informative markers at the interspecific level. Using a multi retrotransposon-based marker approach, we conducted an in-depth investigation of the insertion activity of Nana and Divo in several Coffea species and one accession belonging to the closest genus, Psilanthus, and demonstrated the unique behavior of each LTR-RTN family.
The results we obtained using the RBIP approach applied to the 49N24 locus indicated an ancestral insertion of Nana since it was fixed at this locus in all C. canephora accessions, prior to genetic differentiation of the species. In contrast, Divo was absent in all wild accessions, but present in 22 out of the 90 C. canephora accessions tested, indicating a more recent insertion. Regarding the Coffea genus results, the absence of Nana in all Mascarocoffea accessions tested, while being present in African accessions, suggested that its insertion at the investigated site occurred after the African and Malagasy species separation.
In addition to the evolutionary analysis of these results and those discussed hereafter, the RBIP approach appeared to be fairly efficient for isolating relatively old insertions, which occurred in the Coffea genus. Moreover, the co-dominant nature of RBIP markers appeared to be useful for evaluating intra-specific genetic diversity in C. canephora (fixed Divo insertions at the 49N24 site were only noted for cultivated accessions, Table 4). The recent development of RBIP high throughput analysis technology will provide an ideal future tool for evaluating Coffea germplasm collections (Jing et al. 2010).
At the genomic scale, irrespective of the LTR-RTN considered (Nana or Divo), REMAP and SSAP-based markers gave complementary information (nonsignificant Mantel’s test r values for both Nana and Divo). The clearest Coffea genus organization and the best intra-C. canephora differentiation were obtained by combining these markers. Completely different Divo and Nana insertion activities were revealed. For instance, the genetic diversity assessed according to the Divo distribution in 18 Coffea species and one Psilanthus species revealed C. canephora differentiation and C. liberica var liberica divergence (Fig. 4b), while species divergence was mainly observed on the basis of Nana insertion polymorphism.
New insight was obtained at the genus level concerning the relative position of Psilanthus in comparison to Coffea. As the ancestral state of a retrotransposon insertion is known and subsequent changes at the locus are not subject to homoplasy (Schulman 2007), REMAP and SSAP bands shared between accessions have been inherited from a common ancestor. The presence of Divo and Nana in Coffea and Psilanthus genera indicated that a first round of insertion events took place early in the evolution of these genera. These results support their close phylogenetic relationship as reported by Couturon et al. (1998) and Robbrecht and Manen (2006). Nevertheless, there is a clear-cut genetic divergence between the two genera, as noted by the non-amplification with 7 out of the 16 primer combinations (REMAP and SSAP).
Regarding evolution of the Coffea genus, we obtained more precise information concerning the relative status of the two subspecies C. liberica var liberica (LIB) and. C. liberica var dewevrei (DEW) from Divo. Indeed, Divo insertion polymorphism highlighted the “recent” divergence of C. liberica var liberica and supported their classification into two separated species, as suggested by N’Diaye et al. (2005).
Altogether, these results suggest different timing in the two LTR-RTN insertions. The LIB/DEW pair was differentiated from the remaining species during early Nana activity, but at that time LIB and DEW had not yet diverged. Then, more recent Divo activity induced genetic divergence between LIB and DEW and enabled differentiation of LIB. Interestingly, C. humilis and C. stenophylla were well separated on the Nana tree (Fig. 4a) even though they appeared to be closely linked on all former phylogenic trees, including the most complete one constructed by Maurin et al. (2007). The eight non-African species (Mascarocoffea) used in this study appeared to be well grouped and closer to C. racemosa (from Tanzania) than to any other species. Of course, these results did not enable us to conclude on their genetic origin, but they are congruent with their putative eastern African origin, as suggested by Charrier (1978) and Leroy (1980).
Our results strongly suggest that the major transpositional activity of Nana was associated with Coffea speciation, while more recent Divo activity took place during C. canephora differentiation and C. liberica var. liberica divergence. Hence, phylogenetic trees obtained with these two LTR-RTNs showed contrasted species distribution patterns and relationships.
Informativeness and comparison with other marker systems
LTR-RTNs could provide more polymorphic markers than microsatellite markers, especially for genera and/or species characterized by low variation levels. Moreover, direct comparisons between retrotransposon-based and amplified fragment length polymorphism (AFLP) markers have indicated that retrotransposon-based markers are considerably more informative in plants (Waugh et al. 1997; Porceddu et al. 2002; Venturi et al. 2006).
In the present study, different phylogenetic tree topologies were obtained. On one hand, the Nana tree clustered Coffea accessions into eight consistent groups and supported previous reports of Cubry et al. (2008), whereas the Divo tree showed differentiation among C. canephora, as reported by Dussert et al. (1999) using RFLPs and then Gomez et al. (2009) with SSRs. It also revealed strong differentiation among the LIB/DEW pair (LIB well separated). However, contrary to other molecular markers, Nana and Divo were informative on the timing of events. C. canephora differentiation is not yet complete, whereas LIB and DEW appear to have fully diverged. Indeed, inter-C. canephora genetic group hybrids are completely fertile, while only 44.2% fertility has been reported for LIB-DEW hybrids (N’Diaye et al. 2005).
Conclusion
This study indicates that retrotransposon-based molecular marker approaches are informative for studying Coffea genetic diversity. We showed that Nana and Divo, the two first LTR-RTNs detected in Coffea (C. canephora), belong to two ancient families. Their presence was detected in two genera, i.e., Coffea and Psilanthus. We demonstrated that distinct LTR-RTN insertion activities might give different but complementary histories with respect to the evolution of the Coffea genus. Nana-based molecular markers (both SSAP and REMAP) were efficient enough to differentiate most of the species, thus providing adequate markers for assessment at the genus level. In contrast, Divo, which probably belongs to a more recently active LTR-RTN family, gave polymorphic markers that were efficient at the intraspecific level but uninformative at the interspecific level.
Information generated by Divo insertion polymorphism turned out to resemble that obtained via microsatellites regarding C. canephora; most of the genetic groups obtained by Gomez et al. (2009) were also obtained in this study. However, Divo provided new insight into the timing of LIB/DEW pair divergence relative to C. canephora intraspecific differentiation.
Finally, combining LTR-RTNs characterized by different timing in their transpositional activities provided higher resolution for studying complex species relationships.
The ongoing complete sequencing of the C. canephora genome will permit exhaustive identification of LTR-RTN families, while facilitating characterization of those with diverse transpositional activities. In the future, several additional LTR-RTNs will be necessary to be able to completely resolve the genetic diversity and relationships within the Coffea genus. These new LTR-RTNs will allow a more in-depth and precise analysis in terms of the contribution to genome size variation and Coffea genus evolution.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Bennetzen JL (2000) Transposable element contributions to plant gene and genome evolution. Plant Mol Biol 42(1):251–269
Bousios A, Saldana-Oyarzabala I, Valenzuela-Zapatab GA, Wooda C, Pearce RS (2007) Isolation and characterization of Ty1-copia retrotransposon sequences in the blue agave (Agave tequilana Weber var. azul) and their development as SSAP markers for phylogenetic analysis. Plant Sci 172(2):291–298
Bustamante-Porras J, Campa C, Poncet V, Noirot M, Leroy T, Hamon S, de Kochko A (2007) Molecular characterization of an ethylene receptor gene (CcETR1) in coffee trees, its relationship with fruit development and caffeine content. Mol Genet Genomics 277:701–712
Charrier A (1978) La structure génétiques des caféiers spontanés de la région malgache (Mascarocoffea). Leurs relations avec les caféiers d’origine africaine (Eucoffea). Thèse d’état Université Paris-Sud Orsay, p 221
Chevalier A (1947) Les caféiers du globe. III Systématique des caféiers et faux caféiers. Maladies et insectes nuisibles. Lechevalier P ed Paris
Coulibaly I, Revol B, Noirot M, Poncet V, Lorieux M, Carasco-Lacombe C, Minier J, Dufour M, Hamon P (2003) AFLP and SSR polymorphism in a Coffea interspecific backcross progeny [(C. heterocalyx × C. canephora) × C. canephora]. Theor Appl Genet 107(6):1148–1155
Couturon E, Lashermes P, Charrier A (1998) First intergeneric hybrids (Psilanthus ebracteolatus Hiern × Coffea arabica L.) in coffee trees. Can J Bot 76(3):542–546
Cros J, Combes MC, Trouslot P, Anthony F, Hamon S, Charrier A, Lashermes P (1998) Phylogenetic analysis of chloroplast DNA variation in Coffea L. Mol Phylogenet Evol 9(1):109–117
Cubry P, Musoli P, Legnate H, Pot D, de Bellis F, Poncet V, Anthony F, Dufour M, Leroy T (2008) Diversity in coffee assessed with SSR markers: structure of the genus Coffea and perspectives for breeding. Genome 51(1):50–63
D’Onofrio C, Lorenzis G, Giordani T, Natali L, Cavallini A, Scalabrelli G (2010) Retrotransposon-based molecular markers for grapevine species and cultivars identification. Tree Genet Genomes 6:451–466
Davis AP, Govaerts R, Bridson DM, Stoffelen P (2006) An annotated taxonomic conspectus of the genus Coffea (Rubiaceae). Bot J Linn Soc 142(4):465–512
de Kochko A, Akaffou S, Andrade AC, Campa C, Crouzillat D, Guyot R, Hamon P, Ming R, Mueller LA, Poncet V, Tranchant-Dubreuil C, Hamon S (2010) Advances in coffea genomics. Adv Bot Res 53:23–63
Dice LR (1945) Measures of the amount of ecologic association between species. Ecology 26:297–302
Dussert S, Lashermes P, Anthony F, Montagnon C, Trouslot P, Combes BC, Berthaud J, Noirot M, Hamon S (1999) Coffee (Coffea canephora). In: Hamon, P Seguin, M Perrier, X Glaszmann, JC (eds) Genetic diversity of cultivated tropical plants. CIRAD, Montpellier, p 376
FAO (ed) (2006) Global Forest Resources Assessment 2005. Progress towards sustainable forest management. FAO Forestry Paper No. 147, Rome
Flavell AJ, Knox MR, Pearce SR, Ellis TH (1998) Retrotransposon-based insertion polymorphisms (RBIP) for high throughput marker analysis. Plant J 16(5):643–650
Gardner TA, Barlow J, Chazdon R, Ewers RM, Harvey CA, Peres CA, Sodhi NS (2009) Prospects for tropical forest biodiversity in a human-modified world. Ecol Lett 12(6):561–582
Gomez C, Dussert S, Hamon P, Hamon S, de Kochko A, Poncet V (2009) Current genetic differentiation of Coffea canephora Pierre ex A. Froehn in the Guineo-Congolian African zone: cumulative impact of ancient climatic changes and recent human activities. BMC Evol Biol 9:167
Guyot R, de la Mare M, Viader V, Hamon P, Coriton O, Bustamante-Porras J, Poncet V, Campa C, Hamon S, de Kochko A (2009) Microcollinearity in an ethylene receptor coding gene region of the Coffea canephora genome is extensively conserved with Vitis vinifera and other distant dicotyledonous sequenced genomes. BMC Plant Biol 9(1):22
Hamon P, Siljak-Yakovlev S, Srisuwan S, Robin O, Poncet V, Hamon S, de Kochko A (2009) Physical mapping of rDNA and heterochromatin in chromosomes of 16 Coffea species: a revised view of species differentiation. Chromosome Res 17(3):291–304
Hill P, Burford D, Martin DM, Flavell AJ (2005) Retrotransposon populations of Vicia species with varying genome size. Mol Genet Genomics 273(5):371–381
Jing R, Vershinin A, Grzebyta J, Shaw P, Smykal P, Marshall D, Ambrose MJ, Ellis TH, Flavell AJ (2010) The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymorphism (RBIP) marker analysis. BMC Evol Biol 10:44
Joshi SP, Gupta VS, Aggarwal RK, Ranjekar PK, Brar DS (2000) Genetic diversity and phylogenetic relationship as revealed by inter simple sequence repeat (ISSR) polymorphism in the genus Oryza. Theor Appl Genet 100:1311–1320
Kalendar R, Grob T, Regina M, Suoniemi A, Schulman A (1999) IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet 98:704–711
Kalendar R, Tanskanen J, Immonen S, Nevo E, Schulman AH (2000) Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc Natl Acad Sci USA 97(12):6603–6607
Konovalov FA, Goncharov NP, Goryunova S, Shaturova A, Proshlyakova T, Kudryavtsev A (2010) Molecular markers based on LTR retrotransposons BARE-1 and Jeli uncover different strata of evolutionary relationships in diploid wheats. Mol Genet Genomics 283(6):551–563
Kumar A, Hirochika H (2001) Applications of retrotransposons as genetic tools in plant biology. Trends Plant Sci 6(3):127–134
Lashermes P, Combes MC, Trouslot P, Charrier A (1997) Phylogenetic relationships of coffee-tree species (Coffea L.) as inferred from ITS sequences of nuclear ribosomal DNA. Theor Appl Genet 94(6–7):947–955
Leroy JF (1980) Evolution et taxogenèse chez les caféiers. Hypothèse sur leur origine. Comptes Rendus Hebdomadaires des Séances de l’Académie des Sciences. Série D 291(6):593–596
Leroy T, Marraccini P, Dufour M, Montagnon C, Lashermes P, Sabau X, Ferreira LP, Jourdan I, Pot D, Andrade AC, Glaszmann JC, Vieira LG, Piffanelli P (2005) Construction and characterization of a Coffea canephora BAC library to study the organization of sucrose biosynthesis genes. Theor Appl Genet 111(6):1032–1041
Louarn J (1992) La fertilite des hybrides interspécifiques et les relations génomiques entre caféiers diploïdesd’origine africaine (Genre Coffea L., sous-genre Coffea). Ph.D. Thesis, Paris XI Univ, p 200
Lynch M, Milligan BG (1994) Analysis of population genetic structure with RAPD markers. Mol Ecol 3(2):91–99
Mahesh V, Rakotomalala JJ, Le Gal L, Vigne H, de Kochko A, Hamon S, Noirot M, Campa C (2006) Isolation and genetic mapping of a Coffea canephora phenylalanine ammonia-lyase gene (CcPAL1) and its involvement in the accumulation of caffeoyl quinic acids. Plant Cell Rep 25(9):986–992
Maurin O, Davis AP, Chester M, Mvungi EF, Jaufeerally-Fakim Y, Fay MF (2007) Towards a phylogeny for Coffea (Rubiaceae): identifying well-supported lineages based on nuclear and plastid DNA sequences. Ann Bot (Lond) 100(7):1565–1583
Melayah D, Bonnivard E, Chalhoub B, Audeon C, Grandbastien MA (2001) The mobility of the tobacco Tnt1 retrotransposon correlates with its transcriptional activation by fungal factors. Plant J 28(2):159–168
Musoli P, Cubry P, Aluka P, Billot C, Dufour M, De Bellis F, Pot D, Bieysse D, Charrier A, Leroy T (2009) Genetic differentiation of wild and cultivated populations: diversity of Coffea canephora Pierre in Uganda. Genome 52(7):634–646
N’Diaye A, Poncet V, Louarn J, Hamon S, Noirot M (2005) Genetic differentiation between Coffea liberica var liberica and C. liberica var. Dewevrei and comparison with C. canephora. Plant Syst Evol 253:95–104
Natali L, Giordani T, Buti M, Cavallini A (2007) Isolation of Ty1-copia putative LTR sequences and their use as a tool to analyse genetic diversity in Olea europaea. Mol Breed 19(3):255–265
Noirot M, Poncet V, Barre P, Hamon P, Hamon S, de Kochko A (2003) Genome size variations in diploid African Coffea species. Ann Bot (Lond) 92(5):709–714
Pearce SR, Knox M, Ellis TH, Flavell AJ, Kumar A (2000) Pea Ty1-copia group retrotransposons: transpositional activity and use as markers to study genetic diversity in Pisum. Mol Gen Genet 263(6):898–907
Petit M, Lim KY, Julio E, Poncet C, Dorlhac de Borne F, Kovarik A, Leitch AR, Grandbastien MA, Mhiri C (2007) Differential impact of retrotransposon populations on the genome of allotetraploid tobacco (Nicotiana tabacum). Mol Genet Genomics 278(1):1–15
Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, Collura K, Brar DS, Jackson S, Wing RA, Panaud O (2006) Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res 16(10):1262–1269
Porceddu A, Albertini E, Barcaccia G, Marconi G, Bertoli FB, Veronesi F (2002) Development of S-SAP markers based on an LTR-like sequence from Medicago sativa L. Mol Genet Genomics 267(1):107–114
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277
Robbrecht E, Manen J-F (2006) The major evolutionary lineages of the coffee family (Rubiaceae, angiosperms). Combined analysis (nDNA and cpDNA) to infer the position of Coptosapelta and Luculia, and supertree construction based on rbcL, rps16, trnL-trnF and atpB-rbcL data. A new classification in two subfamilies, Cinchonoideae and Rubioideae. Syst Geogr Pl 76:85–146
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16(10):944–945
Sanz AM, Gonzalez SG, Syed NH, Suso MJ, Saldana CC, Flavell AJ (2007) Genetic diversity analysis in Vicia species using retrotransposon-based SSAP markers. Mol Genet Genomics 278(4):433–441
Schulman A (2007) Molecular markers to assess genetic diversity. Euphytica 158:313–321
Sonnhammer EL, Durbin R (1995) A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167(1–2):GC1–GC10
Tam SM, Causse M, Garchery C, Burck H, Mhiri C, Grandbastien MA (2007) The distribution of copia-type retrotransposons and the evolutionary history of tomato and related wild species. J Evol Biol 20(3):1056–1072
Venturi S, Dondini L, Donini P, Sansavini S (2006) Retrotransposon characterisation and fingerprinting of apple clones by S-SAP markers. Theor Appl Genet 112(3):440–444
Vitte C, Panaud O (2005) LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenet Genome Res 110(1–4):91–107
Waugh R, McLean K, Flavell AJ, Pearce SR, Kumar A, Thomas BB, Powell W (1997) Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP). Mol Gen Genet 253(6):687–694
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8(12):973–982
Acknowledgments
The authors thank Mr. Francis Richard for maintaining the plants in the IRD greenhouses in Montpellier (France), and Dr. Pascal Musoli (NARO/COREC, Uganda) and Dr. Thierry Leroy (CIRAD, France) for kindly providing the Ugandan C. canephora DNAs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M.-A. Grandbastien.
Rights and permissions
About this article
Cite this article
Hamon, P., Duroy, PO., Dubreuil-Tranchant, C. et al. Two novel Ty1-copia retrotransposons isolated from coffee trees can effectively reveal evolutionary relationships in the Coffea genus (Rubiaceae). Mol Genet Genomics 285, 447–460 (2011). https://doi.org/10.1007/s00438-011-0617-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-011-0617-0