
Abstract
Human strongyloidiasis is a deleterious gastrointestinal disease mainly caused by Strongyloides stercoralis infection. Strongyloides stercoralis is a soil-transmitted helminthiasis that is distributed around the globe. Although definitive diagnosis is carried out through the detection of parasite objects in human stool samples, the development of reliable immunological assays is an important alternative approach for supportive diagnosis. We characterized the two sensitive and specific bands of S. stercoralis filariform larvae that reacted with human strongyloidiasis sera based on immunoblot analysis. Serum samples obtained from strongyloidiasis patients showed a sensitivity of 90 and 80 % at the approximate molecular mass of 26 and 29-kDa polypeptide bands, respectively. The reactive specificity of the 26-kDa band was 76.5 % while for the 29-kDa band was 92.2 %. Proteomic analysis identified the 26-kDa band protein was 14-3-3 protein zeta, while the 29-kDa band protein was ADP/ATP translocase 4. The results provided a basic framework for further studies regarding the potential of the S. stercoralis recombinant antigen to become a leading to diagnostic tool.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human strongyloidiasis is a serious gastrointestinal disease mainly caused by Strongyloides stercoralis infection. It is a soil-transmitted helminthiasis that is distributed throughout the world, particularly in Southeast Asia, Latin America, Sub-Saharan Africa, and parts of the southeastern USA (Siddiqui and Berk 2001; Olsen et al. 2009; Casavechia et al. 2016), where there are an estimated 30 to 100 million people infected (Bethony et al. 2006; Olsen et al. 2009). This might be an underestimate of the true number of infections, due to the low sensitivity of diagnostic methods and the fact that diagnosis is often delayed. There are three major factors responsible for delayed diagnosis, including the presence of subclinical cases, the fact that parasite load is usually low or displays irregular larvae output, and the lack of a gold standard diagnostic test (Olsen et al. 2009; Valerio et al. 2013). Strongyloidiasis is primarily diagnosed by detecting larvae in stool samples using method such as direct fecal smear examination, Baermann concentration, formalin-ethyl acetate concentration, Harada-Mori filter paper culture, and agar plate cultures (Olsen et al. 2009). The development of reliable immunological assays is an important alternative approach for strongyloidiasis diagnosis (Levenhagen and Costa-Cruz 2014; Eamudomkarn et al. 2015). Immunoblot analysis has been reported as a method for detection of the specific antibody against various antigenic polypeptide bands (Sato et al. 1990; Northern and Grove 1990; Conway et al. 1993, 1994; Atkins et al. 1999; Sudre et al. 2007). Recently, proteomic analysis for helminth proteins, such as those of S. stercoralis third-stage larvae (L3), have become available (Marcilla et al. 2010). However, there is a lack of knowledge regarding antigenic proteins related to human strongyloidiasis sera. In this research, we characterize the diagnostic polypeptides of S. stercoralis L3 that reacted with human strongyloidiasis sera based on immunoblot analysis and followed by their proteomic analysis. The results provided a basic framework for further studies regarding the potential of the S. stercoralis recombinant antigen to become a leading to diagnostic tool.
Materials and methods
Strongyloides stercoralis antigens
Strongyloides stercoralis L3 was obtained from fecal samples from an infected patient using the filter paper culture technique (Harada and Mori 1955). The L3 samples were washed several times in distilled water and then stored at −20 °C until use.
One hundred microliters of the frozen-packed S. stercoralis L3 was homogenized with a tissue grinder directly into 300 μl of sample preparation solution consisting of 7 M urea, 2 M Thiourea, 2 % (w/v) CHAPS and supplemented with protease inhibitor (Roche Diagnostic GmbH, Mannheim, Germany). The extraction was then sonicated with an ultrasonic disintegrator and centrifuged at 10000×g for 30 min at 4 °C. The supernatant was assayed for protein concentration using the Quick Start Bradford Protein Assay (Bio-Rad Laboratories Inc, CA). The sample was kept at −70 °C until further analysis.
Human sera
A total of 61 human serum samples were supplied by the frozen bank (−70 °C). The sera samples which were confirmed using parasitological methods were divided into three groups as follows: (i) the strongyloidiasis group (N = 10), which were found to positive Strongyloides objects by agar plate culture method (Koga et al. 1991); (ii) the other parasitic infections group (N = 45); and (iii) the negative control group (N = 6). The other parasitic infections group consisted of serum samples from patients with other parasitic infections than strongyloidiasis. Their infections were confirmed by parasitological methods (Elkins et al. 1986), including hookworm infection, opisthorchiasis viverrini, ascariasis, taeniasis, trichuriasis, paragonimiasis, and capillariasis excepted trichinellosis, fascioliasis, angiostrongyliasis, and gnathostomiasis cases were the proven cases whose diagnosis were confirmed by worm recovery. The negative control group was composed of samples from healthy adult volunteers from non-endemic areas whose stool examinations were found to be negative for intestinal parasitic infection using the formalin-ethyl acetate concentration method (Elkins et al. 1986) (Table 1). Pooled positive reference sera were prepared by combining equal volumes of all those strongyloidiasis sera, whereas pooled negative reference sera were combined by combining equal volumes of sera from healthy control individuals. These were used for observing the day-to-day variation in the immunoblotting analysis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
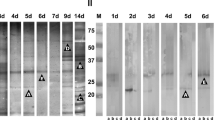
The S. stercoralis L3 extract (150 μg per lane of 6.5 cm width) was resolved separately using 12 % SDS-PAGE under reducing conditions (Laemmli 1970). After electrophoresis, the resolved polypeptides were electrophoretically transferred to a nitrocellulose membrane for immunoblotting (Towbin et al. 1979). For subject to mass spectrometric analysis, S. stercoralis L3 extract (10 μg protein) was loaded into 12 % SDS-PAGE well (0.5 cm width). After electrophoresis, gel was stained with Colloidal Coomassie Blue. The high sensitive and specific polypeptides bands were excised into 0.1 × 0.5 cm for protein identification by mass spectrometry (Fig. 1).

Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of the Strongyloides stercoralis third-stage larval extract (left) and their representative immunoblotting reaction patterns (right). Lane M, molecular mass markers and lane Ss protein, the S. stercoralis extract was stained with Colloidal Coomassie Blue. Blots developed with sera from (1), pooled healthy control (2), pooled positive reference (3–5), strongyloidiasis (6), hookworm infection (7), opisthorchiasis viverrini (8), ascariasis (9), taeniasis (10), gnathostomiasis (11), trichinellosis (12), capillariasis (13), fascioliasis (14) paragonimiasis (15), angiostrongyliasis (16) trichuriasis, and (17–18), individuals healthy control sera. Molecular mass markers are indicated on the left. The arrows indicate the protein (on the left) and immunoreactive bands (on the right) at approximately 26 and 29 kDa
Immunoblot analysis
The antigen-blotted nitrocellulose membranes were blocked with 3 % skim milk for 30 min at room temperature [3 % skimmed milk in 0.1-M phosphate-buffered saline, pH 7.5, containing 0.1 % Tween 20 (Sigma, St. Louis, MO) (PBST)] and washed twice with the washing solution (1 % skim milk in PBST). They were then cut vertically into strips of 0.3 × 5.5 cm. One strip was incubated with one serum sample for 2 h (1:100 dilution in the washing solution) with gentle rocking, washed five times with washing solution, and then incubated for 2 h at room temperature with a horseradish peroxidase-conjugated goat anti-human antibody (Invitrogen Corporation, Camarillo, CA) (1:10000 dilution in the washing solution). Antibody reactions were visualized using 3,3-Diaminobenzidine tetrahydrochloride hydrate (Sigma) and hydrogen peroxide were used as chromogenic substrates.
Mass spectrometry and data analysis
Colloidal Coomassie Blue stained of the interested specific bands that reacted with human strongyloidiasis sera via immunoblotting were manually excised and subjected to in-gel tryptic digestion (Promega, Madison, WI) and mass spectrometric analysis. The tryptic digested samples were analyzed using a nano-liquid chromatography system (EASY-nLC II, Bruker) coupled with a QTOF mass spectrometer (MicrOTOFQ-II, Bruker) equipped with an ESI nano-sprayer. LC–MS/MS spectra were analyzed using Compass Data Analysis v.4.0. Compound lists were exported as Mascot generic files (mgf) for further reference using the MASCOT program (www.matrixscience.com). Protein identification was performed by searching against the protein database from NCBI (All entries) using MASCOT MS/MS Ion Search program with the initial search parameters—enzyme: none; carbamidomethylation (C) as fixed modification, and oxidation (HW) and oxidation (M) as variable modification; peptide mass tolerance of 0.5 Da and fragment mass tolerance of 0.6 Da; a peptide charge state of +1, +2, +3. The identified proteins were assigned to the UniProt database (http://www.uniprot.org/) and described in terms of gene ontology (GO) based on molecular function, cellular component, and biological process. The equipment was provided by Khon Kaen University Research Instrument Center, Thailand.
Results
The Colloidal Coomassie Blue staining of the S. stercoralis L3 extract is shown in Fig. 1. The immunoblotting results are shown in Table 1 and Fig. 1. Serum samples obtained from strongyloidiasis patients showed a sensitivity of 90 and 80 % when reacted with the polypeptide bands at approximately 26-kDa and 29-kDa, respectively. The reactive specificity of the 26-kDa band was 76.5 % while that of the 29-kDa band was 92.2 % (Table 1).
The 26-kDa and 29-kDa specific bands were analyzed using mass spectrometry. The proteins were identified using the Mascot program (Table 2). Since there was limited information regarding the S. stercoralis gene sequences, identification of the respective proteins was based on matches to homologous proteins from Strongyloides ratti. The 26-kDa band protein identified was 14-3-3 protein zeta. While, the 29-kDa band protein identified was ADP/ATP translocase 4.
Discussion
Previous studies have presented various antigenic polypeptide bands from S. stercoralis L3 extract that react with human strongyloidiasis sera (Sato et al. 1990; Conway et al. 1993; Atkins et al. 1999; Sudre et al. 2007). The polypeptide bands at the approximate molecular mass of 26, 41, 66, and 97 kDa from S. stercoralis larvae were frequently recognized by the strongyloidiasis patients’ sera (Sato et al. 1990). The major immunodominant antigens, which were composed of 29, 30, 31, 33, 33.5, 36, 44, and 56 kDa, were reacted with the IgG1, IgG4, and IgE antibodies from human strongyloidiasis sera (Atkins et al. 1999). Conway et al. (1993) reported that the immunodominant bands at the approximate molecular mass of 28, 31, and 41 kDa were reactive with human strongyloidiasis sera, while Sudre et al. (2007) reported that the prominent 21, 26, and 33 kDa protein bands were reactive against human strongyloidiasis serum samples with a high level of frequency.
Our study showed the protein bands at the approximate molecular mass of 26 and 29 kDa were the sensitive and specific bands for diagnosis of human strongyloidiasis. The evidence led us to conduct a proteomic characterization of these two antigenic bands by mass spectrometry for identification of the peptide fragmentation information and gene ontology. The aim is to use this knowledge to develop stable mass production of the sensitive and specific antigenic proteins for serodiagnosis of human strongyloidiasis using recombinant technology.
Each 26 and 29-kDa diagnostic polypeptide of S. stercoralis L3 was characterized using mass spectrometry, the 26-kDa band was identified as 14-3-3 protein zeta whereas the 29-kDa band was identified as ADP/ATP translocase 4.
The 14-3-3 proteins are a large family of widely expressed 24 to 33-kDa acidic proteins, as a key molecule in physiology of parasites (Siles-Lucas Mdel and Gottstein 2003). Their function is in binding specific proteins and they play a major role in basic cellular events related to cellular proliferation, including signal transduction, cell-cycle control, cell differentiation, and cell survival (Siles-Lucas et al. 2008). These proteins have been reported to be important in several parasites, and have mostly been studied in Echinococcus granulosus and Echinococcus multilocularis (Siles-Lucas et al. 2001; Nunes et al. 2004; Pan et al. 2010). They also play an important role in nematode larval development in plant-parasitic nematodes (Jaubert et al. 2004). Siles-Lucas Mdel and Gottstein (2003) have suggested these proteins offer a good target for vaccines and anthelmintic drug development. Interestingly, a related protein, Schistosoma bovis 14-3-3 protein zeta (Sb14ζ), has been used as a vaccine against S. bovis and Schistosoma mansoni in mice (Siles-Lucas et al. 2007; Uribe et al. 2007).
The ADP/ATP translocases are composed of various isoforms (Dolce et al. 2005; Hu et al. 2007; Leung et al. 2013) which belong to the mitochondrial carrier family and play a central role in aerobic eukaryotic cells. This function is related to the transport of ADP into the mitochondrial matrix and ATP out from the matrix for cell utilization (Dahout-Gonzalez et al. 2006; Leung et al. 2013). Recently, Hu et al (2007) isolated and characterized Tv-ant-1 gene encoding of an ADP/ATP translocase of Trichostrongylus vitrinus, an important parasitic round worm found in small ruminants. They suggested that ADP/ATP translocase proteins play a key role in the development and/or apoptotic processes in T. vitrinus (Hu et al. 2007).
Characterization of the two diagnostic polypeptides of S. stercoralis L3 described in this study could be used as supporting data in the production of recombinant antigenic protein for the development of diagnostic methods. Moreover, it could potentially be used in the production of vaccine candidates or for drug target design. This is a novel study of the S. stercoralis proteins based on immunoblot analysis and a proteomic approach.
References
Atkins NS, Conway DJ, Lindo JF, Bailey JW, Bundy DA (1999) L3 antigen-specific antibody isotype responses in human strongyloidiasis: correlations with larval output. Parasite Immunol 21:517–526
Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, Hotez PJ (2006) Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet 367:1521–1532
Casavechia MT, Lonardoni MV, Venazzi EA, Campanerut-Sá PA, da Costa Benalia HR, Mattiello MF, Menechini PV, Dos Santos CA, Teixeira JJ (2016) Prevalence and predictors associated with intestinal infections by protozoa and helminths in southern Brazil. Parasitol Res. doi: 10.1007/s00436-016-4980-y
Conway DJ, Bailey JW, Lindo JF, Robinson RD, Bundy DA, Bianco AE (1993) Serum IgG reactivity with 41-, 31-, and 28-kDa larval proteins of Strongyloides stercoralis in individuals with strongyloidiasis. J Infect Dis 168:784–787
Conway DJ, Lindo JF, Robinson RD, Bundy DA, Bianco AE (1994) Strongyloides stercoralis: characterization of immunodiagnostic larval antigens. Exp Parasitol 79:99–105
Dahout-Gonzalez C, Nury H, Trézéguet V, Lauquin GJ, Pebay-Peyroula E, Brandolin G (2006) Molecular, functional, and pathological aspects of the mitochondrial ADP/ATP carrier. Physiology (Bethesda) 21:242–249
Dolce V, Scarcia P, Iacopetta D, Palmieri F (2005) A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett 579:633–637
Eamudomkarn C, Sithithaworn P, Sithithaworn J, Kaewkes S, Sripa B, Itoh M (2015) Comparative evaluation of Strongyloides ratti and S. stercoralis larval antigen for diagnosis of strongyloidiasis in an endemic area of opisthorchiasis. Parasitol Res 114:2543–2551
Elkins DB, Haswell-Elkins M, Anderson RM (1986) The epidemiology and control of intestinal helminths in the Pulicat Lake region of Southern India. I. Study design and pre- and post-treatment observations on Ascaris lumbricoides infection. Trans R Soc Trop Med Hyg 80:774–792
Harada U, Mori OA (1955) A new method for culturing hookworm. Yonago Acta Med 1:177–179
Hu M, Campbell BE, Pellegrino M, Loukas A, Beveridge I, Ranganathan S, Gasser RB (2007) Genomic characterization of Tv-ant-1, a Caenorhabditis elegans tag-61 homologue from the parasitic nematode Trichostrongylus vitrinus. Gene 397:12–25
Jaubert S, Laffaire JB, Ledger TN, Escoubas P, Amri EZ, Abad P, Rosso MN (2004) Comparative analysis of two 14-3-3 homologues and their expression pattern in the root-knot nematode Meloidogyne incognita. Int J Parasitol 34:873–880
Koga K, Kasuya S, Khamboonruang C, Sukhavat K, Ieda M, Takatsuka N, Kita K, Ohtomo H (1991) A modified agar plate method for detection of Strongyloides stercoralis. Am J Trop Med Hyg 45:518–521
Laemmli UK (1970) Cleavage of structural protein during the assembly of the head of bacteriophage T4. Nature 227:680–685
Leung WY, Hamazaki T, Ostrov DA, Terada N (2013) Identification of adenine nucleotide translocase 4 inhibitors by molecular docking. J Mol Graph Model 45:173–179
Levenhagen MA, Costa-Cruz JM (2014) Update on immunologic and molecular diagnosis of human strongyloidiasis. Acta Trop 135:33–43
Marcilla A, Sotillo J, Pérez-Garcia A, Igual-Adell R, Valero ML, Sánchez-Pino MM, Bernal D, Muñoz-Antolí C, Trelis M, Toledo R, Esteban JG (2010) Proteomic analysis of Strongyloides stercoralis L3 larvae. Parasitology 137:1577–1583
Northern C, Grove DI (1990) Strongyloides stercoralis: antigenic analysis of infective larvae and adult worms. Int J Parasitol 20:381–387
Nunes CP, Zaha A, Gottstein B, Müller N, Mdel Siles-Lucas M (2004) 14-3-3 gene characterization and description of a second 14-3-3 isoform in both Echinococcus granulosus and E. multilocularis. Parasitol Res 93:403–409
Olsen A, van Lieshout L, Marti H, Polderman T, Polman K, Steinmann P, Stothard R, Thybo S, Verweij JJ, Magnussen P (2009) Strongyloidiasis- the most neglected of the neglected tropical diseases? Trans R Soc Trop Med Hyg 103:967–972
Pan D, Bera AK, De S, Bandyopadhyay S, Das SK, Manna B, Sreevatsava V, Bhattacharya D (2010) Relative expression of the 14-3-3 gene in different morphotypes of cysts of Echinococcus granulosus isolated from the Indian buffalo. J Helminthol 84:394–397
Sato Y, Inoue F, Matsuyama R, Shiroma Y (1990) Immunoblot analysis of antibodies in human strongyloidiasis. Trans R Soc Trop Med Hyg 84:403–406
Siddiqui AA, Berk SL (2001) Diagnosis of Strongyloides stercoralis infection. Clin Infect Dis 33:1040–1047
Siles-Lucas Mdel M, Gottstein B (2003) The 14-3-3 protein: a key molecule in parasites as in other organisms. Trends Parasitol 19:575–581
Siles-Lucas M, Nunes CP, Zaha A (2001) Comparative analysis of the 14-3-3 gene and its expression in Echinococcus granulosus and Echinococcus multilocularis metacestodes. Parasitology 122:281–287
Siles-Lucas M, Uribe N, López-Abán J, Vicente B, Orfao A, Nogal-Ruiz JJ, Feliciano AS, Muro A (2007) The Schistosoma bovis Sb14-3-3zeta recombinant protein cross-protects against Schistosoma mansoni in BALB/c mice. Vaccine 25:7217–7223
Siles-Lucas M, Merli M, Gottstein B (2008) 14-3-3 proteins in Echinococcus: their role and potential as protective antigens. Exp Parasitol 119:516–523
Sudre AP, Siqueira RC, Barreto MG, Peralta RH, Macedo HW, Peralta JM (2007) Identification of a 26-kDa protein fraction as an important antigen for application in the immunodiagnosis of strongyloidiasis. Parasitol Res 101:1117–1123
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354
Uribe N, Siles-Lucas M, López-Abán J, Esteban A, Suarez L, Martínez-Fernández A, del Olmo E, Muro A (2007) The Sb14-3-3zeta recombinant protein protects against Schistosoma bovis in BALB/c mice. Vaccine 25:4533–4539
Valerio L, Roure S, Fernández-Rivas G, Basile L, Martínez-Cuevas O, Ballesteros ÁL, Ramos X, Sabrià M, North Metropolitan Working Group on Imported Diseases (2013) Strongyloides stercoralis, the hidden worm. Epidemiological and clinical characteristics of 70 cases diagnosed in the North Metropolitan Area of Barcelona, Spain, 2003-2012. Trans R Soc Trop Med Hyg 107:465–470
Acknowledgments
We wish to acknowledge the support of the English Consultation Clinic at the Khon Kaen University Faculty of Medicine Research Affairs Division and the Khon Kaen University Publication Clinic at the Research and Technology Transfer Affairs Division for their assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All procedures performed in studies involving human participants were in accordance with ethical standards of the Khon Kaen University Ethics Committee for Human Research (HE591192) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict of interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by a TRF Senior Research Scholar Grant, Thailand Research Fund grant number RTA5880001; the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, Thailand, through the Health Cluster (SHeP-GMS); the Faculty of Medicine, Khon Kaen University (grant no. TR57201) through WM and PM. RR was supported by the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (grant no. PHD/0053/2556). OS were supported by Post-Doctoral Training Program of Graduate School and Khon Kaen University (grant no. 58101).
Rights and permissions
About this article

Cite this article
Rodpai, R., Intapan, P.M., Thanchomnang, T. et al. Strongyloides stercoralis diagnostic polypeptides for human strongyloidiasis and their proteomic analysis. Parasitol Res 115, 4007–4012 (2016). https://doi.org/10.1007/s00436-016-5170-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-016-5170-7