
Abstract
Legume seeds represent a major source of proteins for human and livestock diets. The model legume Medicago truncatula is characterized by a process of seed development very similar to that of other legumes, involving the interplay of sets of transcription factors (TFs). Here, we report the first expression profiling of over 700 M. truncatula genes encoding putative TFs throughout seven stages of seed development, obtained using real-time quantitative RT-PCR. A total of 169 TFs were selected which were expressed at late embryogenesis, seed filling or desiccation. The site of expression within the seed was examined for 41 highly expressed transcription factors out of the 169. To identify possible target genes for these TFs, the data were combined with a microarray-derived transcriptome dataset. This study identified 17 TFs preferentially expressed in individual seed tissues and 135 corresponding co-expressed genes, including possible targets. Certain of the TFs co-expressed with storage protein mRNAs correspond to those already known to regulate seed storage protein synthesis in Arabidopsis, whereas the timing of expression of others may be more specifically related to the delayed expression of the legumin-class storage proteins observed in legumes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
More than 70% of the average human diet is derived from seeds (essentially cereals and legumes). Seeds of cereal species, such as maize (Zea mays) and wheat (Triticum aestivum) are a major source of starch and contain less than 16% protein content in their main storage tissue, the endosperm. Legume species such as pea (Pisum sativum), soybean (Glycine max) or fava bean (Vicia faba) represent a richer protein source than the major cereals, with seed protein content ranging from 20% to as much as 40%, depending on species, genotype and environment. In most legumes, the storage compounds accumulate essentially in the embryo which develops surrounded by endosperm and enclosed in the maternal integument layers. Legume seed development commences with embryogenesis during which the embryo undergoes a programme of cell divisions, acquires polarity along the shoot-root axis, and the shoot and root apical meristems are established. In the subsequent seed maturation or seed filling period, embryo cell divisions cease and cell expansion occurs. This stage corresponds to the deposition of storage molecules, lipids, carbohydrates and proteins. The economically and nutritionally important legume storage proteins consist principally of the legumin (11S) and vicilin (7S) globulin classes.
The extent of transcriptional regulation in the developing M. truncatula seed has been evaluated by a comparison between transcriptome and proteome profiles during seed development. This suggested that the abundances of approximately 50% of the proteins identified, including the seed storage proteins, were mainly regulated at the transcriptional level (Gallardo et al. 2007). In plants, gene regulation at the level of transcription is fundamental to many aspects of development. Transcription factors (TFs) account for nearly 6% of the gene complement of the Arabidopsis genome sequence, with many of them being plant-specific (Riechman and Ratcliffe 2000).
Profiling of TF gene expression during seed development has been carried out for Arabidopsis and cereals (Ruuska et al. 2002; De Folter et al. 2004; Druka et al. 2006), and several TFs have been independently identified by mutant analyses as playing key roles in different steps of seed development. Examples include regulators of the maternal contribution to seed size (TTG2, Garcia et al. 2005), homeotic mutants of embryo development (MONOPTEROS, Hardtke and Berleth 1998), factors governing the deposition of seed storage proteins (OPAQUE2, Unger et al. 2002; LEAFY COTYLEDON2, Santos Mendosa et al. 2005), and lipids (WRINKLED1, Cernac and Benning 2004), and those directing other aspects of seed maturation (e.g., desiccation tolerance, ABSCISIC ACID-INSENSITIVE3, Parcy et al. 1995).
The aim of the present study was to provide an overview of transcription factor genes expressed during legume seed development. We have chosen to use the model legume Medicago truncatula for which extensive sequence information is available from both EST and genome sequencing programmes (Cook 1999; Ané et al. 2008). The strategy we employed involved a large-scale profiling of transcription factor gene expression during seed development, using quantitative real-time RT-PCR. Temporal changes in gene expression of more than 700 M. truncatula TF transcripts were identified and catalogued during seed development. A combination of our data with a microarray transcriptome dataset (Gallardo et al. 2007) has allowed us to assign TFs to key phases and processes during seed development. About 80 TF genes were preferentially expressed during storage compound accumulation, most of which could be attributed to one of the three principal seed tissues. We have further identified amongst these TF candidates for regulating the sequential expression of vicilin and legumin storage protein classes observed in legumes.
Materials and methods
Plant growth
Two independent series of M. truncatula plants (cultivar Jemalong, line A17) representing two biological replicates were grown and tagged according to Gallardo et al. (2007). ∼5000 seeds [1.5 g of seeds for each of the seven developmental stages ranging from 10 to 36 Days After Pollination (DAP)] were collected, on ice to prevent dehydration, per biological replicate. For seed tissue analysis, seed coat, endosperm and embryo were dissected from 14 DAP seeds under a magnifying glass (magnification, ×3.5) on Petri dishes placed on ice and immediately frozen in liquid nitrogen after tissue dissection. Flowers, leaves, stems, and roots were collected in growth chamber at flowering.
Total RNA isolation and cDNA synthesis
Total RNA was isolated from developing seeds according to Chang et al. (1993). 200 μg of total RNA was treated with RNAse free DNAse RQ ITM (Promega, Madison, USA) for 45 min according to the manufacturer’s instructions. The absence of genomic DNA contamination was confirmed by real-time RT-PCR on non-reverse-transcribed RNA. RNA was purified and concentrated using a centrifugal filter (Microcon YM-30, Millipore, Bedford, USA) before use in first strand cDNA synthesis. RT reactions were performed using a mix of polyT(23) and random primers with SuperScriptTM III reverse transcriptase (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. The efficiency of cDNA synthesis was assessed by real-time PCR amplification of a control gene encoding Ubiquitin. Only cDNA preparations, that yielded similar threshold cycle (Ct) values (i.e. 24±1) and that showed a 3′/5′ ratio in the range of 1–1.5 (using formula 2(Ct 5′ – Ct 3′)) for the Ubiquitin gene, were used for comparing TF transcript levels. cDNA from the three seed tissues was synthesised using iScript one-step reverse-transcriptase kit (Bio-Rad, CA, USA) according to the manufacturer’s instructions.
M. truncatula primer collection of transcription factors
A M. truncatula primer collection representing all annotated M. truncatula transcription factors was generated using available genomic databases (Udvardi et al. 2007). This resource was constituted by 712 primer pairs corresponding to 712 putative transcription factors in M. truncatula, and including representatives of all the major TF families. Oligonucleotide primers were designed according to common real-time RT-PCR criteria to ensure maximum specificity and efficiency during PCR amplification of cDNA (Czechowski et al. 2004). The set of 712 putative TFs with corresponding BAC and TC accession numbers as well as sequences of the primer pairs used for amplification is listed in Table S1 of the Supplementary Material.
Real-time PCR conditions and analysis
Polymerase Chain Reactions were performed in an optical 384-well plate with an ABI PRISM® 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA) using SYBR® Green to monitor dsDNA synthesis. Reactions, containing 2.5 μl 2X SYBR® Green master mix reagent (Applied Biosystems, Foster City, CA, USA), 0.5 μl cDNA and 200 nM of each gene specific primer in a final volume of 5 μl, were incubated at: 50°C for 2 min; 95°C for 10 min; and 45 cycles of 95°C for 15 s and 60°C for 1min. Immediately after thermal cycling, a dissociation curve was performed by heating PCR products from 60°C to 95°C, to reveal the presence of non-specific amplicons including primer dimerization.
Data were analysed using the SDS 2.0 software (Applied Biosystems). All amplification plots were analysed with a threshold of 0.3 to obtain Ct values. PCR efficiency (E) was estimated using LinReg software with data obtained from the exponential phase of each individual amplification plot and the equation (1 + E) = 10slope (Ramakers et al. 2003). To profile TFs in the three seed tissues, we used iCyclerTM IQ thermal cycler with 5 μl of 2X IQ SYBR Green Supermix detection (Bio-Rad, Hercules, USA), 100 nM of each gene specific primer, 2 μl of cDNA in a final volume of 10 μl. Data were analysed using iCyclerTM IQ Optical system software v.3.00 (Bio-Rad, Hercules, USA).
Selection of reference genes constitutively expressed during Medicago truncatula seed development realized for Real-Time RT-PCR normalization
Out of the 10 housekeeping genes [UBIQUITIN, ACTIN, GAPDH (Glyceraldéhyde 3 Phosphate Dehydrogenase), PTB (Polypyrimidine Tract-Binding), PDF2 (Protein Phosphatase 2A Subunit A3), PPRre (pentatricopeptide repeat-containing), EF1ά (Elongation factor 1 alpha), TUBULIN, bHLH (Basic helix-loop-helix transcription factor) and HELICASE] present on each plate, a selection was performed based on two methods.
Firstly, by comparison of coefficient of variation (CV) values during seed development, and secondly by analysis of gene expression stability (M) using geNORM software v3.4 (Vandesompele et al. 2002). This software computes the expression ratio between all pairwise control genes for all stages. For an ideal control gene this ratio is the same for all stages. The four genes with the lowest M values, GAPDH (MtC00030_GC), ACTIN (TC107326), PDF2 (TC107161; Slabas et al. 1994) and PTB (TC111751; Salanoubat et al. 2000) were selected as controls (Supplementary Table S2).
For qualitative analysis in seed and non seed tissues, a single housekeeping gene MSC27, a translationally controlled tumor protein homolog (TC85211) (Bouton et al. 2005), was used to normalize gene expression, the objective being to estimate distribution of expression of the gene.
Statistical analyses and clustering of TFs
To analyse gene profiles between the two biological replicates, a one-way ANOVA (P < 0.05), using Statistica software (Statsoft, Tulsa, OK), was performed on each TF. In order to calculate relative TF expression levels, the efficiency values of each amplification reaction were taken into account using LinReg software (Ramakers et al. 2003). Amplification reactions of efficiencies lower than 1.6 were considered as missing data (approximately 11.7% of the total reactions (2332/19852). Differences in transcript abundance during seed developmental stages were also evaluated by a one-way ANOVA test (P < 0.05) and a Student–Newman–Keuls test for each TFs using SAS software package (SAS Institute 1999). To compare expression profiles, expression values were adjusted by a normal distribution. K-means clustering were performed using Genesis software (Sturn et al. 2002) and Cluster v3.0 (Eisen et al. 1998) with the average linkage method and visualized using TreeView v1.0.5 software.
Combination of the real-time RT-PCR profiling data with a Mt16kOLI1 microarray dataset
To combine data from the real-time RT-PCR transcript profiling and from microarray experiments previously performed (Gallardo et al. 2007), all normalized real-time RT-PCR data were expressed in log2 ratios of relative expression of the reference sample (12 DAP). Genes present on the microarrays and real-time RT-PCR data were hierarchically clustered using Cluster v3.0 software (Eisen et al. 1998) according to the average linkage method, and visualized using Java TreeView v.1.0.5. To identify genes co-expressed with different TFs, we carried out a correlation analysis between the gene expression profiles from microarray dataset and different TF profiles.
Results
Strategy used to identify TFs expressed during seed development
The profiles of expression during seed development of a comprehensive collection of Medicago truncatula TF gene sequences were determined at seven developmental stages chosen based on earlier studies of seed development (Gallardo et al. 2003, 2007). The first stage, 10 Days After Pollination (DAP), corresponds to late embryogenesis. The next stage, 12 DAP, is the transition between embryogenesis and the onset of seed filling. The following stages correspond to the onset of vicilin and legumin expression (14 DAP and 16 DAP, respectively) and the peaks of vicilin and legumin expression (20 DAP and 24 DAP). The last stage, 36 DAP, marks the end of seed filling and the entry into quiescence associated with desiccation (Fig. 1a).

(a) Time course of M. truncatula seed development, with the timing of accumulation of the main Seed Storage Protein (SSP) classes. (b) Averaged Profiles of mRNA abundance during seed development for TF clusters I–VI. The log2 of the normalized expression values (i.e. adjusted by a normal distribution) was calculated for classification into clusters. For each cluster, the median of these values at each stage is represented in graphic form (±SD), showing that each cluster has a characteristic peak of expression at a particular developmental stage. (c) Heat-maps for the two independent biological replicates (Rep1 and Rep2) of TF expression profiles from each cluster during seed development (10, 12, 14, 16, 20, 24 and 36 days after pollination, respectively). Down-regulation is indicated in green, up-regulation in red, normalized expression values close to the mean are in black and no detectable expression is in grey. The number of TFs present in each cluster is indicated. The identity of the TFs present in different clusters and their expression profiles are given in Supplementary Table S4
The mRNA abundances of putative TFs were determined at each stage using real-time quantitative reverse transcription PCR (real-time RT-PCR). A primer pair collection was designed to amplify each of the 712 TFs annotated in the genomic IMGAG database (the International Medicago Genome Annotation Group) and assigned to different families (Fig. 2). The representation of different TF families in the collection is similar to that observed for the Arabidopsis genome (Riechman 2002; Udvardi et al. 2007).

Overview of the M. truncatula TF families present in the primer set collection. (a) All 712 genes were classified into TF families. The Number of TF members of each family is indicated. (b) The subgroups belonging to the Zinc-Finger superfamily are represented with the number of genes in each subfamily
To measure expression levels accurately, we have normalized data to the geometric mean of the relative quantity of transcript for several housekeeping genes which display the least varying expression profiles throughout the stages studied (Supplementary Table S2).
The threshold cycle values of the housekeeping genes, loaded on each plate, were used to calculate the coefficients of variation (CVs) between plates. CVs calculated as the ratio between standard deviation and mean threshold cycle value, were always below 0.01 (Data not shown). More than 95% of TF transcripts (681/712) showed reproducible profiles throughout seed development between the two biological replicates (Pearson’s correlation coefficient, r = 0.93). This linear correlation was observed for expression values above 10−4 (Supplementary Figure S1). The 72% (512/712) of genes, whose expression was detected above this threshold for at least one stage during seed development, and which were consistent between the two biological replicates, were selected for further analysis (Supplementary Table S3).
Analysis of TF expression profiles during seed development
To group the TFs expressed at specific stages of development, and hence to relate them to possible functions, we performed a hierarchical clustering of the profiles, along with real-time RT-PCR expression data for genes encoding the major M. truncatula seed storage proteins of the globulin-type. Using one-way ANOVA and Student-Newman-Keuls tests, we divided TFs into two different groups: those differentially expressed and those constitutively expressed during seed development. Approximately 67% of the TFs (343/512) are expressed throughout seed development. A k-means clustering was performed on the remaining 33% of TFs (169/512) that are differentially expressed during seed development. This cluster analysis revealed six different clusters of genes which correspond to expression profiles at different developmental stages (Fig. 1b and Supplementary Table S4). We observe major differences in TF family distributions between different clusters and therefore in the proportions of different TF families expressed at different stages of seed development (Fig. 3).

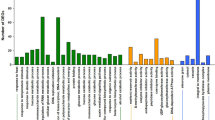
Frequency analyses of different families of transcription factor classes expressed at different stages of seed development. The 169 TFs present in different clusters (Cluster I–VI) were classified into TF families
The first cluster (Cluster I), corresponding to late embryogenesis, contains 41 genes which are preferentially expressed at 10 DAP. 14 of these genes are Zn-Finger family members, including sequences related to five WRKY (Eulgem et al. 2000), and notably three C2H2 type factors (Takatsuji 1999). In addition, five AP2/ERFs, four MADS and four MYB factors, one E2F-type cell proliferation factor (Sozzani et al. 2006) and a putative orthologue of the homeodomain MERISTEM LAYER 1 protein (Lu et al. 1996) were expressed at this stage. The following clusters (Clusters II–V) contain 80 TFs, with peaks of expression during seed filling (between 12 and 36 DAP). Cluster II (12–14 DAP) contains genes which are upregulated just before storage protein synthesis, marking the switch between embryogenesis and seed filling. This cluster includes candidate orthologues of genes already implicated in this transition phase such as LEAFY COTYLEDON1-LIKE (Kwong et al. 2003) and CUP-SHAPED COTYLEDON LIKE (CUC-LIKE) (Aïda et al. 1999). After 14 DAP, embryo cell divisions cease, and cellular expansion phase begins, during which the principal seed reserves accumulate. The three following clusters, III, IV, and V, correspond to genes concomitantly expressed with vicilin (TC100302) (16–20 DAP), legumin K (TC100253) (20–24 DAP) and legumin A (TC100252) (24–36 DAP). In M. truncatula, a clear phasing in the accumulation of transcripts for different globulin classes is observed (Gallardo et al. 2003, 2007). Vicilin mRNA synthesis starts around 14 DAP, with maximum accumulation around 20 DAP, whereas legumin K transcript accumulation begins and peaks around 4 days later than vicilin (Fig. 4). Corresponding differences in the timing of transcript accumulation of the different legumin classes, legumin K and legumin A, are also observed. TFs from clusters III to V include possible orthologues of regulators of seed storage protein synthesis in other species including B3 domain, bZIP, DOF and MYB family members (Fig. 4 and Supplementary Table S4).

Expression profiles of the three major M. truncatula seed storage protein genes and of the corresponding TF cluster. The continuous line represents storage protein mRNA profiles from 10 to 36 DAP, determined by real-time RT-PCR [(a) vicilin (TC100302), (b) legumin K (TC100253) and (c) legumin A (TC100252)]. The bars represent the median (±SD) of normalized expression values (i.e. adjusted by a normal distribution) of TFs from the corresponding clusters (cluster III, IV and V, respectively related to vicilin, legumin K and legumin A synthesis) (Standard deviation values for the storage protein genes are close to zero). TF expression profiles related to corresponding globulin expression are available in Supplementary Table S4
Finally, cluster VI contains genes expressed during the dessication phase (36 DAP). A total of 48 genes are expressed at this stage constituting the largest group of differentially-expressed TFs. Most of the TFs in this group belong to the Zn-Finger and MYB families. The bulk of the mRNA accumulated at this stage is stored and used for protein synthesis during subsequent germination, and certain of these TFs may represent potential regulators of germination performance, which is largely programmed during seed maturation (Rajjou et al. 2003).
Distribution of the TFs in the three component seed tissues during seed maturation
To get a better understanding of the roles played by individual TFs, we have analyzed their expression profiles by real-time RT-PCR in seed coat, endosperm and embryo isolated from seed collected at 14 DAP. This stage represents the onset of seed filling and is also characterized by remobilization of endogenous reserves from endosperm to the embryo. We have also analyzed expression in roots, flowers and leaves, to assess overall cell-type specificities. The data were normalized to the gene MSC27 (TC85211) (Bouton et al. 2005), a translationally controlled tumor protein homolog, previously validated in a M. truncatula comparative study using three other housekeeping genes. Among the 80 TFs preferentially expressed during seed filling, the 41 highest expressed candidates were selected for further analysis of their seed tissue specificity. We have taken as a working definition of tissue-type specificity a greater than 20-fold difference in transcript abundance between the expressing and the non-expressing cell type examined (Czechowski et al. 2004).
Three of the 41 TFs profiled by real-time RT-PCR gave no detectable transcript in any seed tissue examined, probably due to their lack of expression at 14 DAP. Six TFs were concomitantly expressed in all three seed tissues and are expressed throughout seed filling (Table 1). The remaining factors were classified according to their preferential sites of expression in seeds, (Table 1), although some are also highly expressed in other tissues.
TF genes preferentially expressed in seed coat or seed coat plus endosperm
Six TFs were expressed in the seed coat (group 1 in Table 1). They belong to the MYB, B3 domain, AP2/ERF, bZIP and MADS box families. Two of them displayed a seed coat-specific expression: a MYB R2R3 factor (1212.m00019) and an AP2/ERF member (1162.m00011). The remaining four TFs are expressed in the seed coat but also in other plant organs. They include a MADS Box factor (1274.m00002) closely related to AGAMOUS LIKE2 (AGL2, Flanagan and Ma 1994) (Tblastx, score = 1e−62), which displays a similar expression level in flower and seed coat, and a second MYB factor (727.m00020) with a R2R3 domain. Among this group were also an Auxin Response Factor (1372.m00012) closely related to auxin-responsive protein in Arabidopsis and a bZIP factor (732.m00026) which is a putative orthologue of ATB2 (Tblastx, score = 5e-43) (Weber et al. 1995). This group is made up of genes expressed at stages which extend throughout seed filling. Our study also revealed six TFs expressed in both seed coat and endosperm of which three are seed-specific (group 2 in Table 1). This group includes essentially Zn-finger TFs (2 CCHC, 1 DOF and 1 RING type), none of which are obvious orthologues to Arabidopsis genes. The majority of these TFs are expressed early during seed filling when the seed coat and endosperm are active in supplying nutrients to the developing embryo.
TF genes preferentially expressed in embryo and/or endosperm
Six TF genes were expressed in endosperm tissue of the seed, of which three are endosperm-specific (group 3, Table 1). Whereas nine TFs were co-expressed in endosperm and embryo (group 4, Table 1), including sequences closely related to TFs of known function in Arabidopsis such as a CCAAT-box binding factor (1347.m00026) related to LEAFY COTYLEDON 1 LIKE (LEC1-LIKE or L1L) (Tblastx, score = 6e−53) and an AP2 factor (1376.m00008) related to ABSCISIC ACID INSENSITIVE-4 (ABI4) (Tblastx, score = 3e−33) (Finkelstein et al. 1998). Only two of the nine TFs displayed seed-specific expression (LEC1-LIKE and ABI4-LIKE). Most of the TFs in this group are preferentially expressed at 12–14 DAP (i.e. at the onset of filling).
Finally, five TFs are preferentially expressed in the embryo (group 5 in Table 1). Four of these are seed- and embryo- specific, and include bHLH, bZIP and two B3 domain types. Interestingly, all five embryo-expressed mRNAs of this group were expressed either in the cluster associated with vicilin (14–16 DAP) or with that associated with legumin K (20–24 DAP) genes. This group also contains a WRKY type TF sequence (920.m00011) also highly expressed in roots and flowers.
Identification of genes co- expressed with TFs in the seed tissues
To combine data from the real-time RT-PCR transcript profiling and from microarray experiments previously performed (Gallardo et al. 2007), we have first confirmed by 2D SDS-PAGE that the two sets of seed samples used displayed identical timing of protein accumulation (Data not shown). Further, all normalized real-time RT-PCR data were expressed as log2 ratios of relative expression of the 12 DAP reference sample. A comparison of TF expression profiles obtained by real-time RT-PCR and microarray was then performed. Of the 169 TFs identified by real-time RT-PCR as being differentially expressed throughout seed development, approximately 40 were also present in the microarray dataset. Although the microarray dataset is less exhaustive, broadly similar expression patterns were observed for both experiments, thus validating our real-time RT-PCR results based on a different technique and normalization procedure. To propose hypotheses about the roles of individual TFs, we combined the expression profiles of the transcription factors preferentially expressed in one seed tissue, obtained by real-time RT-PCR, with profiles of seed expression of 16,000 mRNA sequences obtained by microarray-based transcriptomics (Gallardo et al. 2007). We selected 17 genes from the RT-PCR experiment which exhibit expression in only one seed tissue, disregarding any expression in other plant tissues. Of these, six genes were expressed in seed coat, six in endosperm and five in embryo. Of the microarray-derived gene list, 2,496 showed seed tissue-specific expression with less than 20% of missing data throughout seed development, 1,694 being specifically expressed in the embryo, 431 in the endosperm and 371 in the seed coat. A hierarchical cluster analysis was performed between expression profiles of these genes and those of the TFs.
To identify genes co-expressed with different TFs, gene expression profiles were considered as co-expressed when showing a correlation higher than 0.9. This analysis revealed that whereas for certain TFs many sequences are co-expressed [e.g. a WRKY factor, (920.m00011), which is co-expressed with 50 genes], for others only a few co-expressed genes were identified [e.g. a B3 domain (1299.m00024) which co-clustered with 6 genes] (Fig. 5 and Supplementary Table S5). Four TFs [MYB (1212.m00019), bZIP (732.m00026), DOF (1147.m00025) and a homeodomain factor (951.m00006)] co-clustered with only one gene. Moreover, three TFs [Zn Finger HD (783.m00019), MYB (727.m00020) and B3 domain (1372.m00012)] did not correlate with any gene expression profiles from the non-exhaustive microarray dataset.

Functional classification of genes co-expressed with different transcription factors in the three seed tissues (embryo, endosperm and seed coat). All TFs expressed in one seed tissue at 14 DAP are represented along with the corresponding cluster of co-expressed genes (microarray dataset from Gallardo et al. 2007). The number of co-expressed genes is indicated along with the cluster, and the annotation into functional classes according to Mapman (Usadel et al. 2005). Co-expressed TFs having the same co-expressed genes are listed together. Seven TFs have not been represented in this figure: for three (783.m00019, 727.m00020 and 1372.m00012), there were no co-expressed gene profiles in this microarray dataset and four TFs, (1212.m00019, 732.m00026, 1147.m00025 and 951.m00006), are only co-expressed with one target. A list of co-expressed genes belonging to the “development, storage and LEA proteins” functional class is detailed for three TFs (1299.m00024, 1018.m00015 and 920.m00011). The complete list of co-expressed genes (with TC and name) for each TF is given in Supplementary Table S5
Figure 5 shows putative functions of the co-expressed genes based on the Mapman ontology (Usadel et al. 2005). First, three of the six TFs preferentially expressed in seed coat are co-expressed with genes involved in amino acid and secondary metabolism. Two of the six TFs expressed in endosperm (WRKY, 853.m00019 and MYB, 814.m00021) are co-expressed with the same set of genes, mainly related to sugar metabolism (e.g. a sucrose synthase, TC85400), signalling and transport, and one (1235.m00014) is co-expressed with genes of lipid metabolism and late embryogenesis abundant proteins.
Of the five TFs preferentially expressed in the embryo, two groups could be identified. One is composed of two TFs (bZIP, 1240.m00018 and bHLH, 762.m00015) associated with numerous biological processes (e.g. metabolism, DNA/RNA and protein regulation, cell organization, transport) but not with seed storage proteins. The other group contains three TFs co-expressed with seed storage protein genes: a WRKY factor (920.m00011) and two B3 domain factors, (1018.m00015 and 1299.m00024).
Discussion
Large scale profiling of transcription factors during seed development
This study represents the first high throughput real-time quantitative RT-PCR profiling of legume transcription factors throughout a developmental process and the most comprehensive profiling study of TFs in developing seeds. In total, of 712 M. truncatula putative TFs examined, 512 (72%) were expressed at least at one stage during seed development. This result supports earlier observations that a majority of TFs are expressed constitutively throughout the plant but at very low levels in most cell types (Walling et al. 1986) which may be overlooked in DNA microarray analyses (Chen et al. 2002; Czechowski et al. 2004). The high sensitivity and broad linear response range of this technology has allowed us to identify the low abundance TF transcripts including those that could be associated with developmental transitions (Putterill et al. 1995; Long et al. 1996; Mayer et al. 1998). However, among the 512 TFs expressed during seed development, 343 were not differentially expressed from 10 to 36 DAP. The remaining 169 were classified into six clusters covering major developmental stages. Based on profiling data, TFs were associated with late embryogenesis processes (41 TF genes); the transition between embryogenesis and seed filling (28); the synthesis of vicilin (15), legumin K (15) and legumin A (22), and with desiccation (48) (Figs. 1, 4).
We have carried out a more detailed analysis of 41 TFs which showed the highest relative expression, on the assumption that they are less likely to be substitutable by other genes, and that functions would be more readily attributable to them. First, by determining the seed tissue distribution of the 41 TF transcripts, 17 were found to be preferentially expressed in a particular seed tissue at one stage (14 DAP). Second, a comparison of this sub-group of 17 TFs with the expression profiles of 16,000 M. truncatula genes, (Mt16kOLI1 microarray dataset, Gallardo et al. 2007), allows us to propose hypotheses about biological roles for these factors.
A comparison between expression profiles of these M. truncatula TFs and their putative orthologues in Arabidopsis suggests conserved and distinct elements of transcriptional regulation during seed development.
Conservative transcriptional regulation between legume and Arabidopsis seed development
Out of 41 TFs, we found 19 putative orthologues in Arabidopsis according to amino acid sequence alignments. Using the ATH1 22K array data (www.genevestigator.ethz.ch), we observed that the majority of these (15/19) show similar expression patterns to the closely related M. truncatula sequences (Table 1 Supplementary data Table S6). Two of them are known to regulate seed size in Arabidopsis (Garcia et al. 2005) and are preferentially expressed in the seed coat (Table 1). One encodes an Auxin Response Factor-type (1372.m00012, Table 1) which could be involved in determining seed size (Schruff et al. 2006) and the second is a bZIP factor (732.m00026, Table 1), related to ATB2 (Rook et al. 1998), could influence hexose concentration, known to control seed size, by activation of cell wall invertase. Its expression during the early stages of seed filling supports a role for this factor in the control of assimilate supply to the embryo with invertases as possible targets.
Two further M. truncatula TF genes expressed during seed filling encode a CCAAT binding factor (1347.m00026, Table 1) closely related to LEAFY COTYLEDON 1(LEC1), which coordinates embryo morphogenesis and maturation phases in Arabidopsis (Lothan et al. 1998; Kagaya et al. 2005) and an ABI4-related sequence (1376.m00008, Table 1). ABI4 is a regulator of sugar and ABA–mediated gene repression, with effects on seed maturation in combination with LEAFY COTYLEDON genes (Brocard-Gifford et al. 2003). Another embryo-expressed TF encodes a B3 domain transcription factor, putatively orthologous to FUSCA3 (FUS3) (1299.m00024, Table 1) (Tblastx, score = 1e−67), which is a direct regulator of seed storage protein gene transcription in Arabidopsis (Parcy et al. 1997). It may be significant that this gene is co-expressed during seed filling with another B3 domain factor (1018.m00015, Table 1), as in Arabidopsis embryo two B3 domain factors, FUSCA3 and ABI3, act in synergy. A hierarchical clustering of expression profiles of these B3 domain genes with the Mt16kOLI1 microarray dataset identified four encode vicilin-type seed storage proteins and an oleosin among the six co-expressed genes (Fig. 5).
Legume specificity in transcriptional regulation during seed development and differences in the fine regulation of legume and Arabidopsis seed storage protein gene expression
All the TFs identified as preferentially expressed in the seed coat identified have putative orthologues (i.e. sequence homology plus similar expression profile) in Arabidopsis, suggesting similar mechanisms of regulation. In contrast, for 63% (26/41) of the other M. truncatula TFs, there was no unambiguous corresponding orthologue in Arabidopsis (Supplementary Table S6), despite finding putative orthologues in other legumes, raising the possibility of “legume-specific” TFs (Graham et al. 2004).
As an example, three out of the five embryo TF genes lack obvious orthologues among Arabidopsis sequences or other NCBI accessions. These TFs, belonging to three different families (bHLH, B3 domain and bZIP), are expressed during seed filling with an embryo and seed specificity. Whereas in Arabidopsis the two major storage globulins and the albumins are co-ordinately transcribed (data from the ATH1 22K array available at www.genevestigator.ethz.ch), implying a common transcriptional regulation, in legumes vicilins and legumins accumulate successively (Meinke et al. 1981; Walling et al. 1986; Gatehouse et al. 1986; Boulter et al. 1987). Although this phasing has been widely observed in legumes, the mechanism of regulation involved has not been investigated, but may involve different TFs.
Models for transcriptional regulation of vicilin and legumin genes
Previous studies on other dicotyledonous species, essentially on Arabidopsis, suggest a model for coordinate transcriptional regulation of seed storage protein synthesis (Fig. 6a). M. truncatula factors corresponding in tissue-specificity and timing of expression to the main players in this model have been indicated. Before seed filling storage protein promoters are thought to be repressed by a mechanism involving the bZIP factor ROM1 (Chern et al. 1996b) [see M. truncatula candidate gene 1010.m00004 in Fig. 6(b) A]. During seed filling, storage protein gene transcription is promoted by the combined action of the B3 domain proteins LEAFY COTYLEDON2 (LEC2) [Fig. 6(b) B], FUS3 [Fig. 6(b) C] and ABSCISIC ACID INSENSITIVE-3 (ABI3) [Fig. 6B, Reidt et al. 2000 and Kroj et al. 2003], the latter in a complex with a bZIP [Fig. 6(b) D, Lara et al. 2003]. The synthesis of these factors is in turn activated by LEC1 and L1L [Fig. 6(b) E, Kwong et al. 2003; Kagaya et al. 2005].

(a) Regulatory elements and interacting factors implied in regulating Seed Storage Protein gene promoters: A synthetic model for the transcriptional regulation of Seed Storage Protein (SSP) in dicotyledonous plants, based on data from Arabidopsis and Phaseolus vulgaris (Chern et al. 1996a, b; Reidt et al. 2000; Diaz et al. 2002; Kroj et al. 2003; Kwong et al. 2003; Lara et al. 2003; Bensmihen et al. 2005; Kagaya et al. 2005; Suzuki et al. 2007; Tsukagoshi et al. 2007). Boxes represent conserved sequence motifs: AACA motifs, RY boxes and G boxes. Putative transcription factors and binding sites are linked by arrows. (b) Possible M. truncatula orthologues identified by the data presented here are indicated with letters (from A to H) corresponding to transcription factors present in model with their corresponding names and normalized expression profiles (colour code for the expression profiles is indicated on the top right and is explained in Fig. 1)
An interaction between the AACA box and a MYB factor [Fig. 6(b) F] is also essential for seed storage protein gene activation in cereals (Suzuki et al. 1998) and proposed for Arabidopsis (Vicente-Carbajosa and Carbonero 2005), and DOF factors [Fig. 6(b) G] are known to regulate storage protein accumulation in cereal endosperm in association with bZIP factors (Yamamoto et al. 2006). Finally, at the end of seed filling, further bZIP factors, ROM2 (Chern et al. 1996a) or ABI5/EEL (Bensmihen et al. 2005) [Fig. 6(b) H] have been implicated in shutting down seed storage protein synthesis during seed desiccation. In parallel, the VAL B3 proteins that act to repress LEC1 or L1L genes (Suzuki et al. 2007) or other B3-domain genes may repress seed filling genes in conjunction with the CHD3 chromatin-remodelling factors (Tsukagoshi et al. 2007).
All TFs identified in the present study to be closely related to putative regulators of storage protein gene expression were expressed just before or during vicilin synthesis. The later expression of legumin genes may therefore involve other mechanisms. This could be a specific post-transcriptional activation of a TF, or chromatin remodelling, as shown for phaseolin gene regulation (Li et al. 2001). A further possibility might be the need for an additional late-expressed factor, possibly from among the 37 TFs identified in this study (e.g. from cluster IV and/or V) (Fig. 6 and Supplementary Table S4), which are co-expressed with legumin genes.
References
Aida M, Ishida T, Tasaka M (1999) Shoot apical meristem and cotyledon formation during Arabidopsis embryogenesis: interaction among the CUP-SHAPED COTYLEDON and SHOOT MERISTEMLESS genes. Development 126:1563–1570
Ané JM, Zhu H, Frugoli J (2008) Recent advances in Medicago truncatula genomics. Int J Plant Genomics Article ID 256597, doi:10.1155/2008/256597
Bensmihen S, Giraudat J, Parcy F (2005) Characterization of three homologous basic leucine zipper transcription factors (bZIP) of the ABI5 family during Arabidopsis thaliana embryo maturation. J Exp Bot 56:597–603
Boulter D, Evans MI, Ellis RJ, Shirsat A, Gatehouse JA, Croy RRD (1987) Differential gene expression in the development of Pisum sativum. Plant Physiol Biochem 25:283–289
Bouton S, Viau L, Lelievre E, Limami A (2005) A gene encoding a protein with a proline-rich domain (MtPPRD1), revealed by suppressive subtractive hybridization (SSH), is specifically expressed in the Medicago truncatula embryo axis during germination. J Exp Bot 56:825–832
Brocard-Gifford IM, Lynch TJ, Finkelstein RR (2003) Regulatory networks in seeds integrating developmental, abscisic acid, sugar, and light signaling. Plant Physiol 131:78–92
Cernac A, Benning C (2004) WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. Plant J 40:575–85
Chang S, Puryear J, Cairney J (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Rep 11:113–116
Chen W, Provart NJ, Glazebrook J, Katagiri F, Chang HS, Eulgem T, Mauch F, Luan S, Zou G, Whitman SA (2002) Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 14:559–574
Chern MS, Bobb AJ, Bustos MM (1996a) The regulator of MAT2 (ROM2) protein binds to early maturation promoters and represses PvALF-activated transcription. Plant Cell 8:305–321
Chern MS, Eiben HG, Bustos MM (1996b) The developmentally regulated bZIP factor ROM1 modulates transcription from lectin and storage protein genes in bean embryos. Plant J 10:135–148
Cook RD (1999) Medicago truncatula—A model in the making! Curr Opin Plant Biol 2:301–304
Czechowski T, Bari RP, Stitt M, Scheible WR, Udvardi MK (2004) Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root and shoot specific genes. Plant J 38:366–379
De Folter S, Busscher J, Colombo L, Losa A, Angenent G (2004) Transcript profiling of transcription factor genes during silique development in Arabidopsis. Plant Mol Biol 56:351–366
Diaz I, Vicente-Carbajosa J, Abraham Z, Martínez M, Isabel-La Moneda I, Carbonero P (2002) The GAMYB protein from barley interacts with the DOF transcription factor BPBF and activates endosperm-specific genes during seed development. Plant J 29:453–464
Druka A, Muehlbauer G, Druka I, Caldo R, Baumann U, Rostoks N, Schreiber A, Wise R, Close T, Kleinhofs A, Graner A, Schulman A, Langridge P, Sato K, Hayes P, McNicol J, Marshall D, Waugh R (2006) An atlas of gene expression from seed to seed through barley development. Funct Integr Genomics 6:202–211
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 99:10611–10616
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY super family of plant transcription factors. Trends Plant Sci 5:199–206
Finkelstein RR, Wang ML, Lynch TJ, Rao S, Goodman HM (1998) The Arabidopsis abscisic acid response locus ABI4 encodes an APETALA 2 domain protein. Plant Cell 10:1043–1054
Flanagan CA, Ma H (1994) Spatially and temporally regulated expression of the MADS-box gene AGL2 in wild-type and mutant Arabidopsis flowers. Plant Mol Biol 26:581–595
Gallardo K, Le Signor C, Vandekerckhove J, Thompson RD, Burstin J (2003) Proteomics of Medicago truncatula seed development establishes the time frame of diverse metabolic processes related to reserve accumulation. Plant Physiol 133:664–682
Gallardo K, Firnhaber C, Zuber H, Héricher D, Belghazi M, Henry C, Küster H, Thompson R (2007) A combined proteome and transcriptome analysis of developing Medicago truncatula seeds: Evidence for metabolic specialization of maternal and filial tissues. Mol Cell Proteomics 6(12):2165–2179
Garcia D, Fitz Gerald JN, Berger F (2005) Maternal control of integument cell elongation and zygotic control of endosperm growth are coordinated to determine seed size in Arabidopsis. Plant Cell 17:52–60
Gatehouse JA, Evans IM, Croy RRD, Boulter D (1986) Differential expression of genes during legume seed development. Philosophical Transactions of the Royal Society of London. Biol Sci 314:367–384
Graham MA, Silverstein KA, Cannon SB, VandenBosch KA (2004) Computational identification and characterization of novel genes from legumes. Plant Physiol 135:1179–1197
Hardtke CS, Berleth T (1998) The Arabidopsis gene MONOPTEROS encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J 17:1405–1411
Kagaya Y, Toyoshima R, Okuda R, Usui H, Yamamoto A, Hattori T (2005) LEAFY COTYLEDON1 controls seed storage protein genes through its regulation of FUSCA3 and ABSCISIC ACID INSENSITIVE3. Plant Cell Physiol 46:399–406
Kroj T, Savino G, Valon C, Giraudat J, Parcy F (2003) Regulation of storage protein gene expression in Arabidopsis. Development 130:6065–6073
Kwong RW, Bui AQ, Lee H, Kwong LW, Fischer RL, Goldberg RB, Harada JJ (2003) LEAFY COTYLEDON1-LIKE defines a class of regulators essential for embryo development. Plant Cell 15:5–18
Lara P, Oñate-Sánchez L, Abraham Z, Ferrándiz C, Díaz I, Carbonero P, Vicente-Carbajosa J (2003) Synergistic activation of seed storage protein gene expression in Arabidopsis by ABI3 and two bZIPs related to OPAQUE2. J Biol Chem 278:21003–21011
Li G, Chandrasekharan MB, Wolffe AP, Hall TC (2001) Chromatin structure and phaseolin gene regulation. Plant Mol Biol 46:121–129
Long JA, moan EI, Medford JI, Barton MK (1996) A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 404:66–69
Lotan T, Ohto M, Yee KM, West MA, Lo R, Kwong RW, Yamagishi K, Fischer RL, Goldberg RB, Harada JJ (1998) Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 93:1195–1205
Lu P, Porat R, Nadeau JA, O’Neill SD (1996) Identification of a meristem L1 layer-specific gene in Arabidopsis that is expressed during embryonic pattern formation and defines a new class of homeobox genes. Plant Cell 8:2155–2168
Mayer KF, Schoof H, Haecker A, Lenhard M, Jurgens G, Laux T (1998) Role of WUSCHEL in regulating stem cell fate in Arabidopsis shoot meristem. Cell 95:805–815
Meinke DW, Chen J, Beachy RN (1981) Expression of storage-protein genes during soybean seed development. Planta 153:130–139
Parcy F, Valon C, Raynal M, Gaubier-Comella P, Delseny M, Giraudat J (1995) Regulation of gene expression programs during Arabidopsis seed development: roles of the ABI3 locus and of endogenous abscisic acid. Plant Cell 6:1567–1582
Parcy F, Valon C, Kohara A, Miséra S, Giraudat J (1997) The ABSCISIC ACID-INSENSITIVE3, FUSCA3, and LEAFY COTYLEDON1 loci act in concert to control multiple aspects of Arabidopsis seed development. Plant Cell 9:1265–1277
Putterill J, Robson F, Lee K, Simon R, Coupland G (1995) The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 80:847–857
Rajjou L, Gallardo K, Debeaujon I, Vandekerckhove J, Job C, Job D (2003) The effect of ά-amanitin on the arabidopsis seed proteome highlights the distinct roles of stored and neosynthesized mRNAs during germination. Plant Physiology 134:1598–1613
Ramakers C, Ruijtera JM, Lekanne Deprez RH, Moormana AFM (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66
Reidt W, Wohlfarth T, Ellerström M, Czihal A, Tewes A, Ezcurra I, Rask L, Bäumlein H (2000) Gene regulation during late embryogenesis: the RY motif of maturation-specific gene promoters is a direct target of the FUS3 gene product. Plant J 21:401–408
Riechman JL (2002) Transcriptionnal regulation: a genomic overview (September, 30, 2002). In Sommerville CR and Meyerowitz EM (eds) Arabidopsis Book, American Society of Plant Biologists, Rockville, MD (http://www.aspb.org/publications/Arabidopsis/)
Riechman JL, Ratcliffe OJ (2000) A genomic perspective on plant transcription factors. Curr Opin Plant Biol 3:423–434
Rook F, Gerrits N, Kortstee A, van Kampen M, Borrias M, Weisbeek P, Smeekens S (1998) Sucrose-specific signalling represses translation of the Arabidopsis ATB2 bZIP transcription factor gene. Plant J 15:253–263
Ruuska SA, Girke T, Benning C, Ohlrogge JB (2002) Contrapuntal networks of gene expression during Arabidopsis seed filling. Plant Cell 14:1191–1206
Salanoubat M, Lemcke K, Rieger M, Ansorge W, Unseld M, Fartmann B, Valle G, Blocker H, Perez-Alonso M, Obermaier B et al (2000) Sequence and analysis of chromosome 3 of the plant Arabidopsis thaliana. Nature 408:820–822
Santos Mendoza M, Dubreucq B, Miquel M, Caboche M, Lepiniec L (2005) LEAFY COTYLEDON 2 activation is sufficient to trigger the accumulation of oil and seed specific mRNAs in Arabidopsis leaves. FEBS Lett 579:4666–4670
Schruff MC, Spielman M, Tiwari S, Adams S, Fenby N, Scott RJ (2006) The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development 133:251–61
Slabas AR, Fordham-Skelton AP, Fletcher D, Martinez-Rivas JM, Swinhoe R, Croy RR, Evans IM (1994) Characterisation of cDNA and genomic clones encoding homologues of the 65 kDa regulatory subunit of protein phosphatase 2A in Arabidopsis thaliana. Plant Mol Biol 26:1125–1138
Sozzani R, Maggio C, Varotto S, Canova S, Bergounioux C, Albani D, Cella R (2006) Interplay between Arabidopsis activating factors E2Fb and E2Fa in cell cycle progression and development. Plant Physiol 140(4):1355–1366
Sturn A, Quackenbush J, Trajanoski Z (2002) Genesis: cluster analysis of microarray data. Bioinformatics 18:207–208
Suzuki A, Wu CY, Washida H, Takaiwa F (1998) Rice MYB protein OSMYB5 specifically binds to the AACA motif conserved among promoters of genes for storage protein glutelin. Plant Cell Physiol 39:555–559
Suzuki M, Wang HH, McCarty DR (2007) Repression of the LEAFY COTYLEDON1/B3 regulatory network in plant embryo development by VP1/ABSCISIC ACID INSENSITIVE 3-LIKE B3 genes. Plant Physiol 143:902–11
Takatsuji H (1999) Zinc-finger proteins: the classical zinc finger emerges in contemporary plant science. Plant Mol Biol 39:1073–1078
Tsukagoshi H, Morikami A, Nakamura K (2007) Two B3 domain transcriptional repressors prevent sugar-inducible expression of seed maturation genes in Arabidopsis seedlings. Proc Natl Acad Sci USA 104:2543–2547
Udvardi MK, Kakar K, Wandrey M, Montanari O, Murray J, Andriankaja A, Zhang JY, Benedito V, Hofer JM, Chueng F, Town CD (2007) Legume transcription factors: global regulators of plant development and response to the environment. Plant Physiol 144:538–549
Unger E, Parsons RL, Schmidt RJ, Bowen B, Roth BA (2002) Dominant negative mutants of Opaque2 suppress transactivation of a 22-kD zein promoter by Opaque2 in maize endosperm cells. Plant Cell 5:831–841
Usadel B, Nagel A, Thimm O, Redestig H, Blaesing OE, Palacios-Rojas N, Selbig J, Hannemann J, Piques MC, Steinhauser D, Scheible WR, Gibon Y, Morcuende R, Weicht D, Meyer S, Stitt M (2005) Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol 138:1195–1204
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3
Vicente-Carbojosa V, Carbonero P (2005) Seed maturation: developing an intrusive phase to accomplish a quiescent state. Int J Dev Biol 49:645–651
Walling L, Drews GN, Goldberg RB (1986) Transcriptional and post-transcriptional regulation of soybean seed protein mRNA levels. Proc Natl Acad Sci USA 83:2123–2127
Weber H, Borisjuk L, Heim U, Buchner P, Wobus U (1995) Seed coat-associated invertases of fava bean control both unloading and storage functions: cloning of cDNAs and cell type-specific expression. Plant Cell 7:1835–1846
Yamamoto MP, Onodera Y, Touno SM, Takaiwa F (2006) Synergism between RPBF Dof and RISBZ1 bZIP activators in the regulation of rice seed expression genes. Plant Physiol 141:1694–1707
Acknowledgements
We are grateful to all the members of laboratory, particularly Vincent Savois for his very valuable technical support with bioinformatics analyses; Judith Burstin and Michael Bourgeois for helpful discussions; Delphine Hericher and Françoise Moussy for useful assistance with plant growth. In addition, we would also like to acknowledge Dr. Hugues Barbier (Max Planck Institute for Plant Breeding Research MPIZ, Koeln, Germany) for advice and help with RNA extraction; Helge Küster and Christian Firnhaber (Bielefeld University, Germany) who have developed the Mt16kOLI1 microarrays for fruitful collaboration. Research was supported by the FP6 EU project Grain Legumes (FOOD-CT-2004-506223).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11103_2008_9320_MOESM1_ESM.ppt
Experimental variation and assessment of reproducibility of 19,000 real-time RT-PCR reactions. Comparisons of log10 relative expression of all 19,000 real-time RT-PCR reactions obtained between the two biological replicates. The relative expression values from the first biological replicate (horizontal axis) were plotted against the relative expression values from the second biological replicate (vertical axis). The dotted line represents the cut off limit of the relative expression values taken for subsequent analyses, values with a lower relative expression being considered as missing data. Pearson’s correlation coefficient (R) was calculated and is indicated. (PPT 685 kb)
Rights and permissions
About this article
Cite this article
Verdier, J., Kakar, K., Gallardo, K. et al. Gene expression profiling of M. truncatula transcription factors identifies putative regulators of grain legume seed filling. Plant Mol Biol 67, 567–580 (2008). https://doi.org/10.1007/s11103-008-9320-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-008-9320-x