
Abstract
The root apex contains meristematic cells that determine root growth and architecture in the soil. Specific transcription factor (TF) genes in this region may integrate endogenous signals and external cues to achieve this. Early changes in transcriptional responses involving TF genes after a salt stress in Medicago truncatula (Mt) roots were analysed using two complementary transcriptomic approaches. Forty-six salt-regulated TF genes were identified using massive quantitative real-time RT-PCR TF profiling in whole roots. In parallel, Mt16K+ microarray analysis revealed 824 genes (including 84 TF sequences) showing significant changes (p < 0.001) in their expression in root apexes after a salt stress. Analysis of salt-stress regulation in root apexes versus whole roots showed that several TF genes have more than 30-fold expression differences including specific members of AP2/EREBP, HD-ZIP, and MYB TF families. Several salt-induced TF genes also respond to other abiotic stresses as osmotic stress, cold and heat, suggesting that they participate in a general stress response. Our work suggests that spatial differences of TF gene regulation by environmental stresses in various root regions may be crucial for the adaptation of their growth to specific soil environments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Abiotic stresses impact plant growth and development to diminish stress exposure (Potters et al. 2007) leading to stress-induced morphogenetic responses such as the adaptation of root architecture to specific soil conditions. The root system is the primary site of perception of the soil environment. Development of the root system depends on the activity of the apical meristem, which produces all cell types in a highly defined pattern of cell divisions (Benfey and Scheres 2000; Brady et al. 2007). Complex regulatory mechanisms in these cell types have been identified by analysing expression patterns in 15 different zones of the Arabidopsis thaliana root (Birnbaum et al. 2003). Among 577 transcription factor (TF) transcripts detected in the root, 331 were differentially expressed in different zones, allowing tentative links between gene activity and cell fate/tissue specialization. This suggests that spatial expression of TF genes is a critical aspect of the networks involved in transcriptional regulations (Lee et al. 2006). Moreover, a recent study demonstrates that developmental parameters highly constrained the transcriptional response to high salinity of different cell layers and developmental stages of the Arabidopsis root. This reveals the crucial role the environment plays in defining the transcriptional outcome of cell-fate decisions (Dinneny et al. 2008).
One critical step controlling stress responses involves transcriptional regulation (Wang et al. 2003). Plant genomes dedicate a large number of their coding sequences to TFs reaching about 6% (>1,500 TF genes) in the fully sequenced Arabidopsis genome (Riechmann et al. 2000). Among these TFs, a number of them, such as AP2/EREBP (Apetala2 / Ethylene Response Element Binding Protein), bZIP/HD-ZIP (basic leucine ZIPper/HomeoDomain—leucine ZIPper), MYB (MYeloBlastosis), HSF (Heat Shock Factor) and WRKY TF families, have been implicated in plant stress responses (Shinozaki and Yamaguchi-Shinozaki 2000; Jiang and Deyholos 2006). Very few TFs have been implicated in abiotic stress tolerance in legumes. Among them, the soybean SCOF-1 TF belonging to the C2H2 zinc-finger family was induced by low temperature and abscisic acid but not by dehydration or high salinity (Kim et al. 2001). The Medicago MtZpt2-1 gene from the same family was shown to be required for both symbiotic nodule development and root growth recovery after salt stress (Frugier et al. 2000; Merchan et al. 2003, 2007). Furthermore, overexpression of the C2H2 zinc-finger alfalfa Alfin1 and the chickpea AP2-type TF CAP2 in transgenic plants enhanced growth and tolerance to salt (Winicov 2000; Shukla et al. 2006). Nevertheless, the molecular links between perception of the environment and activation of developmental adaptive responses remain largely unknown.
Transcriptomic studies have been used to identify TFs regulated by abiotic stresses potentially linked to specific genetic programs activated to cope with stress (Chen et al. 2002; Seki et al. 2002). In model legumes, extensive sequencing highlighted around 2,000 TFs per genome, of which less than 1% have been genetically characterized (reviewed by Udvardi et al. 2007). In Medicago truncatula, Mt16K+ microarrays covering 16,086 tentative consensus sequences derived mainly from 164,000 M. truncatula ESTs collected in the TIGR M. truncatula Gene Index 5 (http://www.tigr.org/tdb/mtgi) were recently used to monitor changes in the transcriptome of desiccation-sensitive radicles of M. truncatula seeds (Buitink et al. 2006). In this work, more than 1,300 genes were differentially expressed during embryo desiccation and several regulatory genes (including TFs) were up-regulated during maturation. This showed a partial overlapping between ABA-dependent and independent regulatory pathways in both drought stress and desiccation tolerance in seeds. Homologs of up-regulated TFs during the re-establishment of desiccation tolerance were shown to be involved in both abiotic stress and seed development in other species (Buitink et al. 2006). Recently, transcription profiling revealed 912 genes differentially expressed during salt acclimation in Lotus japonicus (Sanchez et al. 2008). These transcriptional changes were presumably coordinated by specific TFs modulated during this process such as AP2/ERF and MYB (24 and 20% of the total number of TFs, respectively). An alternative high throughput approach for profiling TF genes is massive real-time RT-PCR (qRT-PCR). The sensitivity of this approach has been validated in A. thaliana revealing novel root- and shoot-specific lowly expressed TF genes not previously linked to these organs (Czechowski et al. 2005), as well as TF genes that respond to sugars and nutrients (Scheible et al. 2004; Osuna et al. 2007). Recently, this technology was exploited to identify putative TF genes expressed at different stages of seed development in M. truncatula (Verdier et al. 2008).
In root cells, overlapping stress perception and signal transduction pathways may be most critical at very early times after stress exposure (Swindell 2006). To this end, we have identified, in this work, TFs rapidly regulated in root apexes by salt stress in the model legume M. truncatula. A massive real time RT-PCR approach allowed us to identify 46 TF genes responding after a 1-h salt treatment in roots. Several of these TFs displayed even higher salt-inductions in root apexes. We then performed a microarray expression profiling directly on root apexes submitted to salt stress and identify 84 regulated TF sequences. We propose that these TF genes, strongly regulated in root meristematic regions, are good candidates to be involved in transcriptional networks related to the adaptation of root architecture to the soil environmental conditions.
Materials and methods
Plant material, growth conditions, and stress treatments
Medicago truncatula cv. Jemalong A17 seeds (Barker et al. 1990) were scarified, surface-sterilised by immersion for 20 min in Bayrochlore (3.75 g/L active Chlore, Novelty Chlor, France), and washed six times with sterilised water. Seeds were sown on 1% water-agar plates and stored for 2 days at 4°C before incubating overnight at 24°C in the dark to ensure uniform germination.
For analysing stress responses in root apexes, germinated seedlings were transferred to pots without bottom containing perlite:sand (3:1, v/v) placed on a grid in a container filled with liquid medium ([Soluplant 18.6.26]:water 1:1, Duclos, France). Nine pots containing nine seedlings each were used per experiment. Seedlings were grown for 10 days at 22°C on a 16-h light/8-h dark cycle in a growth chamber with 60% relative humidity until roots passed through the grid and were immersed in the medium. Then, liquid plant growth media imbibing the root apexes was replaced by fresh medium with or without salt (100 mM NaCl). Four biological replicates per salinity treatment were performed (nine plants/replicate). Root tips (1 cm) were harvested after a 1-h salt-stress treatment, immediately frozen in liquid nitrogen and stored at −80°C for RNA extraction.
For the analysis of stress responses in whole roots, 40 germinated seedlings were grown in a container filled with perlite:sand (3:1) for 2 weeks at 24°C on a 16-h light/8-h dark cycle in a greenhouse. Then, plants were immersed into a glass container containing plant growth media, with or without 100 mM NaCl supplement, for 1 h under agitation (to prevent anoxia). Whole roots were collected, immediately frozen in liquid nitrogen and stored at −80°C for RNA extraction.
For the analysis of temporal expression patterns, 30 germinated seedlings were grown in Magenta pots and placed for 3–4 days on a grid (to allow only the immersion of the roots in liquid plant growth medium), under agitation in a growth chamber (24°C and a 16 h-light period). In this set-up, plants were submitted to a 100-mM NaCl stress and roots were collected at various time points (0, 15, 30, 60, and 360 min). For the different abiotic stress treatments, similarly grown seedlings were treated for 1 h with 100 mM NaCl or 200 mM mannitol, whereas alternatively the pots were placed for 1 h at 4 or 37°C. Root tips (1 cm) were harvested after different treatments, immediately frozen and stored at −80°C for RNA extraction. Two biological replicates per treatment were performed.
Real-time RT-PCR
Total RNA was isolated with Trizol reagent (Gibco-BRL), treated with DNase (RQ1 RNase-free DNase, Promega, France), purified using the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions and stored at −80°C. RNA quality was controlled using a bioanalyzer and RNA was quantified using a Nanodrop spectrophotometer. RNA was tested for the absence of genomic DNA by PCR before proceeding to the RT step to avoid background PCR signal due to genomic DNA.
Massive real time RT-PCR TF profiling was carried out using a set of 710 RT-PCR primer pairs targeting TF genes derived from M. truncatula genome annotation (Kakar et al. 2008). Consistent values were selected as those having less than a cycle of difference between technical replicates.
For other real-time RT-PCR, two micrograms of total RNA were retrotranscribed for 1 h at 42°C using the “SUPERSCRIPT® II first-strand synthesis system” (Invitrogen, Cergy Pontoise, France) and subsequently denatured for 10 min at 75°C. These cDNAs was diluted to 60 μL in sterile water and 2.5 μL was amplified using the “LC FastStart DNA Master SYBR Green I” (Roche Diagnostics, Meylan, France) according to the manufacturer’s instructions. Real time PCR were done on a Light Cycler apparatus, using the following conditions: 95°C for 10 s, 50 amplification cycles at 95°C for 5 s, 60°C for 5 s and 72°C for 15 s. A negative control without cDNA template was always included for each primer combination. The Mt UBI, Mt H3L and Mt RBP1 genes were used as internal controls and behave similarly all through the salt treatment kinetic. Mt UBI was then selected to normalize the expression data for each gene in all figures. Primer sequences for the different TFs and internal control analysed are given in Electronic supplementary material Table S6.
Syntheses of two independent cDNA preparations from the same RNA sample (technical duplicates) were performed for each experiment. Ratios against a constitutive control (Mt UBI gene) were used to normalize gene expression between different biological conditions. The ratio value of the experimental control condition was set up to 1 as a reference to determine relative induction/repression factors. Except for Fig. 6, a representative example out of the two biological replicates performed is shown for each figure.
Hybridization of Mt16K+ microarrays
Four independent biological replicates of salt-treated root apexes and controls were used. Two micrograms of total RNA from each sample was extracted as described for RT-PCR experiments and used to synthesize Cy5/Cy3-labelled cDNA using the Amino Allyl MessageAmp™ aRNA Kit (Ambion, TX, USA) according to the manufacturer’s instructions.
Cy5/Cy3-labelled cDNA were hybridized with the 70-mer Mt16K+ oligonucleotide microarrays (http://www.ebi.ac.uk/arrayexpress; accession number A-MEXP-138) for 16 h at 60°C. Microarray hybridizations have been deposited and are described at http://www.ebi.ac.uk/arrayexpress (ArrayExpress accession: E-MEXP-1441).
Microarray data analysis and statistics
Microarray slides were scanned with a Genepix two-laser scanner (Axon Instruments, Inc.) and the resulting images were analysed with the GenePix 6.0 software (Molecular Devices). Data transformations and normalization, performed with the MAnGO R script (version 1.0; Marisa et al. 2007), consisted in a local background correction, omitting flagged spots, and successively an intensity-dependent print-tip loess normalization and a scale between array normalization (Yang et al. 2002). Differential analysis was based on an empirical Bayes moderated t test adjusted with the false discovery rate (FDR, Benjamini and Hochberg 1995) multiple test correction. Differentially expressed genes were defined based on thresholds on adjustedP-values (P < 0.001), fold-change (|FC| > 1.8), and mean intensity of the two channels (A-mean > 7 on a log2 scale; threshold defined using negative controls available on the microarray).
Bioinformatic analyses
Annotation of differentially expressed TFs was systematically verified based on BLASTX searches on the TIGR database (http://www.tigr.org/tdb/tgi/mtgi/; release 8.0, January, 2005) for ESTs, and on the NCBI database (http://www.ncbi.nlm.nih.gov/BLAST/) for available genomic sequences.
MapMan display
Data were visualized using the MapMan software (downloadable at http://gabi.rzpd.de/projects/MapMan/; Thimm et al. 2004; Tellström et al. 2007), following instructions provided on the website. To predict BINs that exhibit a different bias of expression profile among all constituting BINs, we applied the statistical analysis provided in Mapman (Benjamini and Hochberg corrected Wilcoxon rank sum test or BHW test). Significant differences were defined based on a p-value < E-02 and the BIN size (element number > 10).
Results
Identification of transcription factor genes differentially expressed in M. truncatula roots under salt stress
An analysis based on a massive qRT-PCR TF profiling approach was carried out using set of 710 PCR primer pairs targeting TF genes. As large quantities of RNA were required for this approach, whole root systems, either untreated or submitted to a 1-h salt treatment (100 mM NaCl) were used. We retained 401 TF transcripts (56%) having consistent values in the two technical replicates for each condition. Among these, 46 TF transcripts were salt-regulated and showed a difference of at least one PCR cycle between normalized untreated control and 1-h salt-treated samples (a cut-off threshold of 1.8-fold was considered; Electronic supplementary material Table S1). To further validate these expression analyses, the efficiency and specificity of 26 primer pairs (corresponding to various TF gene families) were tested and 15 (58%) of them gave efficient and specific amplification for the expected TF gene (Electronic supplementary material Table S2). Using these oligonucleotides, the results obtained were validated in 80% of the cases using a replicated biological experiment.
Refined temporal expression patterns were analysed for six TF genes (randomly selected from ESM Table S2) and named AP/EREBP1207, DOF207, MYB119, NAC969, NAC1081 and NAC1126 (based on their best homology to known TF domains), in M. truncatula roots submitted to a 100-mM NaCl treatment for various incubation times (from 15 min to 6 h; Fig. 1). Different patterns of gene expression were observed. AP2/EREBP1207 and MYB119 transcript levels were rapidly and transiently induced, reaching maximum values after 15 or 30 min, respectively, and then decreased after 1 h and 6 h of salt treatment. For NAC1081 and NAC1126, the highest transcript levels were detected after 6 h of salt treatment, indicating a late activation of these genes, whereas NAC969 transcript levels were maximal at 1 h. Hence, several new legume TFs regulated in response to salt stress have been identified in whole M. truncatula roots.

Expression analysis of whole roots submitted to salt stress for different times. Real-time RT-PCR expression analysis of selected M. truncatula transcription factors was performed. Total RNAs were obtained from M. truncatula whole roots treated with 100 mM NaCl for 15 min to 6 h. Histograms represent quantification of the products normalized to the constitutive control Mt UBI. The value for untreated control roots is set to 1 as reference. A representative example out of two biological replicates is shown
Salt regulation of several TF encoding transcripts is enhanced in root apexes
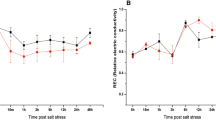
To determine TF genes that might be linked to the activation of specific genetic pathways in root apexes, we analysed the expression of TF genes in root apexes versus whole roots (Fig. 2). We first analysed ten TF genes having specific and optimised real time RT-PCR conditions (Fig. 2a) including seven TFs identified from our initial approach (DOF207, DOF891, HD953, MYB119, NAC969, NAC1126, WRKY1219, Electronic supplementary material Table S1) and three other genes (AP/EREBP1207, and NAC1081 and MYB1070).

Comparison of transcription factor (TF) expression in response to salt stress in whole roots versus root apexes. Real-time RT-PCR analysis was performed for 15 TF genes identified using the two transcriptomic approaches. Total RNAs were obtained from M. truncatula whole roots and root apexes treated with 100 mM NaCl for 1 h. Histograms represent quantification of the products normalized to the constitutive control Mt UBI. a Selected TF genes identified through the massive RT-PCR approach or b through microarray analysis. The value for untreated control roots is set to 1 as reference. A representative example out of two biological replicates is shown
Weak changes of expression levels were observed in whole roots for the ten tested TFs (induction factors were up to 3.4, Fig. 2 black bars) similar to those observed in the massive real time RT-PCR approach albeit their magnitude sometimes differed (ESM Table S1). In contrast, several TF genes such as AP2/EREBP1207 and MYB119 showing minor variation in whole root RNA samples were strongly induced in root apexes (with induction factors > 30; Fig. 2 white bars). Similar trends in favour of higher transcript levels in root apexes were observed for NAC1081, WRKY1219 and DOF207, with induction factors ranging between 5 and 13. Interestingly, larger decreases in transcript levels of DOF891 and HD953 were observed in root apexes in contrast to whole roots. Hence, specific members of TF gene families showed an enhanced transcriptional regulation in root apexes challenged with salt stress. In contrast, MYB1070, NAC969 and NAC1126 transcriptional induction was higher in whole roots compared to root apexes, suggesting ubiquitous role of these TF genes. Hence, large spatial differences of TF gene regulation by salt stress were found between complete root systems and root apexes, allowing us to define a specific set of TF genes rapidly regulated by salt in the root meristematic and elongation zones.
Identification of early salt-regulated transcription factors expressed in M. truncatula root apexes using a microarray-based approach
In order to investigate more broadly changes in gene expression induced by salt stress in Medicago root apexes, a transcriptome analysis based on M. truncatula 16K+ microarrays (Mt16KOLI1) was performed using four independent biological replicates of salt-treated root apexes and controls. Normalization and statistical analysis (P < 0.001, Amean > 7, |FC| > 1.8, see Materials and methods) resulted in 824 genes that were differentially expressed between untreated control and 1-h salt-treated root apexes (Electronic supplementary material Table S3), 558 of them being up-regulated (Fig. 3). The salt-regulated genes were functionally assigned using the GeneBin database of MapMan software (Thimm et al. 2004) initially designed for studying the Arabidopsis transcriptome and adapted for M. truncatula (Tellström et al. 2007). This automatic annotation may limit the relevance of Medicago MapMan assignments, but has been used as an initial approach for Medicago transcriptomes.

Functional categories distribution of the 824 salt-regulated genes in M. truncatula root apexes based on Mapman BIN assignments. A complete list of the classified genes is given in ESM Table S3. N denotes the number of regulated genes (Amean > 7, P < 0.001, |FC| > 1.8). Upper and lower distributions correspond to induced or repressed genes, respectively
Global analysis of transcriptome changes indicated that the largest group of up-regulated genes (besides the non-assigned category representing about 30% of total number of differentially regulated genes), corresponded to genes related to secondary metabolism genes (13 and 2% of the up- and down-regulated genes respectively, Figs. 3, S1). Benjamini Hochberg corrected Wilcoxon rank sum test (BHW test) was used to define precisely the statistical significance of the differential expression of this functional category (BHW test; P = 2.6 E-11, Electronic supplementary material Table S4). Within this category, genes involved in phenylpropanoid, flavonoid, and isoprenoid pathways were significantly induced (BHW test, P = 9.96 E-05, P = 6.27 E-05 and P = 2.68 E-02, respectively; Electronic supplementary material Table S4), indicating likely activation of cellular protection mechanisms in M. truncatula root apexes in response to salt stress.
In addition, respectively, 7 and 3% of the up- and down-regulated genes were significantly linked to environmental stress responses (biotic and abiotic, BHW test, P = 9.81 E-05, ESM Table S4, Fig. S2), most of them being related to biotic stresses (BHW test, P = 1.30 E-05, ESM Table S4, Fig. 3). Surprisingly, few up-regulated genes (11/42) were specifically related to abiotic stress and only three of them were annotated as regulated by drought or salt stress in Mapman GeneBins. This probably reflects the limits of the current annotation in the GeneBin database for Medicago based on analysis of abiotic stress responses detected, mainly in Arabidopsis. For example, GSTs are annotated as miscellaneous genes although they are well-known defence-related genes.
Another significant group included, respectively, 7 and 2% of the salt up- and down-regulated genes (BHW test, P = 2.29 E-04, ESM Table S4) linked to phytohormonal pathways (Figs. 3, ESM S3). Within this group, ABA-related genes were found in our analysis as not significantly regulated at these early time points in root apexes (based on BHW test). However, genes involved in jasmonate and ethylene pathways were significantly regulated (BHW test, P = 9.96 E-05 and P = 5.86 E-02, respectively, ESM Table S4). This suggests a role of these phytohormones in adaptation of root apexes to external environmental conditions.
Finally, the TF genes represented an important salt-regulated group in root apexes as visualized on the MapMan pictorial diagram referred as “regulation overview” (Fig. S3, ESM Table S3). Their MapMan GeneBin annotation was verified by BLAST against the M. truncatula TIGR database (release 8.0). From the 824 salt-regulated genes corresponding to stringent statistical criteria, 84 were found to be TFs distributed into 72 salt-induced and 12 salt-repressed genes after a 1-h NaCl treatment (Fig. 4, Electronic supplementary material Table S5). Whereas the largest TF set is associated to “transcription regulation” (37 and 10 genes; salt-induced and salt-repressed, respectively), several other salt up-regulated TF genes are linked to “phytohormones” (mainly ethylene; nine genes), “stress” (six genes) or “development” (four genes) categories. As automatic annotations are derived from best homology with genes from other plants, these classifications should be interpreted with caution in the context of M. truncatula roots, particularly as certain gene families contain many members (e.g. the AP2/EREBP family) carrying a homologous DNA-binding domain.

Number of TF genes belonging to different functional categories based on Mapman BIN assignment. The 84 TF genes (Amean > 7, P < 0.001, |FC| > 1.8) regulated in M. truncatula root apexes after a 1 h salt-treatment were classified accordingly to Mapman BINs. Complete list is given in ESM Table S5
In conclusion, functional assignment of the 824 differentially expressed genes revealed extensive modifications of M. truncatula transcriptome in root apexes, including 84 TFs linked to the activation of transcriptional networks, in response to salt stress.
Characterization of early salt-regulated transcription factor families of M. truncatula
TF MapMan GeneBin annotation and putative functional domains of the 84 salt-regulated TF genes expressed in root apexes were systematically determined using BLAST comparisons (Fig. 5). The overall view highlighted a distribution of 16 TF families grouped into three categories according to their regulation. A first category exhibited exclusively salt-induced TF genes and is constituted by eight TF families [named WRKY, GAI, RGA, SCR (GRAS), Zinc-finger protein expressed in Inflorescence Meristem (ZIM), basic Helix-Loop-Helix (bHLH), HD, HSF, NAM, ATAF1, CUC2 (NAC) and MYeloCytomatosis (MYC)]. In the second group, four TF families [named AP2/EREBP, Zn-finger (Zinc-finger), MYB and bZIP] showed a majority of salt-induced but a few salt-repressed TF members. Among them, the predominant AP2/EREBP family included 15 members of which 14 were up-regulated by salt and a salt-repressed DREB-like gene (Dehydration Responsive Element Binding, MT004883, Supplementary Table S5). The Zn-finger TF family included nine members salt-induced and one down-regulated. The third category grouped four TF families showing gene repression after a salt treatment [response regulator (RR), lateral organ boundaries (LBD), Bric-a-brac, tramtrack, and broad/POxvirus zinc finger (BTB/POZ) and TB1, CYC, PCF (TCP)]. Only the WRKY TF family was statistically significantly regulated by salt (BHW test, P = 7.52 E-02, ESM Table S4).

Number of gene family members with a known functional TF domain. The collection of 84 salt-regulated TF gene identified in M. truncatula root apexes was individually analysed to score the number of members for each gene family that is either up-regulated (white) or down-regulated (black). Names are: AP2/EREBP apetala2/ethylene response element binding protein, AS2 asymetric 2, bHLH basic helix-loop-helix, bZIP basic leucine ZIPper, Zn-finger zinc-finger, BTB/POZ bric-a-brac, tramtrack, and broad/POxvirus zinc finger, GRAS GAI, RGA, SCR, HD HomeoDomain, HSF heat shock factor, LBD lateral organ boundaries, MYB myeloblastosis, MYC MyeloCytomatosis, NAC NAM, ATAF1, CUC2, RR Response regulator, SBP squamosa binding protein, TCP TB1, CYC, PCF, ZIM zinc-finger protein expressed in inflorescence meristem, Zn-finger zinc-finger
The spatial regulation of salt-regulated TFs identified in root apexes was similarly analysed as the previously identified TFs (in the massive real time RT-PCR approach, Fig. 2). Four highly induced [HD1374 (MT008226), HSF045 (MT016090), MYB634 (MT014860) and MYB636 (MT014600)] and one salt-repressed [bZIP316 (MT015835)] TF genes were chosen to evaluate their expression in root apexes versus whole roots submitted to salt stress (Fig. 2b). HD1374, MYB634 and MYB636 were strongly transcriptionally induced in root apexes (induction factors of 52, 98 and 88, respectively) whereas changes in transcript abundance using whole root RNA samples were very weak (induction factors of 1.1, 1.3 and 1.4, respectively). Less differential induction was observed for HSF045 (Fig. 2b). In contrast, the salt-repressed bZIP316 showed similar transcript levels in both RNA samples indicating a salt-regulation independent of the root zone. These results suggest that rapid increase in transcriptional response of some specific TF genes in meristematic regions after a salt stress may be crucial for adaptation of root growth in response to changes in the soil environment.
To evaluate the specificity of the salt response, eight selected salt-induced TFs (DOF207, HD1374, MYB634, MYB636, MYB1070, NAC969, NAC1081 and NAC1126) in Medicago root apexes were submitted to three additional stresses: osmotic stress (mannitol 200 mM), cold (4°C) and heat stress (37°C) for 1 h (Fig. 6). These other stresses also induce the expression of the TF genes at variable levels. Furthermore, differential regulation by these stresses was observed among the three MYB or the three NAC genes belonging to the same TF family. Globally, these genes seem to be general stress-responsive. Nevertheless, several TF genes (e.g. NAC1081 and NAC 969) are induced at much higher levels by salt stress than by any of the other treatments, suggesting certain specificity of the salt response.

Expression of TF genes in response to different abiotic stresses in Medicago root apexes. Real-time RT-PCR analysis was performed for eight selected TF genes in root apexes subjected to four abiotic stresses [NaCl 100 mM, mannitol 200 mM, cold (4°C) and heat (37°C)] for 1 h. Total RNAs were obtained from root apexes after the different treatments. Histograms represent quantification of the products normalized to the constitutive control Mt UBI. The mean of two biological replicates is shown
Discussion
The development of genomic resources for M. truncatula has enabled to combine Mt16k+ microarrays and massive quantitative real-time PCR approaches to identify a large number of differentially expressed TF genes in early salt stress conditions. The massive TF profiling approach may be more sensitive to monitor changes in TF expression when compared to arrays (Czechowski et al. 2005; Kakar et al. 2008), but may require large amounts of RNA, a limitation to analyse expression specifically in root apexes. Another difference between both transcriptomic approaches is that the genes targeted by the RT-PCR resource were selected from IMGAG (International Medicago Genome Annotation Group) annotated-genes derived from genome sequences (including potential annotation errors), while the microarray contained probes designed mostly from TC/ESTs generally corresponding to highly expressed genes. Further, oligo-based microarray hybridization is more prone to cross-hybridization than PCR-derived techniques, even though in our case we were able to confirm by RT-PCR the microarray results in all cases. Finally, gene expression patterns in whole roots and root apexes revealed large differences between these regions, preventing a direct comparison of the identified TF genes from these different approaches. Although globally a large overlapping of TF genes exist, these methodologies are complementary for TF gene identification to dissect regulatory pathways involved in salt stress responses in legume roots.
As an initial step in responding to salt stress, plants must sense and transduce appropriate stress-derived signals into physiological processes leading to changes in root growth and development. It has been speculated that the early responsive genes may provide initial protection and amplification of signals (Ramanjulu and Bartels 2002). Consistent with this idea, our results revealed global transcriptional changes in response to salt stress in the secondary metabolism including phenylpropanoid, flavonoid, and isoprenoid pathways in agreement with the stress-triggered production of defence-related secondary metabolites previously reported for rice (Walia et al. 2005). Moreover, large-scale genomic studies realized in the legume L. Japonicus subjected to long-term salt acclimatization also reinforce this observation (Sanchez et al. 2008). In addition, phytohormones such as abscisic acid, jasmonic acid, ethylene and salicylic acid appear to be critical components of complex signalling networks and are being incorporated into current models of stress responses (Zhu 2002). In Medicago root apexes, we significantly identified early salt-regulated genes mainly linked to jasmonate and ethylene signalling pathways. Multiple interferences between both pathways may occur when tissues are submitted to environmental stresses (Devoto and Turner 2005; Ludwig et al. 2005). Moreover, microarray experiments in A. thaliana have previously demonstrated that the majority of ethylene- and jasmonic acid- related transcripts were responsive to salt treatment (Jiang and Deyholos 2006; Ma et al. 2006).
Stress perception and signalling pathways are critical components of plant adaptative response to survive under environmental constraints. General-stress responses may arise from the activities of various TFs interacting among distinct abiotic stress response pathways (Chen et al. 2002; Chen and Zhu 2004). We have identified a collection of TF genes that are rapidly induced by salt treatment in roots of the model legume M. truncatula. This finding is consistent with large-scale genomic studies realized in several plant species as A. thaliana (Kreps et al. 2002; Seki et al. 2002; Jiang and Deyholos 2006), tomato (Ouyang et al. 2007), rice (Chao et al. 2005), L. japonicus (Sanchez et al. 2008) and moss (Cuming et al. 2007) which support the strong implication of TF genes in early salt stress responses.
The TFs of M. truncatula identified in this study could be classified into TF families based on their functional domains and fits well with the recent report of Udvardi et al. (2007). Expression of members of these families has been reported in Arabidopsis roots (Shinozaki and Yamaguchi-Shinozaki 2000; Jiang and Deyholos 2006; Dinneny et al. 2008). Nevertheless, little is known about spatial control of transcriptional complexity in plant organs in response to stress. Previous studies showed diverse responses in different maize root regions using a kinematic analysis to characterize spatial and temporal patterns of cell expansion within the root growth zone (Sharp et al. 2004). Furthermore, Sun et al. (2008) reported that salt inhibits root elongation, but also greatly affects root tip growth direction in A. thaliana, indicating that salt-induced changes in root apical cells may contribute to alter gravitropism in order to enable plants adapt to saline environments. Recently, stress responses including salt, osmotic, drought, cold and hormone treatments were placed within the context of tissues and cell lineages in the Arabidopsis root using the fuzzy k-means clustering method to identify conditionally co-expressed genes (Ma et al. 2006; Ma and Bohnert 2007). In these studies, the gene expression map previously established for the Arabidopsis root (Birnbaum et al. 2003) was integrated with clusters defining stress specificity of the responses. This analysis provided intersections between stress-responsive and cell-specific profiles and identified distinct cell lineages and developmental stages affected by abiotic stresses. In addition, the diversity of Arabidopsis transcriptional patterns in different cell-types exposed to salinity was recently further analysed (Dinneny et al. 2008). These analyses revealed that the transcriptional state of a cell is largely a reaction to environmental conditions regulated by a small core set of genes that stably determines cell identity. Hence, the environment plays a crucial role in defining the transcriptional outcome of cell-fate decisions in roots. Our data are in agreement with these results, showing large spatial differences of TF gene regulation by salt stress between whole root systems and root apexes in Medicago. TF genes encoding AP2/EREBP1207, HD1374, MYB119, MYB634, MYB636, NAC1081, WRKY1219, DOF207 and HSF045 showed a considerable enhancement of expression in root apexes in response to salt stress when compared to whole roots. In Arabidopsis, most of the TF families examined including AP2-like, HD-ZIP, MYB and WRKY expressed specific members in multiple cell types and tissues of the root at progressive developmental stages (Birnbaum et al. 2003). Furthermore, the analysis of the publicly available Affymetrix Arabidopsis gene-chip data on abiotic stress displayed a cluster of 171 genes only or mostly up-regulated by salt-stress, among which nearly 20% corresponded to TFs (Ma et al. 2006). The regulation of specific members of TF families in Medicago root tips supports the hypothesis that these genes may intersect root developmental pathways and salt-related transcriptional networks also in legumes. The AP2/EREBP TF genes were already reported to be linked to abiotic stress in many plants (Kizis et al. 2001; Yamasaki et al. 2004; Zhang et al. 2005; Shukla et al. 2006; Kumari et al. 2008). In addition, drought and salt tolerance in Arabidopsis are conferred by the related HARDY gene (Karaba et al. 2007). In our study, the AP2/EREBP1207 was one of the most highly salt-induced TF genes in Medicago root apexes, consistent with the involvement of DREB genes of the AP2/EREBP family in abiotic stress tolerance notably in the soybean and chickpea legumes (Li et al. 2005; Shukla et al. 2006). Moreover, spatial differences in the expression of AP2/EREBP in Medicago root meristems and differentiated roots were previously observed under non-stress conditions (Holmes et al. 2008). Our data also revealed different expression levels for two Medicago HD-ZIP members (HD1374 and HD953) in root apexes versus whole roots in response to salt stress. In agreement, Ni et al. (2008) have recently reported that the cotton homeodomain-leucine zipper GhHB1 transcripts accumulated during early root development but decreased to very low levels as roots developed further. The expression of the GhHB1 gene was dramatically increased in roots under salt treatment, further supporting the involvement of HD-ZIP TF members in response to environmental conditions (Ariel et al. 2007). Three Medicago MYB members were highly induced in root apexes, a superfamily known to be involved in numerous processes, including stress responses (Jin and Martin 1999; Ito et al. 2001; Stracke et al. 2001; Chen et al. 2006). Finally, NAC1081 was highly induced in root apexes in contrast the other selected NAC TFs. Members of this TF family have previously been associated with multiple stress responses in Arabidopsis (Tran et al. 2004) and rice (Nakashima et al. 2007). AtNAC2, a NAC-type TF from A. thaliana, has been proposed to integrate environmental and endogenous stimuli into the process of plant lateral root development (He et al. 2005).
The productivity and yield of plant crops is often limited by the joint influence of several stress combinations (Mittler 2006). Identification of genes exhibiting expression responses to several stresses may provide insights into the functional basis of multiple stress tolerance in plants. Genome wide correlations between transcriptional responses to different stress treatments peaked in Arabidopsis roots at 1 h of stress exposure (Swindell 2006), suggesting a general-stress response at this time point. Our analysis of transcriptional changes in Medicago root apexes subjected for 1 h to salinity, osmotic stress, heat and cold shocks is in agreement with this report. Indeed, the selected eight salt-induced TF genes were also induced in Medicago root apexes under one or several additional stresses, even though the salt induction was quantitatively higher in most cases. As these TF genes respond to various environmental stresses, they may represent possible intersection points among otherwise independent stress-response pathways (Swindell 2006). Indeed, osmotic adjustment is a response involved in salt stress, and several TF genes were also regulated by this stress.
In conclusion, we characterized TF genes issued from large-scale transcriptomic analyses of Medicago roots subjected to salt stress. Analysis of salt-stress responses in root apexes versus whole roots linked these TFs to the spatial regulation of gene expression after salt stress. Using four short-term applications of abiotic stresses, we conclude that these TF genes may be part of a general stress response of root apexes even though their induction is much higher under salt stress conditions. These data contribute to elucidate the complex regulatory mechanisms associated with stress responses in legume roots. Further functional studies are, however, required to understand the role of specific TF networks activated. It will be particularly challenging to decipher how root developmental pathways may intersect specific salt-related transcriptional networks in the root apex to control salt tolerance and lead to the adaptation of root growth to the soil environment.
References
Ariel FD, Manavella PA, Dezar CA, Chan RL (2007) The true story of the HD-Zip family. Trends Plant Sci 12:419–426
Barker DG, Bianchi S, Blondon F, Datté Y, Duc G, Flament P, Gallusci R, Génier R, Guy R, Muel X, Tourneur J, Dénarié J, Huguet T (1990) Medicago truncatula, a model plant for studying the molecular genetics of the Rhizobium-legume symbiosis. Plant Mol Biol Rep 8:40–49
Benfey PN, Scheres B (2000) Root development. Curr Biol 10:R813–R815
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B57:289–300
Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN (2003) A gene expression map of the Arabidopsis root. Science 302:1956–1960
Brady SM, Orlando DA, Lee JY, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U, Benfey PN (2007) A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318:801–806
Buitink J, Leger JJ, Guisle I, Vu BL, Wuillème S, Lamirault G, Le Bars A, Le Meur N, Becker A, Küster H, Leprince O (2006) Transcriptome profiling uncovers metabolic and regulatory processes occurring during the transition from desiccation-sensitive to desiccation-tolerant stages in Medicago truncatula seeds. Plant J 47:735–750
Chao DY, Luo YH, Shi M, Luo D, Lin HX (2005) Salt-responsive genes in rice revealed by cDNA microarray analysis. Cell Res 15:796–810
Chen W, Provart NJ, Glazebrook J, Katagiri F, Chang H-S, Eulgem T, Mauch F, Luan S, Zou G, Whitham SA, Budworth PR, Tao Y, Xie Z, Chen X, Lam S, Kreps JA, Harper JF, Si-Ammour A, Mauch-Mani B, Heinlein M, Kobayashi K, Hohn T, Dangl JL, Wang X, Zhu T (2002) Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 14:559–574
Chen WQJ, Zhu T (2004) Networks of transcription factors with roles in environmental stress response. Trends Plant Sci 9:591–596
Chen YH, Yang XY, He K, Liu MH, Li JG, Gao ZF, Lin ZQ, Zhang YF, Wang XX, Qiu XM, Shen YP, Zhang L, Deng XH, Luo JC, Deng XW, Chen ZL, Gu HY, Qu LJ (2006) The myb transcription factor superfamily of Arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol Biol 60:107–124
Cuming AC, Cho SH, Kamisugi Y, Graham H, Quatrano RS (2007) Microarray analysis of transcriptional responses to abscisic acid and osmotic, salt, and drought stress in the moss, Physcomitrella patens. New Phytol 176:275–287
Czechowski T, Bari RP, Stitt M, Scheible W-R, Udvardi M (2005) Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J 38:366–379
Devoto A, Turner JG (2005) Jasmonate-regulated Arabidopsis stress signalling network. Physiol Plant 123:161–172
Dinneny JR, Long TA, Wang JY, Jung JW, Mace D, Pointer S, Barron C, Brady SM, Schiefelbein J, Benfey PN (2008) Cell identity mediates the response of Arabidopsis roots to abiotic stress. Science 320:942–945
Frugier F, Poirier S, Satiat-Jeunemaître B, Kondorosi A, Crespi M (2000) A kruppel-like zinc finger protein is involved in nitrogen-fixing root nodule organogenesis. Genes Dev 14:475–482
He XJ, Mu RL, Cao WH, Zhang ZG, Zhang JS, Chen SY (2005) AtNAC2, a transcription factor downstream of ethylene and auxin signaling pathways, is involved in salt stress response and lateral root development. Plant J 44:903–916
Holmes P, Goffard N, Weiller GF, Rolfe BG, Imin N (2008) Transcriptional profiling of Medicago truncatula meristematic root cells. BMC Plant Biol 8:21
Ito M, Araki S, Matsunaga S, Itoh T, Nishihama R, Machida Y, Doonan JH, Watanabe A (2001) G2/M-phase-specific transcription during the plant cell cycle is mediated by c-Myb-like transcription factors. Plant Cell 13:1891–1905
Jiang Y, Deyholos MK (2006) Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol 6:1471–2229
Jin H, Martin C (1999) Multifunctionality and diversity within the plant MYB-gene family. Plant Mol Biol 41:577–585
Kakar K, Wandrey M, Czechowski T, Gaertner T, Scheible WR, Stitt M, Torres-Jerez I, Xiao Y, Redman JC, Wu HC, Cheung F, Town CD, Udvardi MK (2008) A community resource for high-throughput quantitative RT-PCR analysis of transcription factor gene expression in Medicago truncatula. Plant Meth 4:18
Karaba A, Dixit S, Greco R, Aharoni A, Trijatmiko KR, Marsch-Martinez N, Krishnan A, Nataraja KN, Udayakumar M, Pereira A (2007) Improvement of water use efficiency in rice by expression of HARDY, an Arabidopsis drought and salt tolerance gene. Proc Natl Acad Sci USA 104:15270–15275
Kim JC, Lee SH, Cheong YH, Yoo C-M, Lee SI, Chun HJ, Yun D-J, Hong JC, Lee SY, Lim CO, Cho MJ (2001) A novel cold-inducible zinc finger protein from soybean, SCOF-1, enhances cold tolerance in transgenic plants. Plant J 25:247–259
Kizis D, Lumbreras V, Pagès M (2001) Role of AP2/EREBP transcription factors in gene regulation during abiotic stress. FEBS Lett 498:187–189
Kreps JA, Wu YJ, Chan HS, Zhu T, Wang X, Harper JF (2002) Transcription changes for Arabidopsis in response to salt, osmotic and cold stress. Plant Physiol 130:2129–2141
Kumari M, Taylor GJ, Deyholos MK (2008) Transcriptomic responses to aluminum stress in roots of Arabidopsis thaliana. Mol Genet Genomics 279:339–357
Lee BJ, Park CJ, Kim SK, Kim KJ, Paek KH (2006) In vivo binding of hot pepper bZIP transcription factor CabZIP1 to the G-box region of pathogenesis-related protein 1 promoter. Biochem Biophys Res Commun 344:55–62
Li F, Wu X, Tsang E, Cutler AJ (2005) Transcriptional profiling of imbibed Brassica napus seed. Genomics 86:718–730
Ludwig AA, Saitoh H, Felix G, Freymark G, Miersch O, Wasternack C, Boller T, Jones JD, Romeis T (2005) Ethylene-mediated cross-talk between calcium-dependent protein kinase and MAPK signaling controls stress responses in plants. Proc Natl Acad Sci USA 102:10736–10741
Ma S, Gong Q, Bohnert HJ (2006) Dissecting salt stress pathways. J Exp Bot 57:1097–1107
Ma S, Bohnert HJ (2007) Integration of Arabidopsis thaliana stress-related transcript profiles, promoter structures, and cell-specific expression. Genome Biol 8:R49
Marisa L, Ichanté JL, Reymond N, Aggerbeck L, Delacroix H, Mucchielli-Giorgi MH (2007) MAnGO: an interactive R-based tool for two-colour microarray analysis. Bioinformatics 23:2339–2341
Merchan F, Breda C, Hormaeche JP, Sousa C, Kondorosi A, Aguilar OM, Megias M, Crespi M (2003) A kruppel-like transcription factor gene involved in salt responses in Medicago spp. Plant Soil 257:1–9
Merchan F, de Lorenzo L, Gonzalez Rizzo S, Niebel A, Manyani H, Frugier F, Sousa C, Crespi M (2007) Identification of regulatory pathways involved in the reacquisition of root growth after salt stress in Medicago truncatula. Plant J 51:1–17
Mittler R (2006) Abiotic stress, the field environment and stress combination. Trends Plant Sci 11:15–19
Nakashima K, Tran L-SP, Van Nguyen D, Fujita M, Maruyama K, Todaka D, Ito Y, Hayashi N, Shinozaki K, Yamaguchi-Shinozaki K (2007) Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-response gene expression in rice. Plant J 51:617–630
Ni Y, Wang X, Li D, Xu W, Li X (2008) Novel cotton homeobox gene and its expression profiling in root development and in response to stresses and phytohormones. Acta Biochim Biophys Sin 40:78–84
Ouyang B, Yang T, Li H, Zhang L, Zhang Y, Zhang J, Fei Z, Ye Z (2007) Identification of early salt stress response genes in tomato root by suppression substractive hybridization and microarray analysis. J Exp Bot 58:507–520
Osuna D, Usabel B, Morcuende R, Gibon Y, Bläsing OE, Höhne M, Günter M, Kamlage B, Trethewey R, Scheible WR, Stitt M (2007) Temporal responses of transcripts, enzyme activities and metabolites after adding sucrose to carbon-deprived Arabidopsis seedlings. Plant J 49:463–491
Potters G, Pasternak TP, Guisez Y, Palme KJ, Jansen MAK (2007) Stress-induced morphogenic responses: growing out of trouble? Trends Plant Sci 12:98–105
Ramanjulu J, Bartels D (2002) Drought- and desiccation-induced modulation of gene expression in plants. Plant Cell Env 25:141–151
Riechmann JL, Heard J, Martin G, Reuber L, Jiang C-Z, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, Creelman R, Pilgrim M, Broun P, Zhang JZ, Ghandehari D, Sherman BK, Yu G (2000) Arabidopsis transcription factors: genome wide comparative analysis among eukaryotes. Science 290:2105–2110
Sanchez DH, Lippold F, Redestig H, Hannah MA, Erban A, Krämer U, Kopka J, Udvardi MK (2008) Integrative functional genomics of salt acclimatization in the model legume Lotus japonicus. Plant J 53:973–987
Scheible W-R, Morcuende R, Czechowski T, Fritz C, Osuna D, Palacios-Rojas N, Schindelasch D, Thimm O, Udvardi MK, Stitt M (2004) Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol 136:2483–2499
Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T, Satou M, Akiyama K, Taji T, Yamaguchi-Shinozaki K, Carninci P, Kawai J, Hayashizaki Y, Shinozaki K (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31:279–292
Sharp RE, Poroyko V, Hejlek LG, Spollen WG, Springer GK, Bohnert HJ, Nguyen HT (2004) Root growth maintenance during water deficits: physiology to functional genomics. J Exp Bot 55:2343–2351
Shinozaki K, Yamaguchi-Shinozaki K (2000) Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signaling pathways. Curr Opin Plant Biol 3:217–223
Shukla RK, Raha S, Tripathi V, Chattopadhyay D (2006) Expression of CAP2, an APETALA2-family transcription factor from chickpea, enhances growth and tolerance to dehydration and salt stress in transgenic tobacco. Plant Physiol 142:113–123
Stracke R, Werber M, Weisshaar B (2001) The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol 4:447–456
Sun F, Zhang W, Hu H, Li B, Wang Y, Zhao Y, Li K, Liu M, Li X (2008) Salt modulates gravity signaling pathway to regulate growth direction of primary roots in Arabidopsis. Plant Physiol 146:178–188
Swindell WR (2006) The association among gene expression responses to nine abiotic stress treatments in Arabidopsis thaliana. Genetics 174:1811–1824
Tellström V, Usadel B, Thimm O, Stitt M, Küster H, Niehaus K (2007) The lipopolysaccharide of Sinorhizobium meliloti suppresses defense-associated gene expression in cell cultures of the host plant Medicago truncatula. Plant Physiol 143:825–837
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) Mapman: a user–driven tool to display genomics data sets diagrams of metabolic pathways and other biological processes. Plant J 37:914–939
Tran LSP, Nakashima K, Sakuma Y, Simpson SD, Fujita Y, Maruyama K, Fujita M, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2004) Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 16:2481–2498
Udvardi MK, Kakar K, Wandrey M, Montanari O, Murray J, Andriankaja A, Zhang JY, Benedito V, Hofer JM, Chueng F, Town CD (2007) Legume transcription factors: global regulators of plant development and response to the environment. Plant Physiol 144:538–549
Verdier J, Kakar K, Gallardo K, Le Signor C, Aubert G, Schlereth A, Town CD, Udvardi MK, Thompson RD (2008) Gene expression profiling of M. truncatula transcription factors identifies putative regulators of grain legume seed filling. Plant Mol Biol 67:567–580
Walia H, Wilson C, Condamine P, Liu X, Ismail AM, Zeng L, Wanamaker SI, Mandal J, Xu J, Cui X, Close TJ (2005) Comparative transcriptional profiling of two contrasting rice genotypes under salinity stress during the vegetative growth stage. Plant Physiol 139:822–835
Wang H, Miyazaki S, Kawai K, Deyholos M, Galbraith DW, Bohnert HJ (2003) Temporal progression of gene expression responses to salt shock in maize roots. Plant Mol Biol 52:873–891
Winicov I (2000) Alfin1 transcription factor overexpression enhances plant root growth under normal and saline conditions and improves salt tolerance in alfalfa. Planta 210:416–422
Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Tomo Y, Hayami N, Terada T, Shirouzu M, Osanai T, Tanaka A, Seki M, Shinozaki K, Yokoyama S (2004) Solution structure of B3 DNA binding domain of the Arabidopsis cold-responsive transcription factor RAV1. Plant Cell 16:3448–3459
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15
Zhang JY, Broeckling CD, Blancaflor EB, Sledge MK, Sumner LW, Wang ZY (2005) Overexpression of WXP1, a putative Medicago truncatula AP2 domain-containing transcription factor gene, increases cuticular wax accumulation and enhances drought tolerance in transgenic alfalfa (Medicago sativa). Plant J 42:689–707
Zhu JK (2002) Salt and drought stress signal transduction in plants. Ann Rev Plant Biol 53:247–273
Acknowledgments
We thank M. Stitt (MPI, Golm, Germany) for his useful suggestions and support all along this work. We also thank J. M. Prosperi (INRA Montpellier, France) for M. truncatula A17 seeds, L. Troussard (ISV, France) for sequencing, J. L. Ichanté, S. Imbeaud and H. Delacroix (Plateforme GODMAP, CNRS Gif-sur-Yvette, France) for advices in microarray experimental design and data analyses, B. Usadel (MPIMP Golm, Germany) for providing Mapman BINs Mapping file for M. truncatula, H. Kuester (Bielefeld University, Germany) for providing 16KOli + arrays. This work was supported by the GRAIN LEGUMES Integrated Project (GLIP-EEC-FP6) including also a GLIP Fellowship for A. B. and S. B. O.Z. was the recipient of a grant from the Tunisian Government, Tunis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
V. Gruber and S. Blanchet have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2008_392_MOESM1_ESM.ppt
Fig. S1 : Mapman “general metabolism” pathway for the 824 salt-regulated genes in M. truncatula root apexes.Changes in transcript levels of the 824 genes regulated by a 1 hour salt treatment in Medicago root apexes were identified based on micro-array analysis (Amean>7, p<0.001, ¦FC ¦>1.8, see Methods). Changes are displayed on a colour scale (log2 ratios are presented on the side bar). Transcripts absent are shown in grey whereas transcripts that change by less than a given threshold are shown in the colour code bar (increased, red or repressed, blue, respectively).Fig. S2 : Mapman “cellular responses” pathway for the 824 salt-regulated genes in M. truncatula root apexes.Idem as Supplementary Fig. S1.Fig. S3 : Mapman “regulation overview” pathway for the 824 genes salt-regulated in M. truncatula root apexes.Idem as Supplementary Fig. S1 (PPT 239 kb)
438_2008_392_MOESM2_ESM.xls
Supplementary Table S1: List of the 46 transcription factor (TF) transcripts salt-regulated in M. truncatula whole roots. A cut-off threshold of 1.8-fold is considered. E is the PCR efficiency. CTs and CTc represent the threshold cycle for 1h salt-treated roots and 1h control roots, respectively. TF gene expression was normalized to that of the constitutive control Mt UBI by subtracting the CT value of Mt UBI from the CT value of the TF gene of interest (ΔCT). ΔCTs and ΔCTc referred to 1h salt-treated roots and 1h control roots, respectively. ΔΔCT represents ΔCTs minus ΔCTc. S/C expression ratios were obtained from the equation (1+E)-ΔΔCT.Supplementary Table S2: List of the 26 real time RT-PCR primer pairs corresponding to various transcription factor (TF) gene families.Efficient and specific RT-PCR amplification for the expected TF gene is annotated “yes”.Supplementary Table S3: List of the 824 salt-regulated genes in M. truncatula root apexes. Genes identified based on microarray analyses (Amean>7, p<0.001, ¦FC¦>1.8; see Methods) are classified in functional categories based on BIN assignment.Supplementary Table S4: Statistical significance of differential gene expression in functional Mapman BIN categories. The statistical analysis is provided in MapMan software and corresponds to the p value of the Benjamini and Hochberg corrected Wilcoxon rank sum test. “Elements” correspond to element numbers per BIN category.Supplementary Table S5: List of the 84 salt-regulated transcription factors in M. truncatula root apexes.The 84 salt-regulated TF genes in root apexes were classified through their putative functional domains using BLASTX comparisons. Supplementary Table S6: List of primer pairs used in real time RT-PCR analysis.F and R correspond to forward and reverse primers, respectively. (XLS 276 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gruber, V., Blanchet, S., Diet, A. et al. Identification of transcription factors involved in root apex responses to salt stress in Medicago truncatula . Mol Genet Genomics 281, 55–66 (2009). https://doi.org/10.1007/s00438-008-0392-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-008-0392-8