
Abstract
We have developed 85 new markers (50 RFLPs, 5 SSRs, 12 DD cDNAs, 9 ESTs, 8 HSP-encoding cDNAs and one BSA-derived AFLP marker) for saturation mapping of QTL regions for drought tolerance in rice, in our efforts to identify putative candidate genes. Thirteen of the markers were localized in the close vicinity of the targeted QTL regions. Fifteen of the additional markers mapped, respectively, inside one QTL region controlling osmotic adjustment on chromosome 3 ( oa3.1) and 14 regions that affect root traits on chromosomes 1, 2, 4, 5, 6, 7, 8, 9, 10 and 12. Differential display was used to identify more putative candidate genes and to saturate the QTL regions of the genetic map. Eleven of the isolated cDNA clones were found to be derived from drought-inducible genes. Two of them were unique and did not match any genes in the GenBank, while nine were highly similar to cDNAs encoding known proteins, including a DnaJ-related protein, a zinc-finger protein, a protease inhibitor, a glutathione-S-transferase, a DNA recombinase, and a protease. Twelve new cDNA fragments were mapped onto the genetic linkage map; seven of these mapped inside, or in close proximity to, the targeted QTL regions determining root thickness and osmotic adjustment capacity. The gene I12A1, which codes for a UDP-glucose 4-epimerase homolog, was identified as a putative target gene within the prt7.1/brt7.1 QTL region, as it is involved in the cell wall biogenesis pathway and hence may be implicated in modulating the ability of rice roots to penetrate further into the substratum when exposed to drought conditions. RNAs encoding elongation factor 1β, a DnaJ-related protein, and a homolog of wheat zinc-finger protein were more prominently induced in the leaves of IR62266 (the lowland rice parent of the mapping materials used) than in those of CT9993 (the upland rice parent) under drought conditions. Homologs of 18S ribosomal RNA, and mRNAs for a multiple-stress induced zinc-finger protein, a protease inhibitor, and a glutathione-S-transferase were expressed at significantly higher levels in CT9993 than in IR62266. Thus several genes involved in the regulation of DNA structure and mRNA translation were found to be drought-regulated, and may be implicated in drought resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice is the most important staple food crop in the world, and is grown under a broad range of environmental conditions in terms of topography, soil type, water regime, and climatic factors. Drought is one of the important factors that limit rice production in the most rice-growing areas. Physiological studies suggest that drought resistance in rice depends mainly on one or more of the following components: (1) the ability of roots to exploit deep soil water to provide for evapotranspirational demand; (2) the capacity for osmotic adjustment (OA), which allows the plant to maintain turgor and protect meristems from desiccation; (3) the ability to control and reduce water loss (Nguyen et al. 1997).
Maintenance of turgor by osmotic adjustment is an important physiological adaptation for minimizing the detrimental effects of water deficit (Morgan 1984). Osmotic adjustment is therefore considered to be a significant trait that contributes to reducing the effects of drought on grain yield in various crops (Morgan et al. 1991; Moustafa et al. 1996). Quantitative trait loci (QTLs) for OA have been mapped in rice (Lilley et al. 1996), barley (Teulat et al. 1998) and wheat (Morgan and Tan 1996).
Water uptake by the root is a complex character that depends on root structure, root anatomy, and on how the different parts of the root contribute to overall water transport (Cruz et al. 1992). Therefore, the efficiency of water uptake by the root system is a key factor in determining the rate of transpiration and tolerance to drought. Root characteristics such as thickness, depth of rooting, and root length density have been associated with drought avoidance in rice (O’Toole and Chang 1979; Price et al. 2000). QTLs for root morphology and root penetration index have been identified in several rice populations (Champoux et al. 1995; Ray et al. 1996; Price and Tomos 1997; Yadav et al. 1997; Zheng et al. 1999; Ali et al. 2000; Price et al. 2000; Zhang et al. 2001). The introduction of traits which contribute to drought avoidance or drought tolerance should improve the drought resistance of rice, and this strategy therefore has considerable potential for increasing rice production in drought-prone areas (Fukai and Cooper 1995; Nguyen et al. 1997).
At the gene expression level, the accumulation of specific mRNAs in response to water deficit has been well documented in numerous plant species (Claes et al. 1990; Bray 1998). Most of these genes are either involved in, or related to, the production and/or accumulation of compatible osmolytes (Bray 1993; Skriver and Mundy 1990), and the capacity to accumulate osmolytes has been shown to correlate with drought tolerance (Leprince et al. 1993; Quick et al. 1989). Little is known about the genes that contribute to root penetration ability in plants undergoing drought stress. While numerous drought-responsive genes have been identified to date, it has been difficult to pinpoint the key genes contributing to drought tolerance and avoidance in rice.
In this study, we set out to refine the map positions of key QTL regions for drought resistance-related traits, which were previously identified by Zhang et al. (2001). We have combined RFLP, AFLP, SSR, candidate gene, and physical mapping strategies, focusing on the targeted QTL regions, to identify DNA markers that are tightly linked to each QTL. Using differential display (DD) analysis, we have also isolated, mapped and characterized novel cDNAs.
Materials and methods
Plant materials
A subset of 154 doubled haploid lines (DHLs) derived from a cross between the O. sativa accessions CT9993-5-10-1-M and IR62266-42-6-2 (henceforth abbreviated as CT9993 and IR62266, respectively) was used in this study. CT9993 is derived from upland rice breeding materials, has strong root penetration ability and low OA, but is well adapted to rainfed lowland conditions. IR62266 is an elite lowland breeding line, has good osmotic adjustment capacity and a shallow root system.
The initial genetic linkage map was constructed with 315 (145 RFLP, 153 AFLP and 17 SSR) markers at Texas Tech University, Lubbock, Texas (Zhang et al. 2001). The QTLs associated with various traits related to drought resistance, such as osmotic adjustment (oa), root penetration index (rpi), basal root thickness (brt), root pulling force (rpf), total root dry weight (trdw), penetrated root dry weight (prdw), and penetrated root length (prl) were targeted for fine-scale mapping.
Saturation mapping of QTL regions using RFLP, AFLP and SSR markers
The 382 RFLP and 24 SSR markers between or near the targeted QTL regions were selected based on high-density RFLP maps (Causse et al. 1994; Harushima et al. 1998) and 27 drought-related ESTs from the Japanese Rice Research Program were also used. The RFLP and SSR markers were used to screen the 154 DHLs according to standard protocols (Causse et al. 1994; Panaud et al. 1996; Chen et al. 1997). AFLP analysis was undertaken as described by Zabeau and Vos (1993). Bulked segregant analysis was carried out essentially as described Michelmore et al. (1991). Fifteen individual DH lines from the extremes with the highest and lowest scores for OA and root traits were bulked by pooling equal amounts of DNA from each group. Twenty-three primer combinations were employed to amplify fragments from DNA samples obtained from the parent and the bulks of each extreme. The eight primer combinations, which generated putatively linked bands, were then employed to genotype the 154 DHLs.
Differential display analysis
The parental genotypes CT9993 and IR62266 were used for differential display analysis. Rice plants were grown in a growth chamber (Conviron-15) on a 14-h photoperiod (30°C day/25°C night, light intensity 600 μper m2/s. Plants were watered daily and fertilized weekly with a complete nutrient solution for 45 days. Beginning on Day 46, plants were subjected to progressive drought by withholding water for a period of 19 days. Leaf relative water content (RWC) was monitored daily, and leaves that attained 80% and 65% RWC were collected for RNA isolation. Leaf samples from fully watered plants at 98% RWC were used as a control. The RWC was measured in the middle fragment (5 cm) of the fully expanded second leaf, and was calculated as (fresh weight−dry weight)/(fully turgid weight−dry weight). Total RNA was isolated according to Logemann et al. (1987). Differential display was carried out with 96 primer pair combinations as described by Liang et al. (1992), and the analyses were performed using RNAimage kits (GenHunter) according to the manufacturer’s instructions. The cDNA products were resolved on TBE-Urea sequencing gels, which were then dried and exposed to X-ray films. For the purposes of further analysis, we targeted cultivar-specific cDNA fragments for both CT9993 and IR62266 lines, and among them we selected either drought inducible (those for which the corresponding band was amplified only in one and/or both stress RNA samples, but not in the control) or constitutive (those for which corresponding bands were similarly amplified in all three RNA samples from the same cultivar, but with increasing intensity in the samples derived from one or both stages of the drought treatment compared to those derived from the control sample).
Re-amplified fragments were used as probes in Northern analysis. The fragments with confirmed differential expression were cloned in the vector system PCR-TRAP (GenHunter) or pGEM-T-Easy (Promega). After cloning, differential expression of selected fragments was re-confirmed by Northern hybridization. cDNAs were sequenced using a CEQ2000XL DNA sequencer (Beckman Coulter). Sequence data was used for homology searches in the non-redundant set of sequence databases hosted by NCBI. cDNA fragments were then mapped onto the genetic linkage map. Cloning of the full-length cDNAs was done using conventional techniques described elsewhere (Campbell et al. 2001).
Fine-scale mapping of drought resistance QTLs using BACs
A Hin dIII BAC library of cv. Nipponbare ( japonica) with 10.6-fold genome coverage and an average insert size of 130 kb (Clemson University Genome Center) and Bam HI, Eco RI and Hin dIII BAC libraries of Teqing ( indica) with 10.4-fold total genome coverage and an average insert size of 130–150 kb (Texas A and M BAC Center), which consist of 36,864 and 31,488 clones, respectively, were used to screen for overlapping clones. The RFLP markers linked to the QTL regions were used as probes for colony hybridization in order to identify BAC clones derived from the target regions. Each of the labeled probes was added to the pre-hybridization solution and hybridized to high-density BAC filters at 65°C overnight. The filters were washed twice with 0.2 M phosphate buffer containing 0.1% SDS at 65°C for 15 min each with gentle shaking. The filters were then exposed to X-ray films for 2–24 h depending on the signal intensity. Individual candidate BAC clones identified by colony hybridization were then confirmed by Southern hybridization with the DNA markers. Pulsed-Field Gel Electrophoresis (PFGE) was used to determine the size of clone inserts.
The new markers were mapped onto the genetic linkage map using MapMaker Macintosh v. 3.0. To confirm the location of the target QTLs, MapMaker/QTL (Lincoln et al. 1993) and Qgene v. 3.0 (Nelson 1997) were employed.
Results
Saturation mapping of QTLs using RFLP, AFLP, SSR markers and candidate genes
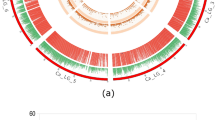
In order to saturate the targeted QTL regions, we selected a total of 382 additional RFLPs, 24 SSRs, 27 drought-related ESTs (from the Japanese Rice Research Program), 51 differential display (DD)-derived cDNAs, heat shock protein (HSP)-related cDNAs, and 20 AFLP markers identified by bulked segregant analysis (BSA). The cDNA and BSA markers were developed at the Plant Molecular Genetics Laboratory, Texas Tech University. These markers were mapped on the 154 doubled haploid lines obtained from the cross between CT9993 and IR62266. The resulting genetic linkage map comprised 400 markers (154 AFLPs, 195 RFLPs, 22 SSRs, 12 DD cDNAs, 9 ESTs and 8 HSP-encoding cDNAs) and was 1686 cM in length, with an average distance between adjacent markers of 4.2 cM (Fig. 1). In comparison with the earlier genetic map (Zhang et al. 2001); the new version contains 85 additional markers (50 RFLPs, 5 SSRs, 12 DD cDNAs, 9 ESTs, 8 HSP cDNAs and one BSA-derived). The map distance was reduced by 102 cM (from 1788 cM to 1686 cM), and the average genetic distance between adjacent markers decreased by 1.5 cM (from 5.7 cM to 4.2 cM). This may be attributed to the fact that with more/additional markers, the degree of genetic interference is also reduced.


High-resolution rice genetic linkage map and positions of QTLs involved in drought resistance in the CT9993/IR62266 DH population. QTL are represented by vertical bars to the right of the chromosomes
In the improved map, a total of 42 QTLs were identified for OA and root traits (Table 1 and Fig. 1). Most of the new QTL positions were consistent with the QTLs reported by Zhang et al. (2001). Four new QTLs— brt4.1, brt7.1, prdw5.1 and prt5.1—were also identified. Two single QTLs controlling penetrated root thickness on chromosomes 1 and 12 were identified instead of a pair of QTLs on each chromosome as reported earlier.
The positions of most of the previously identified chromosome regions controlling osmotic adjustment have not been significantly altered, with a single exception. QTL oa3.1 was shifted from the location EM17_1-C63 (Zhang et al. 2001) to a new location (RG722-RG745) with the same R2 value and length, due to the addition of several markers in its vicinity. The marker RG722 was tightly linked to this QTL (0.0 cM). The positions of most QTLs for root penetration index also did not change, although several new markers were added in these chromosomal regions. However, the QTL rpi4.2 on chromosome 4 was moved from the position RG939-G476 to RG214-R2017, while its R2 was increased from 11.0 to 15.9% (Table 1 and Fig. 1).
Two new QTLs affecting basal root thickness, brt4.1 and brt7.1, were identified. A new RFLP marker, RZ905, was mapped within the QTL region RG939-RG476 for basal root thickness on chromosome 4. The marker RZ572 was located in the basal root thickness QTL region on chromosome 8 ( brt8.1) and replaced the flanking AFLP marker EM14_1 of the QTL. The size of the QTL interval was thus reduced from 10.8 cM to 3.2 cM (Table 1 and Fig. 1).
The RZ19 marker was placed inside a chromosome region harboring a QTL for penetrated root thickness ( prt1.1), and this QTL interval was reduced from 11.2 to 8.0 cM. On chromosome 7 the DD-derived cDNA I12A1 was localized in the QTL prt7.1 and its length was reduced to 4.0 cM. The prt12.1 was relocated from RG9-ME10_1 (8.3 cM) to RG9-ME6_12 (1.5 cM). Several new markers were mapped within a QTL controlling root-pulling force. The DD-derived cDNA C17C2 was localized in the QTL prt2.1, whose length was reduced from 7.5 cM to 1.4 cM. The RFLP marker R2017 was placed inside rpf4.1 (RG214-RG620) and the length of this QTL was reduced from 21.1 cM to 9.7 cM. The rpf5.1 region included the new marker R2217, which replaced the flanking AFLP marker EM15_5. The interval length of this QTL was hence reduced from 30.2 cM to 12.5 cM (Table 1 and Fig. 1).
Two new RFLP markers, R1261 and RG722, were mapped to the QTL trdw1.1; the latter replaced AFLP marker EM11_11. The QTL length here was reduced from 4.8 cM to 0.0 cM. The QTL prdw4.1 was moved and its length reduced from 5.8 cM to 1.9 cM (Table 1 and Fig. 1).
In order to identify markers more closely linked to the QTL regions, we attempted to use Bulked Segregant Analysis (BSA)-based AFLP to screen the isogenic DNA pools composed of all phenotypically extreme lines corresponding to each of the parents’ phenotypes. Out of 20 BSA-AFLP-generated fragments, we were able to map only a single marker on chromosome 3, and this did not co-localize with any QTL region.
Fine mapping of drought resistance QTLs on BACs
The RFLP markers flanking QTLs were used to screen the Nipponbare and Teqing BAC libraries. The RG147 marker flanking the OA QTL on chromosome 1 ( oa1.1) detected five overlapping Nipponbare BAC clones. The other four BAC clones hybridized with RG532, which flanks the other end of this QTL region. Six common BAC clones hybridized with the markers R1394A and G2132, which were linked to oa8.1. Four RFLP markers, which flanked the region controlling major root QTLs ( rpi4.2, rpf4.1 and brt4.1), were selected for screening. Four contigs from Teqing libraries were identified in this region. Three markers (RG214, S5755 and S15741) detected nine overlapping BAC clones from Teqing. The markers RG939 and RZ905, which flanked brt4.1 on each side, detected three and two overlap clones, respectively. The overlapped clones are being used for the identification of BAC ends in order to map them onto the genetic linkage map and walk closer to the targeted QTL.
Differential display-based identification of putative candidate genes for drought resistance traits
The differential display experiment identified 51 cDNA fragments as a ‘candidate’ set. A full-length cDNA corresponding to each selected cDNA fragment was obtained and used for expression analysis and genetic mapping.
First, we confirmed that expression of 11 cDNAs was induced by drought (Fig. 2 and Table 2), while the other 40 cDNAs did not yield a detectable hybridization signal when used as probes on Northern blots. Four of the drought-induced cDNAs (I4G1, I18A1, I23C1, and I28G1) were up regulated in IR62266 as compared to CT9993, while six (C9C1, C11C1, C18G1, C27C1, C27G1, and C30A1) were up regulated in CT9993 relative to IR62266. The cDNA fragment I17A1 hybridized with an mRNA that was up regulated in CT9993 at an earlier stage in the drought response than in IR62266 and, in CT9993, it was expressed at a similar moderate level in mild and severely stressed leaves. In IR62266, this RNA was induced to a low level by the mild stress and further induced by severe stress. Furthermore, several other drought-induced mRNAs differed in their expression levels in mild and severely stressed leaves. The cDNA fragment I4G1 hybridized to a unique mRNA species that was up regulated in leaf tissue exposed to mild drought-stress, and was absent from the severely stressed tissue (Fig. 2). The cDNAs C18G1, C30A1 and I17A1 corresponded to mRNAs that were expressed to higher levels in the severely dehydrated leaves compared to leaves that had been subjected to mild drought conditions.

Northern analysis of RNA in control and drought stressed rice leaf tissues. Lane 1,-CT9993 control; 2,-IR62266 control; 3,-CT9993/mild stress; 4,-IR62266/mild stress; 5,-CT9993/severe stress; 6,-IR62266/severe stress. Total RNA extraction was carried out according to Logemann et al. (1987). Samples containing 30 μg of total RNA were blotted onto Zeta-probe GT membrane. The cDNA fragments were labeled with [α-32P]dATP using a DECA prime II labeling kit (Ambion) and used to probe the filter. Hybridization was performed at 42°C for 18 h in Northern Max prehyb/hyb buffer (Ambion).The filters were then washed in 2×SSPE and exposed to X-ray film
A search for sequence similarities between these cDNA clones and genes in the non-redundant gene database of GenBank resulted in the assignment of putative functions to the proteins encoded by 9 of the 11 cDNAs characterized by Northern analysis (Table 2). Among the cDNAs that were highly expressed in IR62266, the I4G1 cDNA matched the benzothiadiazole and pathogen-inducible mRNA WCI-5 from wheat; cDNA I18A1 was predicted to encode rice elongation factor 1β, I23C1 matched rice oryzain, and the putative product of I28G1 was similar to a DnaJ-related chaperone from maize. Among the CT9993-specific cDNAs, C11C1 matched 18S ribosomal RNA, C18G1 matched a zinc-finger protein, C27C1 and the corresponding full-length cDNA T1-C27C1 matched the senescence-inducible subtilisin-chymotrypsin inhibitor, and C30A1 matched glutathione S-transferase. The mRNA homologous to the drought-inducible cDNA clone I17A1—that differed in its expression profile throughout progressive drought in CT9993 and IR62266—matched recombinases from various bacteria.
Furthermore, we mapped the set of 51 cDNA clones derived from the DD experiment onto the rice genetic map. Twelve partial cDNAs were added to the genetic linkage map of CT9993/IR62266. The remaining 39 fragments that could not been mapped either yielded weak hybridization signals or multiple monomorphic bands. The mapped cDNA fragments were localized on chromosomes 1, 2, 3, 4, 6, 7, 8, 11 and 12 (Table 3). Three of these cDNA clones (I12A1, I23C1, and C17C2) mapped within previously identified QTL regions ( prt 7.1/brt7.1, brt12.1 , and rpf2.1, respectively). Four other cDNAs (I11A1, I29G1, I28C1, and C9A1) were located in the vicinity of the QTL regions trdw6.1, oa3.1, prt6.1, and brt3.1, respectively.
A search for sequence similarities in the GenBank databases resulted in the matching of four of the mapped cDNA fragments to genes encoding proteins with known functions. The cDNA clone I12A1 that mapped within the root thickness QTL region on chromosome 7 showed significant similarity to an UDP-glucose-4-epimerase gene ( OsUGE-1). The cDNA clone I23C1 that mapped in brt12.1 on chromosome 12, and the corresponding full-length cDNA T5-I23C1, matched an oryzain gamma sequence, while the cDNA I28C1 that had been mapped in close proximity to a QTL determining penetrated root thickness on chromosome 6 ( prt6.1) displayed a strong sequence similarity to a homeodomain transcription factor implicated in disease susceptibility (Table 3). Other mapped cDNA fragments did not match with any entry in the GenBank or rice genomic sequence database.
Discussion
The overall aim of this study was to refine the mapping of the targeted QTL regions underlying drought resistance traits in rice. For this purpose, we employed the available molecular markers, and identified several novel markers linked to the QTL regions using the differential display approach. We have placed seven of the novel DD-derived cDNAs inside or in the close vicinity of QTL regions determining root traits and OA capacity. We also characterized a set of cDNA clones derived from transcripts that are up-regulated by drought, and are predicted to encode proteins that function in the modulation of DNA structure, in translational regulation, protein metabolism and folding, and in antioxidant production. These genes add to the pool of putative candidate genes underlying drought stress resistance in rice.
RFLP and SSR markers are very efficient in increasing marker density and saturating targeted genomic regions. However, the degree of polymorphism detected by the existing markers is limited. The aim of this study was to provide additional polymorphic markers for map-based cloning in the targeted QTL regions. In an effort to identify markers tightly linked to the target QTLs, we have selected a set of additional genetic markers from the published maps and used them for the saturation mapping of the QTL regions on our genetic map. Here we report the addition of 85 new markers to the genetic map of rice (Zhang et al. 2001). Of these, 28 markers were mapped within or in close proximity to the targeted QTL regions. New markers were added to the QTL regions on chromosome 1 that control osmotic adjustment ( oa1.1), penetrated root thickness ( prt1.1), and total root dry weight ( trdw1.1). The resolution of QTLs on chromosome 3, which control osmotic adjustment ( oa3.1), root pulling force ( rpf3.2) and root penetration index ( rpi3.1), was significantly improved. In particular, 10 new markers, including seven stress-related ESTs, were mapped within or near the chromosome 4 region controlling major root traits ( rpf4.1, rpi4.2, prt4.1, prdw4.2 and brt4.2). These results indicate that the candidate gene approach is a powerful tool for selecting desirable alleles in the targeted QTL regions.
Notably, the definition of QTL regions on chromosome 5 that affect root pulling force ( rpf5.1), penetrated root thickness ( prt5.1) and penetrated root dry weight ( prdw5.1) was significantly improved by the addition of five new RFLP markers. The new RFLP marker R2217 replaced the AFLP-generated EM15_5 marker and the size of the QTL interval was concomitantly reduced by two-fold. Indeed, the sizes of many of the QTLs were significantly improved in the present study over the previously published genetic map. The RFLP markers that replaced AFLP markers linked to the QTLs can be now used for marker-assisted selection of drought-resistant genotypes. Furthermore, the available RFLP markers closely linked to the QTL regions were used for BAC library screening. We have identified overlapping clones for the markers linked to several QTL regions. Fine-scale QTL mapping of drought resistance QTLs using BACs is now under way, and the overlapping BAC clones are being used to identify markers more closely linked to the QTLs.
Bulked segregant analysis in combination with AFLP detection has proved to be a powerful technique for identifying markers tightly linked to, or co-segregating with, genes underlying monogenic traits (Thomas et al. 1995). We have used AFLP-based BSA to refine map positions of the targeted QTLs. However, in our experiments, only one of the 24 BSA fragments developed—E4M19_3—could be mapped, on chromosome 3 (Fig. 1), and it was not associated with any QTL region. Hence, in our hands this approach was not an efficient tool for dissecting targeted QTL regions for polygenic drought resistance-related traits.
Furthermore, we used DD to identify novel markers more closely linked to the QTL regions for drought resistance trait in rice. We assessed the dynamics of specific mRNA expression under mild and severe drought stress, and assigned putative functions to several isolated genes based on the availability of GenBank matches.
Eleven genes characterized in this study were significantly up regulated following exposure of rice plants to drought. Seven of these genes have orthologs in other species, while two (C9C1 and C27G1) did not match any known genes in the GenBank databases. Of the nine genes with orthologs in other species, six had orthologs that have been characterized as pathogen-, senescence-, or heat stress-inducible (Table 2). However, prior to this work, none of these seven cDNAs had been implicated in responses to drought. The other three cDNAs (I18A1, C11C1, and I17A1) matched orthologous sequences in other species that have not been shown to be stress inducible (elongation factor 1β, 18S ribosomal RNA, and a DNA recombinase).
The cDNA clone I18A1 was predicted to encode translation elongation factor 1β. In our study, the corresponding gene was drought inducible, and the mRNA level increased with the severity of drought stress; the gene was also expressed to a much higher level in the leaves of the drought-tolerant cultivar IR62266 than in CT9993, which is more sensitive to water deficit. This elongation factor may be related to the higher osmotic adjustment capacity of IR62266, perhaps regulating the synthesis of an enzyme (s) mediating osmolyte biosynthesis.
The I17A1 cDNA encodes a homolog of a bacterial integrase/recombinase protein. Interestingly, no matches from eukaryotes have been identified by Blast similarity search. It is unlikely that this cDNA results from bacterial contamination, as it produced a clear hybridization signal with both rice DNA and RNA and it was mapped on chromosome 1 of the genetic linkage map. The corresponding mRNA was significantly induced by drought, and accumulated throughout the development of drought stress in rice leaves (Fig. 2 and Table 2), in a cultivar-specific fashion. A comprehensive sequence analysis of this cDNA with GENSCAN (v1.0, with both Arabidopsis and maize matrix) and the program RiceHMM (at a probability level 9.8e-1), provided by the Rice GAAS program on the MAAF of Japan website, predicted that most of the sequence is derived from an uninterrupted exon that was structurally highly similar to bacterial recombinase genes, indicating a potential retrotransposon origin for this gene. Stress-induced up regulation of retrotransposon-encoded genes has been noted in many eukaryotes, including plants, leading to the hypothesis that abiotic stress may cause genomic DNA rearrangements. Our results indicate that several genes involved in the regulation of DNA structure and mRNA translation can be regulated by drought, and may be implicated in drought resistance.
Three of the genes identified in this study were structurally similar to known genes implicated in pathogen and general stress responses. The cDNA clone I4G1 encodes a protein similar to that specified by a pathogen-inducible cDNA from wheat; C18G1 encodes a zinc-finger protein, while C30A1 encodes glutathione S-transferase (GST). GST genes are known to be induced by a variety of stress conditions, including heavy metal treatment, heat shock and hormones (Ulmasov et al. 1995). Both the GST cDNA C30A1 and pathogen-responsive homologous cDNA C18G1 were expressed at significantly higher levels in CT9993 compared to IR62266 under drought stress, and accumulated to even higher levels under severe drought stress. The I4G1 cDNA was overexpressed in IR62266 relative to CT9993.
Three new cDNAs were mapped within QTL regions. The cDNA I12A1 was mapped within the prt7.1 /brt7.1 QTL region on chromosome 7. This cDNA did not detect any mRNA species on Northern blots, and thus we were unable to analyze the expression pattern of the corresponding mRNA. However, the BLAST search against GenBank sequence databases allowed us to putatively identify the encoded protein as a UDP-glucose 4 epimerase. This enzyme functions in the nucleotide sugar interconversion pathway (converting UDP-glucose to UDP-galactose), and hence is implicated in cell wall biosynthesis as it helps to regulate the monosaccharide pool available for pectin production (Reiter and Vanzin 2001). Thus, this gene could perform a critical function during drought by contributing to cell wall thickening in rice roots.
The I23C1 cDNA was mapped within the brt12.1 QTL region and it was putatively identified as encoding oryzain γ, based on its similarity to the published rice oryzain gene. Oryzains (EC 3.4.22) are cysteine proteinases, which are encoded by a multigene family in rice; in rice seeds these genes are inducible by gibberellins (Watanabe et al. 1991), where they function to promote storage protein utilization upon seed germination. We found that oryzain mRNA was almost undetectable in the leaves of well-watered plants, was up-regulated by drought in both CT9993 and IR62266 and, under mild drought, it accumulated to approximately two-fold higher levels in IR62266 than in CT9993. It is not known whether protease function is critical for root thickening upon exposure to drought. However, proteases are essential for disposing of proteins that have been damaged by osmotic and oxidative stresses.
Only two of the mapped cDNA fragments—I17A1 and I23C1—gave detectable signals when hybridized with RNA on Northern blots. There could be several reasons for this. The isolated cDNAs could represent rare mRNAs, which are difficult to detect using conventional Northern blotting techniques, as the cloned DD fragments were small (between 100 and 600 bp long) and mostly derived from 3′-UTR regions of mRNAs.
The differential display approach and the expression analysis provided us with a novel set of drought-related cDNA clones (Tables 2 and 3). This strategy allowed us (1) to identify two new drought-inducible genes with no sequence database matches (C9C1 and C27G1); (2) to classify nine rice genes with predicted functions (based on GenBank matches) as drought-inducible (I4G1, I17A1, I18A1, I23C1, I28G1, C11C1, C18G1, C27C1 and C30A1); (3) to assign map locations to 11 new cDNAs, and (4) to localize seven novel cDNAs to the vicinity of QTLs determining drought resistance traits, and thus identify them as putative candidates for roles in osmotic adjustment, root thickness, total root dry weight and root pulling force.
It is notable that seven of the 21 novel cDNAs discussed in this paper did not have matches in the GenBank sequence databases, indicating that a significant number of rice genes remain unidentified, and some of these may be involved in drought resistance. Based on the similarity of the new drought-inducible cDNA sequences to mRNAs encoding proteins with known functions, we predict that such cell functions as DNA modification, mRNA translation, protein degradation, protein chaperoning, and signaling may be modified in response to drought stress in rice and/or be involved in drought resistance mechanisms.
Overall, the combination of a number of different approaches, such as using available RFLPs, SSRs, ESTs and developing new DD-derived cDNAs as described here, has provided us with a set of markers that are tightly linked to QTL regions conferring drought resistance in rice. These tightly linked markers are currently being used for marker assisted selection in order to develop near isogenic lines and facilitate map-based cloning of the QTLs using BACs.
References
Ali ML, Pathan MS, Zhang J, Bai G, Sarkarung S, Nguyen HT (2000) Mapping QTLs for root traits in a recombinant inbred population from two indica ecotypes in rice. Theor Appl Genet 101:756–766
Baszczynski CL, Barbour E, Zeka BL, Maddock SE, Swenson JL (1997) Characterization of a genomic clone for a maize DnaJ-related gene, ZmdJ1 , and expression analysis of its promoter in transgenic plants. Maydica 42:189–201
Bray EA (1993) Molecular responses to water deficit. Plant Physiol 103:1035–1040
Bray EA (1998) Plant responses to water deficit. Trends Plant Sci 2:48–54
Campbell JL, Klueva NY, Zheng H, Nieto-Sotelo J, Ho THD, Nguyen HT (2001) Cloning of new members of heat shock protein HSP101 gene family in wheat ( Triticum aestivum (L.) Moench) inducible by heat, dehydration, and ABA. Biochim Biophys Acta 1517:270–277
Causse MA, Fulton TM, Chao YG, Chunwongse J, We K, Xiao J, Yu Z, Ronald PC, Harrington SE, Second G, McCouch SR, Tanksley SD (1994) Saturated molecular map of the rice genome based on an interspecific backcross population. Genetics 138:1251–1274
Champoux MC, Wang G, Sarkarung S, Mackill DJ, O’Toole JC, Huang N, McCouch SR (1995) Locating genes associated with root morphology and drought avoidance in rice via linkage to molecular markers. Theor Appl Genet 90:969–981
Chen XS, Temnykh Y, Xu YG, Cho SR, McCouch SR (1997) Development of a microsatellite map providing genome-wide coverage in rice ( Oryza sativa L.). Theor Appl Genet 95:553–567
Claes B, Dekeyser R, Villarroel R, Vanden Bulcke M, Bauw G, Van Montagu M, Caplan A (1990) Characterization of a rice gene showing organ-specific expression in response to salt stress and drought. Plant Cell 2:19–27
Cruz RT, Jordan WR, Drew MC (1992) Structural changes and associated reduction of hyraulic conductance in root of Sorghum biocolor L. following exposure to water deficit. Plant Physiol 99:203–212
Fukai S, Cooper M (1995) Development of drought resistant cultivars using physio-morphological traits in rice. Field Crop Res 40:67–86
Gorlach J, Volrath S, Knauf-becker G, Hengy G, Beckhove U, Kogel KH, Oostendorp M, Staub M, Ward E, Kessmann H, Ryals J (1996) Benzothiadiazole, a novel class of inducers of systemic acquired resistance, activate gene expression and disease resistance in wheat. Plant Cell 8:629–643
Harushima Y, et al (1998) A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148:479–494
Kleber-Janke T, Krupinska K (1997) Isolation of cDNA clones for genes showing enhanced expression in barley leaves during dark-induced senescence, as well as during senescence under filed conditions. Planta 203:332–340
Leprince O, Hendry GAF, McKersie BD (1993) The mechanisms of desiccation tolerance in developing seeds. Seed Sci Res 3:231–246
Liang P, Pardee AB (1992) Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257:967–971
Lilley JM, Ludlow MM, McCouch SR, O’Toole JC (1996) Locating QTL for osmotic adjustment and dehydration tolerance in rice. J Exp Bot 47:1427–1436
Lincoln SE, Daly MJ, Lander ES (1993) Mapping genes controlling quantitative traits using MapMaker/QTL version 1.1: a tutorial and reference manual. Whitehead Institute for Biomedical Research Technical Report (2nd edn). Whitehead Institute, Cambridge, Mass.
Logemann J, Schell J, Willmitzer L (1987) Improved method for the isolation of RNA from plant tissues. Anal Biochem 163:16–20
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes: a rapid method to detect markers in specific genomic regions by segregating populations. Proc Natl Acad Sci 88:9828–9832
Morgan JM (1984) Osmoregulation and water stress in higher plants. Annu Rev Plant Physiol 35:299–319
Morgan TM, Tan MK (1996) Chromosomal location of a wheat osmoregulation gene using RFLP analysis. Aust J Plant Physiol 23:803–806
Morgan JM, Rodriguez Maribona B, Knight EJ (1991) Adaptation to water-deficit in chickpea breeding lines by osmoregulation: relationship to grain-yield in the field. Field Crops Res 27:61–70
Moustafa MA, Boersma L, Kronstad WE (1996) Response of four spring wheat cultivars to drought stress. Crop Sci 36:982–986
Nelson J (1997) Qgene: software for marker-based genomic and breeding. Mol Breeding 3:239–245
Nguyen HT, Babu RC, Blum A (1997) Breeding for drought resistance in rice: physiology and molecular genetics consideration. Crop Sci 37:1426–1434
O’Toole JC, Chang TT (1979) Drought resistance in cereals. Rice: a case study. In: Mussel H, Staples RC (eds) Stress physiology of crop plants. Wiley InterScience, New York, pp 374–405
Panaud O, Chen X, McCouch SR (1996) Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice ( Oryza sativa L.). Mol Gen Genet 252:597–607
Price AH, Tomos AD (1997) Genetic dissection of root growth in rice ( Oryza sativa L.). II. Mapping quantitative trait loci using molecular markers. Theor Appl Genet 95:143–152
Price AH, Steele KA, Moore BJ, Barraclough PB, Clark J (2000) A combined RFLP and AFLP linkage map of upland rice ( Oryza sativa L.) used to identify QTLs for root-penetration ability. Theor Appl Genet 100:49–56
Quick P, Siegl G, Neuhaus E, Feil R, Stitt M (1989) Short-term water stress leads to stimulation of sucrose synthesis by activating sucrose phosphate synthase. Planta 177:535–546
Ray JD, Yu L, McCouch SR, Champoux MC, Wang G, Nguyen TN (1996) Mapping quantitative trait loci associated with root penetration ability in rice ( Oryza sativa L.). Theor Appl Genet 92:627–636
Reiter WD, Vanzin GF (2001) Molecular genetics of nucleotide sugar interconversion pathways in plants. Plant Mol Biol 47:95–113
Skriver K, Mundy J (1990) Gene expression in response to abscisic acid and osmotic stress. Plant Cell 2:503–512
Teulat B, This D, Khairallah M, Borries C, Ragot C, Sourdille P, Leroy P, Monneveux P, Charrier A (1998) Several QTLs involved in osmotic adjustment trait variation in barley ( Hordeum vulgare L.). Theor Appl Genet 96:688–698
Thomas CM, Vos P, Zabeau M, Jones AD, Norcott KA, Chadwick BP, Jones JDG (1995) Identification of amplified restriction fragment polymorphism (AFLP) markers tightly linked to the tomato Cf-9 gene for resistance to Cladosporium fulvum. Plant J 8:785–794
Ulmasov T, Ohmiya A, Hagen G, Guilfoyle T (1995) The soybean GH2/4 gene that encodes a glutathione S-transferase has a promoter that is activated by a wide range of chemical agents. Plant Physiol 108:919–927
Yadav R, Courtois, Huang N, Melaren G (1997) Mapping genes controlling root morphology and root distribution in a doubled-haploid population of rice. Theor Appl Genet 94:619–632
Watanabe H, Abe K, Emori Y, Hosoyama H, Arai S (1991) Molecular cloning and gibberellin-induced expression of multiple cysteine proteinases of rice seeds (oryzains) J Biol Chem 266:16897–16902
Zabeau M, Vos P (1993) Selective restriction fragment amplification: a general method for DNA fingerprinting. European Patent Application No. 92402629.7. Publication No. 0534858 A1
Zhang J, Zheng HG, Aarti A, Pantuwan G, Nguyen TT, Tripathy TN, Sarial AK, Robin S, Babu RC, Nguyen DB, Sarkarung S, Blum A, Nguyen HT (2001) Locating genomic regions associated with components of drought resistance in rice: comparative mapping within and across species. Theor Appl Genet 103:19–29
Zheng HG, Babu RC, Pathan MS, Ali ML, Huang N, Courtois B, Nguyen TH (1999) Quantitative trait loci for root penetration ability and root thickness in rice: Comparison of genetic backgrounds. Genome 43:53–61
Acknowledgements
We would like to thank Dr. S.D. Tanksley, Dr. S.R. McCouch and Dr. T. Sasaki for their kindly providing the RFLP clones. cDNA sequencing was provided by the Center for Biotechnology and Genomics at Texas Tech University. This work was funded by a grant from the Rockefeller Foundation’s International Program on Rice Biotechnology
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Hagemann
Rights and permissions
About this article
Cite this article
Nguyen, T.T.T., Klueva, N., Chamareck, V. et al. Saturation mapping of QTL regions and identification of putative candidate genes for drought tolerance in rice. Mol Genet Genomics 272, 35–46 (2004). https://doi.org/10.1007/s00438-004-1025-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-004-1025-5