
Abstract
Mutations in titin cap (Tcap), also known as telethonin, cause limb-girdle muscular dystrophy type 2G (LGMD2G). Tcap is one of the titin interacting Z-disc proteins involved in the regulation and development of normal sarcomeric structure. Given the essential role of Tcap in establishing and maintaining normal skeletal muscle architecture, we were interested in determining the regulatory elements required for expression of this gene in myoblasts. We have defined a highly conserved 421 bp promoter proximal promoter fragment that contains two E boxes and multiple putative Mef2 binding sequences. This promoter can be activated by MyoD and myogenin in NIH3T3 fibroblast cells, and maintains the differentiated cell-specific expression pattern of the endogenous Tcap in C2C12 cells. We find that while both E boxes are required for full activation by MyoD or myogenin in NIH3T3 cells, the promoter proximal E box has a greater contribution to activation of this promoter in C2C12 cells and to activation by MyoD in NIH3T3 cells. Together, the data suggest an important role for MyoD in activating Tcap expression through the promoter proximal E box. We also show that myogenin is required for normal expression in vivo and physically binds to the Tcap promoter during embryogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In striated muscle cells, the Z-disc constitutes the borders of individual sarcomeres. The Z-disc is a highly organized structure that is composed of several proteins that are organized in multi protein complexes (Faulkner et al. 2001; Clark et al. 2002). A number of mutations in genes of distinct Z-disc components have been found to be responsible for various forms of muscle disorders (Frank et al. 2006). Titin cap (Tcap), also known as telethonin, is a Z-disc associated protein that is thought to be involved in the regulation and development of normal sarcomeric structure. Titin cap co-localizes with titin, binds to the N-terminal domain of titin (Gregorio et al. 1998) and is a substrate of the titin kinase (Valle et al. 1997). As demonstrated by over expression studies, the interaction with titin is critical for sarcomeric integrity (Gregorio et al. 1998). In that work, it was also shown that Tcap is localized within the Z-line region of adult striated skeletal and cardiac muscles. In the heart, Tcap is necessary for the cardiomyocyte’s stretch sensor and the structural organization of the cardiac sarcomere (Hayashi et al. 2004; Bos et al. 2006).
Titin cap is of clinical importance as mutations in the TCAP gene are associated with the seventh form of autosomal recessive limb-girdle muscular dystrophy, termed LGMD type 2G (Moreira et al. 2000). Patients with TCAP mutations develop a marked weakness in the distal muscles of the legs with proximal involvement and most patients lose the ability to walk by the third or fourth decade of life (Moreira et al. 2000). Tcap appears to exist as a single isoform and is among the 12 most abundant transcripts found in skeletal muscle (Valle et al. 1997). Tcap expression has been found to be greatly up regulated during the differentiation of C2C12 myoblast cells (Mason et al. 1999). Tcap has also been shown to interact with and regulate the secretion of myostatin (MSTN), a negative regulator of muscle growth that inhibits both cell proliferation and differentiation (Nicholas et al. 2002). Knockdown of Tcap by RNA interference in C2C12 myoblast cells inhibits myoblast differentiation and impairs muscle cell growth (Markert et al. 2008). Given the interaction of Tcap with myostatin and the ability of Tcap to modulate myoblast proliferation and differentiation, it has been proposed that Tcap might offer a new therapeutic target for muscular dystrophies. Recently, a Tcap knockout mouse was reported. The Tcap knockout mouse shares many of the features of LGMD type 2G patients, suggesting that the mouse model will provide an important experimental model for potential therapies for LGMD2G patients (Markert et al. 2010). Beyond the interaction with myostatin, Tcap has been shown to interact with additional proteins that influence cell growth and differentiation. These interactions include Ankrd2 (Kojic et al. 2004), potassium channel B-subunit minK (Furukawa et al. 2001), protein kinase D (Haworth et al. 2004), and murine double minute 2 (MDM2) (Tian et al. 2006).
Gene regulation in skeletal muscle is controlled by a family of highly related basic helix loop helix (bHLH) transcription factors, the myogenic regulatory factors (MRFs). The MRF family includes Myf5, MyoD, myogenin, and Myf 6 (also known as Mrf4). The MRFs dimerize with E-proteins and bind E box sequences (CANNTG) in the regulatory regions of muscle genes (Berkes and Tapscott 2005). The MRFs work in conjunction with multiple isoforms of the MADS-box factors, Mef2a, Mef2c, and Mef2d (Blais et al. 2005). Mef2 factors alone do not have myogenic activity, but synergize with the MRFs to enhance gene expression during myogenesis (Molkentin et al. 1995; Wang et al. 2001).
The MRFs play overlapping but non- redundant roles in myogenesis. As revealed by mouse knockouts, Myf5 and MyoD function early in myogenesis to confer a myogenic fate on mesodermal progenitor cells (Rudnicki et al. 1993). Myf6 has roles in both early and late events in myogenesis (Kassar-Duchossoy et al. 2004). Myogenin functions later in myogenesis to stimulate specified myoblasts to differentiate into functional myofibers. Unique among the MRFs, null mutations in myogenin alone cause lethality (Hasty et al. 1993; Nabeshima et al. 1993). In myogenin null mice, myoblasts are specified, but muscle fibers form poorly (Venuti et al. 1995). Myogenin plays a role in regulating the expression of several components of the Z-disc during embryogenesis, including limb domain binding 3, myozenin1, zyxin, and muscle LIM protein (Davie et al. 2007; Ji et al. 2009).
Given the importance of Tcap in maintaining sarcomeric integrity, we were interested in understanding the regulatory elements that govern the expression of this gene. The expression of Tcap in C2C12 cells, a murine myoblast line, has been previously characterized, thus, we felt that C2C12 cells would serve as an appropriate model for these studies. Tcap is not expressed in proliferating myoblasts but is robustly expressed in late differentiation stages (Mason et al. 1999). For this study, we sought to determine the promoter proximal sequence elements that control the expression of Tcap in C2C12 cells. We have identified a 211 bp promoter proximal element in the Tcap promoter that recapitulates the differentiation specific expression of Tcap. The E box contained within this promoter proximal element is required for maximal activation by MyoD and we also show that myogenin contributes to Tcap expression in vivo.
Materials and methods
Cell culture
C2C12 (ATCC) and NIH3T3 (ATCC) cells were grown in Dulbecco’s modified Eagle’s medium containing 10% bovine calf serum, 2% l-glutamine, 1% penicillin–streptomycin according to the standard tissue culture protocols (Pollard and Walker 1997). To induce differentiation into myotubes, C2C12 cells were grown to 70% confluence and the media switched to DMEM supplemented with 2% horse serum (Hyclone). C2C12 cells were grown in differentiation medium (DF) for the number of days indicated in each experiment.
Identification of consensus binding elements
To identify regulatory elements required for expression of Tcap, we identified phylogenetically conserved non-coding sequences containing bHLH protein-binding sites (E boxes) and Mef2 sites using the NCBI Mouse Genome Resource website. To perform this analysis, we used the computational tool regulatory VISTA (rVISTA) to identify clusters of myogenin/MyoD, E-protein and Mef2 consensus binding sites that were conserved between mouse and human genomes (Loots et al. 2002). Vista analysis identified a 421 bp region from −380 to +41, with +1 defined as the start of transcription that contained highly conserved non-coding sequence immediately upstream of the Tcap gene.
Transient transfection and luciferase assays
To address the question of what elements bind to the promoter sequence, we used luciferase reporter assays. The pGL3-Tcap constructs (0.2 μg, this lab) and pRL-CMV (0.01 μg, Promega/#E2261) were co-transfected into cells grown in 96 well plates using calcium phosphate. The positive control in all experiments was the pGL3 control vector (Promega/#E1741), which contains the proximal 202 bp of the SV40 promoter. The negative control in all experiments was the pGL3 basic vector (Promega/#E1751), which lacks a promoter. The level of luciferase and renilla activity in transfected cells was determined using a dual-luciferase reporter assay system (Promega). The plasmids EMSVmyog (provided by Diane Edmondson, The University of Texas Medical School at Houston) and pEMCIIs (provided by Andrew Lassar, Harvard Medical School) were used for expressing myogenin and MyoD, respectively, in transfected NIH-3T3 cells. Expression values for each construct were calculated as a percentage of expression of the pGL3 basic vector. All values were normalized to the renilla values. Standard deviations from the mean were calculated from the mean expression values representing at least three separate experiments. All individual experiments were performed in triplicate.
Cloning of Tcap reporter and deletion constructs
To determine the promoter proximal regions that control Tcap expression, the upstream gene regulatory regions of Tcap were amplified from murine genomic DNA using PCR and cloned into the pGL3 basic luciferase reporter gene vector (Promega/#E1751). Base pair positions are defined with respect to the translational A(TG) start site with A considered to be +1. The 421 promoter proximal promoter fragment of Tcap was amplified from genomic DNA with the following primers: Tcap MluI F 5′ GCACGCGTGAGGGCATCAGTTCCTGCTTCTCC 3′ and Tcap BglII R 5′ GCAGATCTGATTTCTGATTGCTCCCTCTGCT 3′. The resulting PCR fragment was cloned into the pGL3 basic vector (Promega) using standard molecular biology techniques. Resulting clones were sequenced to confirm the insertion. The deletion 1 (D1) construct was generated with the Tcap D1 F 5′ GCACGCGTGACCTCTGACCTGGGAGC 3′ primer and the Tcap BglII R primer described above.
Mutagenesis
To determine the contributions of the two E boxes in the Tcap promoter, the E boxes were individually deleted from the Tcap-pGL3 vector using the QuickChange II site directed mutagenesis kit (Stratagene). Primer sequences used to generate the mutations were Tcap E1F mut 5′ CTTGGCTCTGCTTATAGACGCCAGAGGGGCTG 3′, Tcap E1R mut, 5′ CAGCCCCTCTGGCGTCTATAAGCAGAGCCAAG 3′, Tcap E2F mut, 5′ GCACGAGGCTGCCCCTGGTCCGAGGTG 3′, and Tcap E2R mut 5′ CACCTCGGACCAGGGGCAGCCTCGTGC 3′. Following mutagenesis, plasmids were isolated and sequenced to confirm the presence of the desired mutation. To generate the combined E box deletion mutant (ΔE1,2), the E1 box deletion mutant (ΔE1) was first generated and verified by sequencing. The ΔE1 deletion mutant was then used with the ΔE2 mutant primers to generate the double deletion mutation.
Quantitative reverse-transcriptase polymerase chain reaction
To determine changes in gene expression, RNA was isolated from C2C12 cells or from E15.5 embryonic tongue tissue of Myog wt/wr or Myog null/null mice by Trizol extractions (Invitrogen). Two micrograms of total RNA was reversed transcribed with MultiScribe™ MuLV reverse transcriptase (Applied Biosystems). cDNA equivalent to 40 ng was used for quantitative polymerase chain reaction (qPCR) amplification (Applied Biosystems) with SYBR green PCR master mix (Applied Biosystems). The relative levels of expression of genes were normalized according to the expression levels of 18S rRNA. qPCR data were calculated using the comparative Ct method according to the manufacturer’s instructions (Applied Biosystems). Fold changes in Tcap expression were divided by the fold changes observed for HPRT expression. Standard deviations from the mean of the [Δ]Ct values were calculated from three independent RNA samples. The PCR primers used for detecting Tcap expression are Tcap F 5′ CCTTCTGGGCTGAGTGGAAA 3′ and Tcap R 5′ TCTGTGTATCCTCCTCGTGCAA 3′. The primers for detecting 18S rRNA are 18S rRNA F, 5′-CGCCGCTAGAGGTGAAATTCT-3′ and 18S rRNA R, 5′-CGAACCTCCGACTTTCGTTCT-3′.
Chromatin immunoprecipitation analysis
To establish MRF binding to the Tcap promoter in vivo, chromatin immunoprecipitation (ChIP) assays from embryonic tongue tissue were performed as previously described (Davie et al. 2007) with the following modifications. Approximately, 0.6 g of tissue was processed per immunoprecipitation, protein A agarose beads (Invitrogen) were used to immunoprecipitate the antibody:antigen complexes and the washes following the immunoprecipitation included two RIPA (radio-immunoprecipitation assay buffer) washes and one LiCl containing wash. No high salt wash buffer was used. The antibodies used in the ChIP analysis included an anti-myogenin antibody (F5D, Developmental Studies Hybridoma Bank (DSHB)), an anti-MyoD antibody (5.8A, sc-32758, Santa Cruz Biotechnologies (SCBT)), anti-Myf5 (C-20, sc-302, SCBT) and anti-IgG (sc-2025, SCBT). Two micrograms of antibody was used for each immunoprecipitation with extracts corresponding to approximately 400 μg protein. The PCR primers used for detecting protein-bound DNA are Tcap E1 F 5′ CCCATCACCAC CAGTGAGTCT 3′, Tcap E1 R 5′ GCCCTTTAAATAGCCCCTTCTTC 3′, Tcap E3 F 5′ CTACTGAGGTGTCCCAGGC 3′ and Tcap E3 R 5′ CCACAGGTACCTCAGTAACCC 3′, IgH F 5′ GCCG ATCAGAACCAGAACACCTGC 3′, and IgH R 5′ TGGTGGGGCTGGACAGAGTGTTTC 3′. DNA was amplified by qPCR using SYBR green PCR master mix (Applied Biosystems). The data are presented as the fold change between antibody samples and a mock sample where antibody was not added (no ab) with each comparison normalized to the changes observed at an intergenic region of IgH that contains four non functional E boxes. Input samples were tested for amplification and to compare samples across replicates but were not used for normalization. qPCR data were calculated using the comparative Ct method. Values of [Δ][Δ]Ct were determined by applying the following formula: [Δ]Ct, template (antibody) − [Δ]Ct, reference (no ab) = [Δ][Δ] Ct. Fold enrichments were calculated as 2^−([Δ][Δ]Ct). Fold enrichments for the Tcap promoter were normalized to the fold changes observed at the IgH locus by dividing the fold change observed at the Tcap promoter by the fold change observed at the IgH locus. Standard error from the mean was calculated from replicate [Δ][Δ]Ct values.
Mice
Myogenin null embryos were required to confirm the down regulation of Tcap in these mice. These embryos were obtained by timed breeding of myogenin null/+ mice (gift of William Klein, U.T. M.D. Anderson Cancer Center). The embryos were genotyped with the primers Myog F 5′ GAACAAGCCTTTTCCGACCTGATG 3′ and Myog R 5′ GGTCCACCGACACAGACTTCCTC 3′ to detect wild type alleles and Myog R 5′ GGTCCACCGACACAGACTTCCTC 3′ and Neo F 5′ AGGTGAGATGACAGGAGATC 3′ to detect null alleles. All mouse studies were approved by the Southern Illinois University Institutional Care and Use Committee (IACUC).
Results
Identification of the promoter proximal elements that regulate Tcap expression
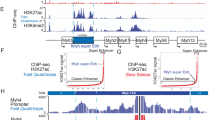
First, we sought to identify the upstream regulatory regions used to regulate expression of the Tcap gene. In both the mouse and the human genomes, Tcap is immediately downstream of another gene, Stard3, and immediately upstream of Pnmt1. Both Stard3 and Pnmt1 are transcribed in the same direction as Tcap. These unrelated genes are separated from Tcap by 2.8 kb in the case of Stard3, and 1.7 kb in the case of Pnmt1. As a first step in understanding the promoter proximal elements that control Tcap expression, we choose to initially analyze the 2.8 kb region upstream of Tcap. One conserved E box was noted 2,067 bp upstream of the Tcap gene (Fig. 1). Immediately upstream of the Tcap gene, we identified a 421 bp region from −380 to +41, with +1 defined as the start of transcription, that contained highly conserved non-coding sequence. This 421 bp region contained two E boxes located at −272 and −103 and Mef2 binding sites (shown in Fig. 1). The transcriptional start of the Tcap gene (indicated in Fig. 1) is very close to the translational start site at position +41, predicting only a 36 bp 5′ UTR. As indicated in Fig. 1, three potential Mef2 binding sites flank the promoter proximal E Box at position −103, termed E1. The promoter distal E box at position −272, termed E2, has one adjacent Mef2 site immediately upstream of the E box. Tcap is encoded by two exons, and no conserved non-coding sequence was observed in the intervening intron (shown in Fig. 1). Conserved non-coding sequence is observed immediately downstream of the Tcap 3′ UTR, but the contribution of this sequence was not tested as part of this work.

DNA sequence analysis of the promoter proximal 5′ regulatory region of the Tcap gene. Shown is a rVISTA plot for the 2.8 kb region upstream of the Tcap gene. Phylogenetically conserved non-coding sequences in mouse and human genomes are depicted as pink regions, conserved translated regions are shown as blue regions, and conserved coding sequence is depicted as purple regions. Green vertical lines indicate conserved Mef2 (MADS-Box) binding sites, and vertical red lines represent the myogenin/MyoD E box binding sites. The gene orientation is indicated by the arrowed line. The region containing clusters of conserved binding sites is indicated by brackets. Mef2 MADS box binding sites are underlined and the myogenin/MyoD E box binding sites are boxed. The promoter proximal E box (E1) and the promoter distal E box (E2) are labeled. The position of the 211 bp deletion is marked with an asterisk. The predicted start of transcription is labeled Txn and the translational start, ATG, is in bold (Color figure online)
Promoter proximal sequences control Tcap expression
A 2.3 kb fragment containing all three E boxes and a 421 bp promoter proximal fragment containing two E boxes were cloned into luciferase reporter vectors and the expression pattern in NIH3T3 fibroblast cells and C2C12 myoblast cells examined. NIH3T3 fibroblast cells were chosen as these cells do not express the MRF family. Immortalized C2C12 cells were used as a model for myogenesis. As anticipated, the Tcap reporter was inactive in the fibroblast cells. However, when either myogenin or MyoD was co-transfected with the Tcap reporter, the reporter was activated (Fig. 2a). The activity of both constructs was very similar, suggesting that much of the regulatory information for the Tcap gene is immediately upstream of the transcription start site. Transfection of MyoD results in higher transcriptional activation than myogenin on both reporters. However, MyoD has shown higher activity than myogenin on every muscle-specific reporter tested to date, so this result may not have functional significance for Tcap regulation in vivo. The result does suggest that both MyoD and myogenin can activate this promoter. When tested in C2C12 myoblast cells, we found that the Tcap luciferase reporters were almost completely inactive in proliferating C2C12 cells, but are strongly up regulated when the C2C12 cells differentiate (Fig. 2b). These results suggest that the 421 bp promoter fragment contains sufficient regulatory information to maintain the normal differentiation specific expression pattern of Tcap.

The 421 bp immediately upstream of the Tcap gene supports transcription by myogenin and MyoD and is only activated under differentiating conditions in myoblast cells. a Luciferase reporter constructs containing a 2.3 kb region or 421 bp region upstream of the Tcap gene were transiently transfected into the NIH3T3 fibroblast line. The Tcap constructs were co-transfected with expression vectors containing myogenin or MyoD or an empty vector. b The Tcap luciferase constructs described in a were transiently transfected into proliferating C2C12 cells and assayed prior to differentiation, or following 2 days of differentiation. For both NIH3T3 and C2C12 cells, the data are expressed as a percentage of the pGL3 basic vector expression levels. Error bars are standard deviations from the mean
To further dissect the regulatory regions, a deletion fragment of the Tcap promoter was tested. The deletion construct removed the distal sequence from −380 to −170 which contained the promoter distal E box, E2, and a potential Mef2 site (shown in Fig. 1). The 211 bp promoter fragment retains the promoter proximal E box (E1) and three associated putative Mef2 sites. The 211 bp promoter fragment was tested in both C2C12 cells and NIH3T3 fibroblasts. This construct could be activated by both myogenin and MyoD in NIH3T3 cells, but the overall stimulation was reduced with respect to the 421 bp reporter (Fig. 3a, b). This result indicates that the second E box and or Mef2 site contributes to the full activity of this promoter in fibroblast cells. Surprisingly, we found that in C2C12 cells, the 211 bp construct retains full activity in differentiating cells (Fig. 3c). In fact, the construct was modestly more active than the full length reporter, indicating that some sequence in the promoter distal region may be slightly inhibitory. The result also demonstrates that the promoter proximal 211 bp region with the promoter proximal E box and surrounding Mef2 sites is sufficient for activity in differentiating C2C12 cells.

A deletion construct of the Tcap promoter retains full activity in C2C12 cells, but not in NIH3T3 cells. a The 221 and 421 bp Tcap regulatory regions were transiently transfected into the NIH3T3 fibroblast line. The Tcap constructs were co-transfected with the empty vector or an expression vector containing myogenin. b The 221 and 421 bp Tcap regulatory regions were transiently transfected into the NIH3T3 fibroblast line co-transfected with the empty vector or an expression vector for MyoD. c The 221 and 421 bp Tcap regulatory regions were transiently transfected into proliferating or differentiated C2C12 cells. For both NIH3T3 and C2C12 cells, the data are expressed as a percentage of the pGL3 control vector expression levels. Error bars are standard deviations from the mean
The promoter proximal E box is required for activity in myoblasts and for activation by MyoD
As the deletion construct with only the promoter proximal E box was partially active with the MRFs in NIH3T3 cells, but fully active in C2C12 cells, we next tested the individual contributions of the two E boxes in both cell lines. Both the promoter proximal (E1) and promoter distal (E2) E boxes were mutated individually, and in combination. The E boxes were fully deleted from the sequence as described in “Materials and methods”. We found that a deletion of either the promoter distal E2 box (ΔE2) or a deletion of the promoter proximal E1 box (ΔE1) reduced the ability of myogenin or MyoD to activate the mutant reporters in NIH3T3 cells (Fig. 4a, b). However, we observed a significant difference in how the E box deletion mutations affected transcriptional activation by myogenin or MyoD. The constructs with ΔE1 or ΔE2 deletion mutations reduced myogenin activity by approximately 50% (Fig. 4a). However, the ΔE1 and ΔE2 deletion mutations had differential effects for activation by MyoD (Fig. 4b). The deletion of E1 (ΔE1) cripples the activation mediated by MyoD to a much greater degree than the deletion of E2 (ΔE2). MyoD activates the reporter with the ΔE2 deletion mutation to approximately 40% of the wild type reporter, while the reporter with the ΔE1 deletion mutation reduces that activation to only 16% of the wild type reporter. This result implies that the E1 box is critically important for activation by MyoD in NIH3T3 cells. We also tested the effect of the combined deletions. When both the promoter proximal (E1) and the promoter distal (E2) boxes were deleted together (ΔE1,2), the promoter had almost no activity in NIH3T3 cells transfected with MyoD or myogenin (Fig. 4a, b).

Both E boxes are utilized in both myoblast and fibroblast cells, but the promoter proximal E box is of greater importance in myoblasts. a Single or combined E box mutations in the 421 bp Tcap regulatory region were transiently transfected into the NIH3T3 fibroblast line. The Tcap constructs were co-transfected with the empty vector or expression vectors containing myogenin or MyoD. E2 refers to the promoter distal E box at position −313. E1 refers to the promoter proximal E box at position −144. b Transient transfections of single or combined E box mutations in the 421 bp Tcap regulatory region of proliferating or differentiated C2C12 cells. For both NIH3T3 and C2C12 cells, the data are expressed as a percentage of the pGL3 basic vector expression levels. Error bars are standard deviations from the mean
In C2C12 cells, we found that deleting the E1 site (ΔE1) dramatically crippled the activation of the reporter in differentiating cells (Fig. 4c). Consistent with our deletion construct results, the deletion of E2 (ΔE2) did not cripple the reporter significantly in differentiating C2C12 cells. Surprisingly, when the construct in which both E1 and E2 were deleted (ΔE1,2) was tested in C2C12 cells, the ΔE1,2 deletion mutation did have a modestly greater effect than the ΔE1 deletion mutation in differentiating C2C12 cells. The construct in which E1 and E2 are deleted (ΔE1,2) fails to show even the small amount of activity seen for the ΔE1 mutant (Fig. 4c). Thus, both E boxes can contribute to the full activation of the Tcap gene, although the E1 box makes a much stronger contribution to the overall activation in differentiating myoblasts.
Myogenin contributes to Tcap expression in vivo
Next, we were interested in understanding the contribution of MyoD or myogenin to the regulation of Tcap expression. Our data show that the E1 site is required for maximal expression in C2C12 cells and that MyoD activity is strongly inhibited by the ΔE1 mutation. Taken together, these data imply that MyoD plays an important role in the regulation of Tcap. Previous expression profiles of Tcap in C2C12 cells have shown that endogenous Tcap expression continues to be up regulated throughout several days of differentiation (Mason et al. 1999). The expression profile is consistent with Tcap being a late differentiation gene and potentially requiring the activity of myogenin, the regulator of terminal differentiation, in its activation. Previously, Tcap was found to be modestly down regulated in myogenin null muscle tissue by microarray analysis (Davie et al. 2007). For this study, we sought to confirm the down regulation of Tcap in myogenin null animals by real time PCR analysis of cDNA samples reverse transcribed for RNA isolated from E15.5 Myogwt/wt and Myognull/null tongue tissue. We found that Tcap is down regulated 5.4-fold in Myognull/null tongue tissue (Fig. 5), confirming that myogenin is required, directly or indirectly, for full activation of this gene in vivo.

Tcap expression is down regulated in myognull/null mice. Shown are the results of qRT-PCR analysis using RNA extracted from E15.5 tongue tissue. Calculations of the relative fold change in gene expression for the myogwt/wt and myognull/null samples are described in Materials and methods. The data are normalized to the fold changes for 18S rRNA. The bar for the myogwt/wt sample represents the average value of three samples normalized relative to 1 as indicated on the Y-axis. The bar for the myognull/null samples represents the average value of three samples normalized relative to the myogwt/wt samples. Error bars represent standard deviations from the mean
MyoD and myogenin bind the Tcap promoter in vivo
As our previous results suggested that the promoter proximal E box is essential for Tcap activity, we next asked if myogenin or MyoD was associated with the promoter proximal E box in vivo. To address this question, we performed ChIP assays on embryonic tongue tissue at E15.5. We observed a robust enrichment of the promoter proximal Tcap region in samples immunoprecipitated with antibodies against myogenin or MyoD, confirming that myogenin and MyoD bind to this promoter element in vivo and play a direct role in the regulation of the Tcap gene during embryogenesis (Fig. 6a). The experiment was also performed with antibodies against Myf5, and we found that Myf5 also binds this promoter at E15.5. This was a surprising result, as Myf5 is considered to be an early differentiation gene and Myf5 transcripts are not detectable beyond E14 (Ott et al. 1991). However, we have observed that Myf5 protein is detectable by western blot in late stage embryos and that Myf5 associates with several differentiation specific genes in differentiated C2C12 cells (Londhe and Davie, submitted). Occupancy of the E box 2.1 kb upstream of the Tcap gene was also assayed, and no association was detected for MyoD, myogenin or Myf5 with the upstream E box in vivo (data not shown). The ChIP assay for myogenin, MyoD, and Myf5 was repeated at E18.5, and we again observed a robust enrichment of each of the MRFs at the Tcap promoter (Fig. 6b). When the ChIP assay was repeated on newborn animals (P0), MyoD, myogenin, and Myf5 were not detected at the Tcap promoter, although we could detect binding of MyoD and myogenin to the Tnni2 promoter (Fig. 6c and data not shown). The ChIP data suggest that each of the MRFs play a transient role in activating the Tcap promoter during embryogenesis.

MyoD, myogenin and Myf5 bind to the Tcap promoter during embryogenesis. a MyoD, myogenin, and Myf5 bind to the promoter at E15.5. Shown is a graph representing qPCR analysis of a ChIP assay using antibodies against MyoD, myogenin, and Myf5 on E15.5 tongue tissue. b The MRFs remain associated at E18.5. Data are represented as in (a). c The MRFs do not bind the promoter in newborn animals. Data are represented as in (a). For all data shown, the ChIP results are graphed as fold enrichment as indicated on the Y-axis for the antibody sample versus the no antibody control sample normalized to the relative enrichments observed on the IgH locus. The calculations are described in Materials and methods
Discussion
Tcap plays an important role in maintaining the integrity of the sarcomere and also appears to have a role in regulating muscle growth through its interaction with myostatin, but the regulatory elements that control Tcap expression were uncharacterized. We have defined the promoter proximal elements required to maintain appropriate expression of this gene and have shown that a single promoter proximal E box contains much of the regulatory information for Tcap in muscle cells. Additional enhancer elements that contribute to Tcap expression could exist, as mammalian enhancers, like the enhancer for the sonic hedgehog gene (Lettice et al. 2003), can be located hundred of kilobases away from the target gene. In this study, we have defined the immediate promoter proximal elements required for Tcap expression as a first step in understanding the regulation of this important regulator of sarcomeric integrity.
DNA sequence analysis of the 5′ regulatory region for the mouse Tcap gene compared to human, chimp, cow, and dog genomes revealed that the only highly conserved region is contained in a 421 bp fragment immediately upstream from the transcription initiation site. This highly conserved region contains several Mef2 sites and two E boxes. We show that the highly conserved 421 bp promoter proximal promoter fragment is sufficient to maintain the differentiated cell-specific expression pattern of Tcap in C2C12 cells. Further, we demonstrate that a 211 bp deletion construct with a single E box and three Mef2 binding sites is sufficient for activity in C2C12 cells. In fact, the 211 bp construct is slightly more active than the 421 bp reporter in muscle cells. However, in fibroblast cells, we find that the 421 bp construct is 50% more active than the 211 bp construct.
These results make two important suggestions regarding the Tcap promoter. First, the result suggests that the vast majority of the regulatory information of the Tcap gene in muscle is contained within a 221 bp promoter proximal sequence with a single E box and associated Mef2 sites. Second, the slightly higher activity of the promoter proximal sequence lacking the distal region implies that the promoter distal region may contain sequences important in mediating repression of the Tcap gene. Both of these results are specific to C2C12 cells and were not observed in NIH3T3 cells. This implies that there are additional muscle-specific proteins that help promote both activation and repression of the Tcap promoter, both of which are likely to be important for the developmental timing of Tcap expression. Many co-factors that cooperate with the MRFs have been described, although the majority of the co-factors described to date are expressed ubiquitously.
We analyzed the 421 bp promoter fragment for potential co-regulators by rVISTA analysis, but were unable to identify consensus binding sites of interest. Our analysis included sites for the ubiquitous homeodomain proteins Pbx and Meis that cooperate with MyoD to activate the myogenin promoter (Berkes et al. 2004); Six1, a DNA bound factor that interacts with Ski to activate transcription at the myogenin locus (Spitz et al. 1998; Li et al. 2003; Zhang and Stavnezer 2009) and MAZ, a transcription factor that activates the muscle creatine kinase (Ckm) gene and other muscle-specific genes (Himeda et al. 2008). Two potential MAZ binding sites were identified in the promoter, but the relevance of these sites is unclear as MAZ sites are based on a GC-rich consensus sequence that is frequent in the genome.
The fragment that was deleted in the 211 bp promoter proximal construct was also analyzed for potential repressor binding sites. We did not observe sites for RP58, a repressor of the Id genes (Yokoyama et al. 2009) whose sites are also frequent at genes that show differentiation decreased MyoD binding profiles (Cao et al. 2010). The binding site for Hey1, a repressor which is a target of Notch signaling, is the canonical E box (Buas et al. 2010). Thus, potential Hey1 sites are present both in the promoter distal and promoter proximal fragments and are unlikely to be the promoter distal repressive element.
The result that myogenin and MyoD responded differently to the loss of the promoter proximal E1 sequence was surprising, as the molecular basis for the specificity of MyoD and myogenin is still unclear. Genome wide binding assays have revealed that MyoD and myogenin largely bind to the same sequences (Blais et al. 2005; Cao et al. 2006). Strikingly, both MyoD and myogenin greatly prefer a CASCTG consensus sequence, where S represents G or C (Cao et al. 2006, 2010). The sequence flanking the E box also contributes to binding affinity (Blackwell and Weintraub 1990). Importantly, sequence function studies have demonstrated that binding affinity does not necessarily correlate with transcriptional activation (Davis et al. 1990; Weintraub et al. 1991).
In the Tcap promoter, the two E boxes differ in their sequence, although neither are the highly preferred binding sites, CAGCTG and CAGGTG/CACCTG (Cao et al. 2010). The far upstream E box at position 2,067 (E3) contains the consensus CAGCTG sequence, although no MRF directed activity or binding was detected for this distal E box. The E2 sequence is CATGTG/CACATG, while the E1 sequence is CATCTG/CAGATG. The sequence flanking E2 contains residues that are favorable for binding, while the sequence flanking the E1 box contains residues that are less favorable for binding. However, we show that MyoD can robustly activate Tcap through the E1 box and, while to a greatly reduced level, MyoD can activate Tcap through the E2 box as well. Myogenin can activate Tcap relatively well through either E1 or E2, suggesting that the functional determinants for MyoD and myogenin activity are distinctly different at this promoter. The lack of tight correlation between preferred binding sites and the transcriptional activity observed at the Tcap promoter again suggests, as discussed above, that additional factors play important roles in modulating MRF binding and activity.
The in vivo ChIP data suggest that MyoD, myogenin, and Myf5 play a direct role in controlling Tcap expression during embryogenesis. The down regulation of Tcap observed in myogenin null/null animals confirms a role for myogenin at this promoter. However, our data also support a role for MyoD on this promoter, as MyoD strongly preferred the E box that retains most of the activity in C2C12 cells. The potential contribution of Myf5 at this promoter remains uncharacterized. MyoD and myogenin have previously been shown to have sequential roles on specific muscle promoters (Cao et al. 2006) and this may be the case at Tcap as well.
An important question in the future will be how Tcap expression is maintained in adult animals. We have shown that MyoD, myogenin, and Myf5 are not present at the Tcap promoter in newborn animals, which implies that either Myf6 (Mrf4), or no MRF, occupies this promoter at this stage. Myf6 was not assayed due to the lack of antibodies against Myf6 suitable for a ChIP assay. Tcap was initially identified as one of the 12 most abundant transcripts in skeletal muscle and the events that maintain this high expression profile in adult tissue are uncharacterized. It is possible that myogenin may again play a transient role on this promoter during postnatal growth and muscle repair and it will be interesting to assay later time points to determine if MRFs reoccupy this promoter during the postnatal growth period and in adult life. It is also possible that the MRFs may direct epigenetic or factor changes at the promoter that maintain expression in the absence of a MRF. Recent work has shown that there is a switch of core transcription complex components during terminal differentiation. In proliferating cells, TBP is bound to promoters, but as cells differentiate, TBP is replaced with TRF3/TAF3 (Deato and Tjian 2007). It is possible that the recruitment of TRF/TAF3, or other basal transcription factors, can maintain expression in the absence of the initiating transcription factor. Future studies will also include the identification of additional muscle-specific co-factors suggested by our data that may help modulate Tcap expression in skeletal muscle.
The absence of myogenin at the Tcap promoter in newborn animals is not entirely surprising, as myogenin is down regulated shortly after birth but can be up regulated in response to diverse stimuli (Grounds et al. 1992), aging (Musaro et al. 1995; Kostrominova et al. 2000) or denervation (Eftimie et al. 1991; Buonanno et al. 1992). A conditional deletion of myogenin has shown that myogenin is not required for post natal growth, although the animals are slightly smaller than wild type littermates (Knapp et al. 2006). Analysis of adult satellite cells with or without myogenin has suggested that myogenin may switch target gene sets, regulating one set of genes during embryogenesis, and an alternative set of genes during satellite cell reactivation and muscle repair (Meadows et al. 2008). These data suggest that while myogenin may be essential in the activation of Tcap during embryonic stages, it may not be required for the continued expression of Tcap, although this hypothesis remains to be tested.
In summary, we have defined a highly conserved 221 bp promoter region of the Tcap gene that maintains the normal expression profile of Tcap in C2C12 cells and confirmed that both MyoD and myogenin contribute to activation of this gene in vivo. Understanding the transcriptional control of this vital regulator of sarcomeric integrity is an essential element in understanding how sarcomere integrity is controlled and maintained. The data also provide information on how the Tcap gene might be experimentally manipulated in potential therapies for LGMD type 2G patients, as we have shown that Tcap expression can be controlled through a very minimal promoter element.
References
Berkes CA, Tapscott SJ (2005) MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 16:585–595
Berkes CA, Bergstrom DA, Penn BH, Seaver KJ, Knoepfler PS, Tapscott SJ (2004) Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell 14:465–477
Blackwell TK, Weintraub H (1990) Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science 250:1104–1110
Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD (2005) An initial blueprint for myogenic differentiation. Genes Dev 19:553–569
Bos JM, Poley RN, Ny M, Tester DJ, Xu X, Vatta M, Towbin JA, Gersh BJ, Ommen SR, Ackerman MJ (2006) Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab 88:78–85
Buas MF, Kabak S, Kadesch T (2010) The Notch effector Hey1 associates with myogenic target genes to repress myogenesis. J Biol Chem 285:1249–1258
Buonanno A, Apone L, Morasso MI, Beers R, Brenner HR, Eftimie R (1992) The MyoD family of myogenic factors is regulated by electrical activity: isolation and characterization of a mouse Myf-5 cDNA. Nucleic Acids Res 20:539–544
Cao Y, Kumar RM, Penn BH, Berkes CA, Kooperberg C, Boyer LA, Young RA, Tapscott SJ (2006) Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J 25:502–511
Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, Gentleman RC, Tapscott SJ (2010) Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 18:662–674
Clark KA, McElhinny AS, Beckerle MC, Gregorio CC (2002) Striated muscle cytoarchitecture: an intricate web of form and function. Annu Rev Cell Dev Biol 18:637–706
Davie JK, Cho JH, Meadows E, Flynn JM, Knapp JR, Klein WH (2007) Target gene selectivity of the myogenic basic helix-loop-helix transcription factor myogenin in embryonic muscle. Dev Biol 311:650–664
Davis RL, Cheng PF, Lassar AB, Weintraub H (1990) The MyoD DNA binding domain contains a recognition code for muscle-specific gene activation. Cell 60:733–746
Deato MD, Tjian R (2007) Switching of the core transcription machinery during myogenesis. Genes Dev 21:2137–2149
Eftimie R, Brenner HR, Buonanno A (1991) Myogenin and MyoD join a family of skeletal muscle genes regulated by electrical activity. Proc Natl Acad Sci USA 88:1349–1353
Faulkner G, Lanfranchi G, Valle G (2001) Telethonin and other new proteins of the Z-disc of skeletal muscle. IUBMB Life 51:275–282
Frank D, Kuhn C, Katus HA, Frey N (2006) The sarcomeric Z-disc: a nodal point in signalling and disease. J Mol Med 84:446–468
Furukawa T, Ono Y, Tsuchiya H, Katayama Y, Bang ML, Labeit D, Labeit S, Inagaki N, Gregorio CC (2001) Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol 313:775–784
Gregorio CC, Trombitas K, Centner T, Kolmerer B, Stier G, Kunke K, Suzuki K, Obermayr F, Herrmann B, Granzier H, Sorimachi H, Labeit S (1998) The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J Cell Biol 143:1013–1027
Grounds MD, Garrett KL, Lai MC, Wright WE, Beilharz MW (1992) Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res 267:99–104
Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364:501–506
Haworth RS, Cuello F, Herron TJ, Franzen G, Kentish JC, Gautel M, Avkiran M (2004) Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ Res 95:1091–1099
Hayashi T, Arimura T, Itoh-Satoh M, Ueda K, Hohda S, Inagaki N, Takahashi M, Hori H, Yasunami M, Nishi H, Koga Y, Nakamura H, Matsuzaki M, Choi BY, Bae SW, You CW, Han KH, Park JE, Knoll R, Hoshijima M, Chien KR, Kimura A (2004) Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol 44:2192–2201
Himeda CL, Ranish JA, Hauschka SD (2008) Quantitative proteomic identification of MAZ as a transcriptional regulator of muscle-specific genes in skeletal and cardiac myocytes. Mol Cell Biol 28:6521–6535
Ji ZX, Du C, Wu GS, Li SY, An GS, Yang YX, Jia R, Jia HT, Ni JH (2009) Synergistic up-regulation of muscle LIM protein expression in C2C12 and NIH3T3 cells by myogenin and MEF2C. Mol Genet Genomics 281:1–10
Kassar-Duchossoy L, Gayraud-Morel B, Gomes D, Rocancourt D, Buckingham M, Shinin V, Tajbakhsh S (2004) Mrf4 determines skeletal muscle identity in Myf5:Myod double-mutant mice. Nature 431:466–471
Knapp JR, Davie JK, Myer A, Meadows E, Olson EN, Klein WH (2006) Loss of myogenin in postnatal life leads to normal skeletal muscle but reduced body size. Development 133:601–610
Kojic S, Medeot E, Guccione E, Krmac H, Zara I, Martinelli V, Valle G, Faulkner G (2004) The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J Mol Biol 339:313–325
Kostrominova TY, Macpherson PC, Carlson BM, Goldman D (2000) Regulation of myogenin protein expression in denervated muscles from young and old rats. Am J Physiol Regul Integr Comp Physiol 279:R179–R188
Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E (2003) A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12:1725–1735
Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG (2003) Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 426:247–254
Loots GG, Ovcharenko I, Pachter L, Dubchak I, Rubin EM (2002) rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res 12:832–839
Markert CD, Ning J, Staley JT, Heinzke L, Childers CK, Ferreira JA, Brown M, Stoker A, Okamura C, Childers MK (2008) TCAP knockdown by RNA interference inhibits myoblast differentiation in cultured skeletal muscle cells. Neuromuscul Disord 18:413–422
Markert CD, Meaney MP, Voelker KA, Grange RW, Dalley HW, Cann JK, Ahmed M, Bishwokarma B, Walker SJ, Yu SX, Brown M, Lawlor MW, Beggs AH, Childers MK (2010) Functional muscle analysis of the Tcap knockout mouse. Hum Mol Genet 19(11):2268–2283
Mason P, Bayol S, Loughna PT (1999) The novel sarcomeric protein telethonin exhibits developmental and functional regulation. Biochem Biophys Res Commun 257:699–703
Meadows E, Cho JH, Flynn JM, Klein WH (2008) Myogenin regulates a distinct genetic program in adult muscle stem cells. Dev Biol 322:406–414
Molkentin JD, Black BL, Martin JF, Olson EN (1995) Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 83:1125–1136
Moreira ES, Wiltshire TJ, Faulkner G, Nilforoushan A, Vainzof M, Suzuki OT, Valle G, Reeves R, Zatz M, Passos-Bueno MR, Jenne DE (2000) Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet 24:163–166
Musaro A, Cusella De Angelis MG, Germani A, Ciccarelli C, Molinaro M, Zani BM (1995) Enhanced expression of myogenic regulatory genes in aging skeletal muscle. Exp Cell Res 221:241–248
Nabeshima Y, Hanaoka K, Hayasaka M, Esumi E, Li S, Nonaka I, Nabeshima Y (1993) Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364:532–535
Nicholas G, Thomas M, Langley B, Somers W, Patel K, Kemp CF, Sharma M, Kambadur R (2002) Titin-cap associates with, and regulates secretion of, Myostatin. J Cell Physiol 193:120–131
Ott MO, Bober E, Lyons G, Arnold H, Buckingham M (1991) Early expression of the myogenic regulatory gene, myf-5, in precursor cells of skeletal muscle in the mouse embryo. Development 111:1097–1107
Pollard JW, Walker JM (1997) Basic Cell Culture Protocols Second Edition. Humana Press, Totawa
Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R (1993) MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75:1351–1359
Spitz F, Demignon J, Porteu A, Kahn A, Concordet JP, Daegelen D, Maire P (1998) Expression of myogenin during embryogenesis is controlled by Six/sine oculis homeoproteins through a conserved MEF3 binding site. Proc Natl Acad Sci USA 95:14220–14225
Tian LF, Li HY, Jin BF, Pan X, Man JH, Zhang PJ, Li WH, Liang B, Liu H, Zhao J, Gong WL, Zhou T, Zhang XM (2006) MDM2 interacts with and downregulates a sarcomeric protein, TCAP. Biochem Biophys Res Commun 345:355–361
Valle G, Faulkner G, De Antoni A, Pacchioni B, Pallavicini A, Pandolfo D, Tiso N, Toppo S, Trevisan S, Lanfranchi G (1997) Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett 415:163–168
Venuti JM, Morris JH, Vivian JL, Olson EN, Klein WH (1995) Myogenin is required for late but not early aspects of myogenesis during mouse development. J Cell Biol 128:563–576
Wang DZ, Valdez MR, McAnally J, Richardson J, Olson EN (2001) The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development 128:4623–4633
Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ (1991) Muscle-specific transcriptional activation by MyoD. Genes Dev 5:1377–1386
Yokoyama S, Ito Y, Ueno-Kudoh H, Shimizu H, Uchibe K, Albini S, Mitsuoka K, Miyaki S, Kiso M, Nagai A, Hikata T, Osada T, Fukuda N, Yamashita S, Harada D, Mezzano V, Kasai M, Puri PL, Hayashizaki Y, Okado H, Hashimoto M, Asahara H (2009) A systems approach reveals that the myogenesis genome network is regulated by the transcriptional repressor RP58. Dev Cell 17:836–848
Zhang H, Stavnezer E (2009) Ski regulates muscle terminal differentiation by transcriptional activation of Myog in a complex with Six1 and Eya3. J Biol Chem 284:2867–2879
Acknowledgments
This work was supported by a grant from the Central Research Committee, Southern Illinois University School of Medicine. The work was also supported by grant #159609 from the American Cancer Society, Illinois Division, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Rights and permissions
About this article
Cite this article
Zhang, S., Londhe, P., Zhang, M. et al. Transcriptional analysis of the titin cap gene. Mol Genet Genomics 285, 261–272 (2011). https://doi.org/10.1007/s00438-011-0603-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-011-0603-6