
Abstract
Gastrointestinal nematodes within the subfamily Ostertagiinae (Teladorsagia, Ostertagia, and Marshallagia et al.) are among the most common infections of domesticated livestock. These parasites are of particular interest, as many of the species within this group are of economic importance worldwide. Traditionally, nematode species designations have been based on morphological criteria. However, this group possesses poorly defined species. There is an urgent need to develop a reliable technique that can distinguish species of Ostertagiinae. DNA barcoding has been proved to be a powerful tool to identify species of birds, mammals, and arthropods, but this technique has not yet been examined for identifying species of Ostertagiinae. In this study, a total of 138 mitochondrial DNA (mtDNA) cytochrome c oxidase subunit I (COI) sequences from individuals representing 11 species of Ostertagiinae were acquired by PCR for the first time. The specimens were collected from pastoral area of northern China. Genetic divergence analyses showed that mean interspecific Kimura two-parameter distances of COI (13.61 %) were about four times higher than the mean value of the intraspecific divergence (3.69 %). Then, the performance of the COI to identify species of Ostertagiinae was evaluated by identification success rates using nearest neighbor (NN) and BLASTn. The results indicated that the rates of correct sequence identification for COI were high (>80 %) when using the NN and BLASTn methods. Besides, the deep lineage divergences are detected in Teladorsagia circumcincta. Meanwhile, the analyses also detected no genetic differentiation between some species such as Ostertagia hahurica and Ostertagia buriatica. These results indicate that the traditional status of species within Ostertagiinae should be closely examined based on the molecular data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ostertagiinae is an important subfamily within the gastrointestinal nematodes (Gibbons and Khalil 1982; Goldfinch et al. 2008; Lancaster and Hong 1981). Ostertagiinae includes Teladorsagia, Ostertagia, and Marshallagia et al. and poses great threats to domesticated livestock (Herd 1988; Lichtenfels and Hoberg 1993; Riggio et al. 2014). Accurate and rapid identification of the species of Ostertagiinae is of fundamental importance to basic biological and epidemiological studies of parasites as well as for their diagnosis, treatment, and control (Blouin et al. 1999; Criscione et al. 2005; Nejsum et al. 2005). Traditionally, most identification focuses on morphological criteria, including the spicules and genital cone of the male tail, the spicules, and gubernacula et al. (Borji et al. 2011; Drożdż 1995; Wu et al. 2001). However, it is often unclear as to whether particular morphological features reflect genuine species differences or are simply morphological variations within the same species (Drożdż 1995; Grillo et al. 2008; Zarlenga et al. 1998). In addition, these criteria tend to be limited to the identification of adult stages, whereas identification of eggs and larvae, found in the more readily available source of material, e.g., feces, can be time-consuming, requires highly trained specialists, and, in some cases, is unreliable and limited to family level (Derycke et al. 2008; Wijova et al. 2005). The limitations of morphological identification have led to a “taxonomy crisis,” and a more integrative approach to taxonomy has been proposed for species delineation (Blaxter 2003; Godfray 2002).
With the rapid development of molecular biology in the 1990s, an increasing number of scientists applied PCR and DNA sequencing technologies to the identification and classification of species (Lee et al. 1993; Snounou et al. 1993). Among these technologies, DNA barcoding is a molecular approach that has gained wide attention (Hebert and Gregory 2005; Ratnasingham and Hebert 2007). This technology aimed to use the sequence of a single piece of mitochondrial DNA (mtDNA) (part of the cytochrome c oxidase subunit I gene (COI)) for the rapid, accurate, and automated identification of most animal species (Folmer et al. 1994; Schindel and Miller 2005). However, rather, few studies based on COI have focused on the identification of Ostertagiinae.
In this study, firstly, the COI sequences of 14 species of Ostertagiinae were recovered by the nematode cocktail primers C_NemF1_t1/C_NemR1_t1 (Prosser et al. 2013) and redesigned primers COI-OF/COI-OR. Then, the performance of COI for identifying species of Ostertagiinae was tested. Genetic divergence of COI was determined using six parameters (average interspecific distance, theta prime, the smallest interspecific distance, average intraspecific distance, theta, and coalescent depth). Finally, species identification efficiencies of COI for Ostertagiinae were analyzed following two methods: the nearest neighbor (NN) and BLASTn.
Materials and methods
Taxon sampling
The 165 specimens of Ostertagiinae used in this study were collected from different areas in northern China. The samples from Inner Mongolia Autonomous Region were collected from the abomasum of sheep on farms in the suburb of Hohhot. The samples from Xinjiang Autonomous Region were collected from the abomasum of sheep and cattle on farms in the suburb of Kashi, Yili, and Urumqiet et al. And, the samples from Qinghai province were collected from the abomasum of sheep on farms in the suburb of Xining.
The species of specimens were identified by the specialist using a combination of morphological data. All specimens were preserved in 100 % ethanol.
DNA extraction and PCR
Ethanol-preserved specimens of Ostertagiinae were rinsed in distilled water, and then, total DNA was extracted using a DNeasy blood and tissue kit (Qiagen GmbH, Hilden, Germany) following the manufacturer’s protocol.
The fragments of the COI genes for analyses were amplified by PCR assay using primer cocktail primers (C_NemF1_t1+C_NemR1_t1) for the COI from vertebrate parasitic nematodes and the new designed primers COI-OF/COI-OR (Table S1).
Each 50-μl PCR solution contained 25 μl of 2× PCR Buffer for KOD FX Neo (1.75-mM final concentration of MgCl2), 10 μl of 2 mM dNTPs, 3 μl of primer mix (0.3-μM final concentration of each primer), 1 μl KOD FX Neo polymerase (1 unit), 2 μl DNA template (about 200 ng genomic DNA), and distilled water. PCR assays were conducted using a GeneAmp PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA, USA). The DNA size marker DL2000 (Takara, Dalian, China) was used to estimate the length of the PCR amplicons.
COI was amplified with COI-OF/COI-OR using a PCR protocol as follows: initial denaturation (94 °C, 5 min); followed by 36 cycles of 94 °C for 30 s, 45 °C for 30 s, and 68 °C for 1 min; followed by a final extension step of 68 °C for 7 min.
COI was amplified with nematode cocktail primers (C_NemF1_t1+C_NemR1_t1) using a PCR protocol as previously described (Prosser et al. 2013).
Data acquisition and sequence alignment
For the field-collected specimens, DNA was amplified and sequences were obtained for the COI gene. The PCR amplicons were sequenced at BGI Tech Inc. (Beijing, China). One hundred thirty-eight COI sequences of specimens were deposited in GenBank under accession numbers KT807578∼KT807715. Two COI numt sequences were deposited in GenBank under accession numbers KT807716 and KT807717.
Amplification success rate was measured based on the proportion of samples that produced sequences of the appropriate length. These sequences were further evaluated for their utility in species identification. Another 37 COI sequences representing five Ostertagiinae species were downloaded from GenBank (http://www.ncbi.nlm.nih.gov/GenBank/).
DNA sequences were assembled and edited in MEGA 5.0 (Tamura 2011). Sequences were aligned using ClustalW (Thompson et al. 1994) in MEGA 5.0 with default parameters (open gap penalty = 10.0, extend gap penalty = 5.0). Analyses were conducted with the sequences of COI that constituted different data sets for each genus.
Genetic divergence analysis through six parameters
Genetic distances were calculated using the Kimura two-parameter (K2P) distance method (Kimura 1980) as implemented in MEGA 5.0. Three parameters were used to characterize interspecific divergence (Chen et al. 2010): (1) average interspecific distance (K2P distance) between all species in each genus with at least two species; (2) average theta prime, where theta prime is the mean pairwise distance within each genus with more than one species, thus eliminating biases associated with different numbers of species among genera; and (3) smallest interspecific distance, i.e., the minimum interspecific distance within each genus with at least two species.
In addition, three parameters were used to determine intraspecific variation (Chen et al. 2010): (1) average intraspecific difference (K2P) between all samples collected within each species with more than one individual; (2) theta, where theta is the mean pairwise distance within each species with at least two representatives; theta eliminates biases associated with unequal sampling among species; and (3) average coalescent depth, which is the maximum intraspecific distance within each species with at least two individuals.
Bayesian analyses
For COI, the Bayesian analyses were performed with MrBayes 3.2 (Ronquist et al. 2012), with the data partitioned into three sets by codon position. Models for each partition were selected using MrModeltest 2.2 (Nylander 2005); these were determined to be GTR+I+G for codon positions 1, GTR+I for codon position 2, and TrN+G for codon position 3. For 16S rDNA and ITS2, the model was determined to be GTR+I+G. For each Bayesian analysis, two runs were performed simultaneously, each with four Markov chains (one cold, three heated) which ran for 1,000,000 generations. The first 250,000 generations were discarded from the analysis (burnin), and every 1000th tree was sampled to calculate a 50 % majority-rule consensus tree with posterior probabilities for nodes.
Data sets utilized in this study
The data set 1 includes 138 sequences acquired from the 138 collected specimens of Ostertagiinae and 37 sequences from GenBank. Finally, data set 1 consists of 175 COI sequences from 15 species (Table 1). Besides, data set 2 consists of 171 COI sequences from 11 species with multiple accessions (irrespective of congeneric species).
Methods utilized for species identification
Nearest neighbor
Genetic distances were calculated based on the K2P model for all pairwise comparisons in the matrix using MEGA 5.0. In the analysis, the query sequence was assigned to the species of the sequence in the reference database which has the smallest genetic distance from the query sequence. In case NNs were from more than one species, the query’s identification was considered uncertain.
BLASTn
Identification based on BLAST was performed using NCBI software version 2.2.28+ (Camacho et al. 2009). Up to 100 hits with at least 80 % identity were returned for each query, which was identified as the species associated with its best hit (highest bit score). In case more than one species were associated, the query’s identification was considered uncertain.
Sequence identification success and species identification success
We assessed relative performance of COI in terms of their identification success with data sets of ticks. Identification success was defined in two ways: (1) “Species identification success” was scored as the number of species for which all query sequences were correctly identified and (2) “sequence identification success” was scored as the number of correctly identified query sequences per data set, which is equivalent to sensitivity (i.e., true positives / [true positives + false negatives]).
Results
Cytochrome c oxidase subunit I amplification and detection of numts
A previous study has described nematode cocktail primers (C_NemF1_t1+C_NemR1_t1) (Prosser et al. 2013), as well as the universal DNA barcoding primers LCO1490/HCO2198 (Folmer et al. 1994), targeting the standard DNA barcode. Firstly, C_NemF1_t1/C_NemR1_t1 was utilized to amplify the 5′ region of COI from 165 specimens of Ostertagiinae, and 111 sequences were acquired. Then, two putative numt (nuclear mitochondrial pseudogene) sequences were found when two fragments from Marshallagia mongolica (380#, 491#) were translated into amino acid alignment, and finally, 109 COI sequences of Ostertagiinae were acquired. When compared with the COI sequences from other M. mongolica (504#, 490#), the numt sequences had codon deletion (two nucleotides disappeared) and a high number of nucleotide substitutions (61/525, 11.6 %).
To improve the efficiency of amplifying COI from Ostertagiinae, the new primers (COI-OF/COI-OR) were designed in GC-rich regions of high intersequence similarity of amplified Ostertagiinae COI sequences, and another 29 COI sequences of Ostertagiinae were acquired (the mtCOIs of 380# and 491# were included). Finally, 138 COI sequences were amplified from 165 Ostertagiinae specimens and the success rate of COI recovery was 83.6 %. More importantly, the problem of numt was avoided. The amplified product by C_NemF1_t1/C_NemR1_t1 was approximately 675 bp while the amplified product by COI-OF/COI-OR was 730 bp. The overlapped segment was about 529 nucleotides, and these DNA sequences were considered for next analysis.
Genetic divergence
Besides the 138 COI sequences representing 11 Ostertagiinae species, another 37 COI sequences representing five Ostertagiinae species in the GenBank were retrieved to our data set. Finally, we constructed a data set consisted of 175 COI sequences representing 15 Ostertagiinae species. A mean of approximately 12 individuals (range 1–32) represented each species, with four species (Teladorsagia trifurcata, Ostertagia wuhingensis, Ostertagia lyrata, Ostertagia gruehneri) represented by a single specimen.
The genetic divergence within and between the species was calculated for this data set. The average intraspecific distance, theta, and coalescent depth were calculated to determine intraspecific variation using a K2P distance matrix. Average interspecific distance, theta prime, and smallest interspecific distances were used to characterize interspecific divergence.
The within-species K2P values based on mtCOI ranged from 0 to 14.2 %, with more than 88 % of values below 6 % (Fig. 1a). Meanwhile, more than 95 % pairwise comparisons among species were distributed from 8 to 22 % K2P (Fig. 1b). Average interspecific K2P (13.61 %) was about four times higher than the average value of the intraspecific K2P (3.69 %). The coalescent depth was about 5.81 % while the smallest interspecific distance was 12.65 % (Table 2). And, there is an overlap between within-species and among-species K2P of COI. These data indicate that COI has the potential as a DNA barcoding locus for Ostertagiinae species, but perhaps, it is not efficient in discriminating some species of Ostertagiinae.

Frequency distributions of K2P distances of cytochrome C oxidase subunit I (COI) within and among species of Ostertagiinae. a Pairwise distance comparisons of the COI within species of Ostertagiinae. b Pairwise distance comparisons of the COI among species of Ostertagiinae
Phylogenetic analyses
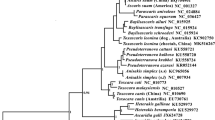
The rate of monophyletic groups in phylogenetic trees is often appropriate to evaluate the discriminatory power of a DNA barcode locus (Maia et al. 2012; Theodoridis et al. 2012; Zhang et al. 2012). In this study, two different phylogenetic methods were used: the neighbor-joining (NJ) and the Bayesian inference (BI) analyses. The NJ tree (Fig. 2) showed that the 175 sequences could be clustered into 12 monophyletic groups. And, the six species identified using morphology, with more than one individual, were obtained as monophyletic lineages with high supports. Besides, the three sequences corresponded to singletons (O. gruehneri, T. trifurcata, and O. wuhingensis) were also flagged as potentially unique in the NJ trees. More notable is that three monophyletic groups separately constitute two species such as Ostertagia dahurica/Ostertagia buriatica, M. mongolica/Marshallagia grossopiculum, and Ostertagia ostertagi/O. lyrata. Obviously, in the NJ tree based on the COI, it is found that some species were not clearly differentiated from other species. The BI analysis yielded a result nearly similar to that of the NJ analysis (Fig. 3). This situation perhaps sleds to the failure of species identification.

Phylogeny of Ostertagiinae species resulting from neighbor-joining analysis of COI. Numbers on branches correspond to bootstrap support (1000 replicates).  , Teladorsagia circumcincta;
, Teladorsagia circumcincta;  , Ostertagia gruehneri;
, Ostertagia gruehneri;  , Teladorsagia trifurcata;
, Teladorsagia trifurcata;  , Teladorsagia davtiani;
, Teladorsagia davtiani;  , Marshallagia mongolica;
, Marshallagia mongolica;  , Marshallagia grossopiculum;
, Marshallagia grossopiculum;  , Ostertagia lanceata;
, Ostertagia lanceata;  , Ostertagia ostertagi;
, Ostertagia ostertagi;  , Ostertagia lyrata;
, Ostertagia lyrata;  , Ostertagia wuhingensis;
, Ostertagia wuhingensis;  , Ostertagia xizangensis;
, Ostertagia xizangensis;  , Ostertagia occidentalis;
, Ostertagia occidentalis;  , Marshallagia hsui;
, Marshallagia hsui;  , Ostertagia dahurica;
, Ostertagia dahurica;  , Ostertagia buriatica;
, Ostertagia buriatica;  , Anisakis simplex
, Anisakis simplex

Phylogeny of Ostertagiinae species resulting from Bayesian analysis of COI. Numbers on branches are posterior probabilities
Performance of cytochrome c oxidase subunit I in species identification of Ostertagiinae
The performance of COI was evaluated by the rates of “the sequence identification success.” In these analyses, the data set 2 was utilized as the query data sets and the data set 1 was taken as the reference data set. The reliability of several different methods of species identification (BLASTn, NN, and tree-based methods) had been tested via simulated or empirical data sets in a previous study (Lv et al. 2014; Ross et al. 2008). And, the results showed that BLASTn and distance methods were equally successful in species identification of ticks. In this study, we followed these two approaches.
The fragment of COI behaves well in the NN and BLASTn approaches, and the rates of the sequence identification success were separately 87.13 % (149/171) and 88.89 % (152/171) (Table 3). For the rates of species identification success, COI behaves similar in the NN and BLASTn approaches: 63.6 % (7/11).
Discussion
The recovery of cytochrome c oxidase subunit I from Ostertagiinae faces two major obstacles
As the standard DNA barcoding marker, the 5′ region of COI has been explored as a potential marker to separate some nematodes into proper species (Derycke et al. 2010; Elsasser et al. 2009; Floyd et al. 2002). However, according to the BOLD system (www.boldsystems.org) (Ratnasingham and Hebert 2007), only two species of Ostertagiinae (O. ostertagi and Teladorsagia circumcincta) had been previously barcoded. To accurately barcode Ostertagiinae, it is important to acquire the mtCOI sequences and construct reliable data sets. In this study, the 11 Ostertagiinae species of northern China were DNA barcoded. But, there are two obstacles to the recovery of COI from Ostertagiinae: the low PCR success rate and the presence of COI numts.
It has been demonstrated that the barcode region of COI has delivered species-level resolution in certain nematode lineages (Derycke et al. 2010), but sequence recovery has proven difficult (De Ley et al. 2005). In this study, the primer cocktail C_NemF1_t1/C_NemR1_t1 (Prosser et al. 2013) was utilized to amplify the 5′ region of COI from 165 samples of Ostertagiinae and only 109 mtCOIs were acquired. The amplification efficiency was 66.1 %. Obviously, the advantage of this primer cocktail is across a broad range of nematodes including members of three orders and eight families but not the high PCR success rate for Ostertagiinae. Then, another 29 mtCOIs were amplified by the new designed primer (COI-OF/COI-OR) though the full-length barcodes (661 bp) were not recovered. It has been inferred that the nematode may be the most species-rich phylum of animals, and the nematode mitochondrial genomes are highly diverse (Hugot et al. 2001). As the PCR success rate was low, we inferred that there are great diversities in the primer binding sites on the mitochondrial genomes of Ostertagiinae. To overcome this limitation, the new primer pair which could efficiently recover the full-length barcodes from Ostertagiinae should be designed.
It has been demonstrated that the presence of COI numts makes the goal difficult to achieve when numts are prevalent and can introduce serious ambiguity into DNA barcoding (Hojun et al. 2008). Until now, it has been proved that >82 eukaryotes, such as Insecta, Mammalia et al., have numts (Bensasson et al. 2001). Though the genome of many nematodes has been determined to have numts, the cases of numts in Ostertagiinae have never been reported before. In our study, the numts in Marshallagia are determined because of the indels within sequences. Obviously, compared to the mtCOI, the numts are more likely to be amplified from Marshallagia by the universal primers. There are two reasons to explain this phenomenon. The first reason is that the number of copies of numt is far more than the mtCOI. The second reason is perhaps that there are sufficient sequence divergences between numt and orthologous mtCOI and conserved primer is more likely to bind numt but not mtCOI.
The genetic characters of numt
Our study showed that the K2P genetic distances between the numts and orthologous mtCOI are over 7 % and a high number of nucleotide substitutions was found (61/525, 11.6 %). But, the number of amino acid substitution was 10/175 (5.7 %), which is less than the nucleotide substitutions. Furthermore, the NJ phylogenetic analyses recovered a large clade of the mtCOI from Ostertagiinae and a small clade of numts from Ostertagiinae (Fig. S1). Then, two clades formed a larger polytomous clade. Based on the above results, it is inferred that if the numts were used in DNA barcoding, the 380# and 491# would be classified to an inexistent species but not M. mongolica and the number of species in Ostertagiinae would be overestimated.
Moreover, there are totally 19 specimens of M. mongolica, and only two numts were amplified while the mtCOI of other 17 specimens could be amplified by universal primer directly. So, it is concluded that the numt is relatively uncommon and limited to a small number of individuals of M. mongolica. And, 380# and 491# should be more close to each other compared with other specimens of M. mongolica, as both of them have similar numts in the genome.
The results support the utility of DNA barcoding for identification of Ostertagiinae
The genetic divergence analyses showed that there is an overlap between within-species and among-species K2P distances of COI from Ostertagiinae. Moreover, the among-species K2P distances ranged from 0 to 14.2 % with the average value as 3.69 %. This indicates that there are deep intraspecific sequence divergences within Ostertagiinae, and this may hinder the effort to species identification of Ostertagiinae using DNA barcodes. In this study, liberal tree-based methods, BLASTn, and NN were examined in species identification of Ostertagiinae using COI.
The NJ phylogenetic analyses (Fig. 2) showed 12 phylogenetic clades, and most of these clades were highly bootstrap supported (>80 %). The NJ tree only recovered monophyly for nine of 14 species with three species represented by just one individual. The Bayesian analyses (Fig. 3) were basically consistent with the NJ analyses and recovered 13 phylogenetic clades. The sequences of T. circumcincta were divided into two phylogenetic clades in the Bayesian analyses, while the two T. circumcincta groups only formed two lineages within the monophyly clade in the NJ analyses. It should be noticed that the monophyletic clade of T. circumcincta in NJ analyses was weakly supported (<50 %). Furthermore, the mean genetic divergences (K2P) between the two T. circumcincta groups reach to 12.1 %, which indicates that T. circumcincta is perhaps species complex. Based on the microsatellite genotyping, a previous study supported the hypothesis that Teladorsagia davtiani and T. trifurcata are morphotypes of T. circumcincta (Grillo et al. 2008). In the NJ analyses, the T. circumcincta, T. davtiani, T. trifurcata, and another species O. gruehneri formed one great phylogenetic clade with very low bootstrap supported (<10 %). In the Bayesian analyses, the four species formed three different phylogenetic clades which separated from each other. Nowadays, molecular genetic tools are increasing using alongside classical taxonomy for species delimitation and the concept of molecular operational taxonomic units (MOTUs) has been utilized to taxonomic surveys of some special biological group with confused species designations. Based on the theory of MOTUs, T. davtiani, T. trifurcata, and T. circumcincta should be treated as three separately species, which is inconsistent with conclusion based on the microsatellite genotyping. We considered that the taxonomic status of the three species should be further examined based on more molecular data.
Our previous data has shown that the NN and BLASTn methods have advantage over the tree-based methods in species identification of ticks (Lv et al. 2014). In this study, the NN and BLASTn methods both performed well in the species identification of Ostertagiinae and the rates of the sequence identification success were separately 87.13 and 88.89 %. The incorrectly identified sequences belong to four species: M. grossopiculum, M. mongolica, O. buriatica, and O. dahurica. Further study showed that two species (M. grossopiculum and M. mongolica) revealed shallow interspecific sequence divergence (<1 %) as well as other two species (O. buriatica and O. dahurica). It is inferred that two reasons could explain this phenomenon. Firstly, the Ostertagiinae is an important subfamily within nematode in which species designations are particularly confused and remain to be completely defined (Borji et al. 2011; Grillo et al. 2008; Leignel and Cabaret 2001). So, perhaps, M. grossopiculum and M. mongolica are simply morphological variations within the same species. Secondly, a large number of bacterial symbionts in the nematodes have been confirmed (Casiraghi et al. 2004; Shapiro-Ilan and Gaugler 2002) and it is inferred that the species of Ostertagiinae also have multiple bacterial symbionts. Previous studies showed that the symbionts can break through the species barrier by hybridization followed by selective sweep, resulting in identical mtDNA sequences among different species (Hurst and Jiggins 2005; Whitworth et al. 2007), which would cause the underestimation of the number of unique species under barcoding.
Conclusions
As the standard DNA barcode, COI is efficient in species identification of Ostertagiinae especially when the methods BLASTn and NN were utilized. Besides, traditional status of species within Ostertagiinae, which was mainly determined by morphology, could be closely examined based on DNA barcoding data. But, nowadays, there are still several problems to be resolved in barcoding Ostertagiinae, such as the difficulty to recover mtCOI from Ostertagiinae and confused species designations within Ostertagiinae.
References
Bensasson D, Zhang DX, Hartl DL, Hewitt GM (2001) Mitochondrial pseudogenes: evolution’s misplaced witnesses. Trends Ecol Evol 16:314–321
Blaxter M (2003) Molecular systematics: counting angels with DNA. Nature 421:122–124. doi:10.1038/421122a
Blouin MS, Liu J, Berry RE (1999) Life cycle variation and the genetic structure of nematode populations. Heredity 83:253–259
Borji H, Raji AR, Naghibi AG (2011) The comparative morphology of Marshallagia marshalli and Ostertagia occidentalis (Nematoda: Strongylida, Trichostrongylidae) by scanning electron microscopy. Parasitol Res 108:1391–1395
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi:10.1186/1471-2105-10-421
Casiraghi M et al (2004) Mapping the presence of Wolbachia pipientis on the phylogeny of filarial nematodes: evidence for symbiont loss during evolution. Int J Parasitol 34:191–203
Chen S et al (2010) Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 5, e8613. doi:10.1371/journal.pone.0008613
Criscione CD, Poulin R, Blouin MS (2005) Molecular ecology of parasites: elucidating ecological and microevolutionary processes. Mol Ecol 14:2247–2257. doi:10.1111/j.1365-294X.2005.02587.x
De Ley P et al (2005) An integrated approach to fast and informative morphological vouchering of nematodes for applications in molecular barcoding. Philos T Roy Soc B 360:1945–1958
Derycke S, Fonseca G, Vierstraete A, Vanfleteren J, Vincx M, Moens T (2008) Disentangling taxonomy within the Rhabditis (Pellioditis) marina (Nematoda, Rhabditidae) species complex using molecular and morphological tools. Zool J Linn Soc-lond 152:1–15
Derycke S, Vanaverbeke J, Rigaux A, Backeljau T, Moens T (2010) Exploring the use of cytochrome oxidase c subunit 1 (COI) for DNA barcoding of free-living marine nematodes. PLoS One 5, e13716. doi:10.1371/journal.pone.0013716
Drożdż J (1995) Polymorphism in the Ostertagiinae Lopez-Neyra, 1947 and comments on the systematics of these nematodes. Syst Parasitol 32:91–99
Elsasser SC, Floyd R, Hebert PD, Schulte-Hostedde AI (2009) Species identification of North American guinea worms (Nematoda: Dracunculus) with DNA barcoding. Mol Ecol Resour 9:707–712. doi:10.1111/j.1755-0998.2008.02393.x
Floyd R, Abebe E, Papert A, Blaxter M (2002) Molecular barcodes for soil nematode identification. Mol Ecol 11:839–850
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3:294–299
Gibbons LM, Khalil LF (1982) A key for the identification of genera of the nematode family Trichostrongylidae Leiper, 1912. J Helminthol 56:185–233
Godfray HC (2002) Challenges for taxonomy. Nature 417:17–19. doi:10.1038/417017a
Goldfinch GM, Smith WD, Imrie L, Mclean K, Inglis NF, Pemberton AD (2008) The proteome of gastric lymph in normal and nematode infected sheep. Proteomics 8:1909–1918
Grillo V, Craig BH, Wimmer B, Gilleard JS (2008) Microsatellite genotyping supports the hypothesis that Teladorsagia davtiani and Teladorsagia trifurcata are morphotypes of Teladorsagia circumcincta. Mol Biochem Parasit 159:59–63. doi:10.1016/j.molbiopara.2008.01.001
Hebert PD, Gregory TR (2005) The promise of DNA barcoding for taxonomy. Syst Biol 54:852–859. doi:10.1080/10635150500354886
Herd RP (1988) Control strategies for ostertagiasis. Vet Parasitol 27:111–123
Hojun S, Buhay JE, Whiting MF, Crandall KA (2008) Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc Natl Acad Sci USA 105:13486–13491
Hugot JP, Morand S, Baujard P (2001) Biodiversity in helminths and nematodes as a field of study: an overview. Nematology 3:199–208
Hurst GD, Jiggins FM (2005) Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc Biol Sci 272:1525–1534. doi:10.1098/rspb.2005.3056
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Lancaster MB, Hong C (1981) Polymorphism in nematodes. Syst Parasitol 3:29–31
Lee IM, Hammond RW, Davis RE, Gundersen DE (1993) Universal amplification and analysis of pathogen 16S rDNA for classification and identification of mycoplasmalike organisms. Phytopathology 83:834–842
Leignel V, Cabaret J (2001) Are Teladorsagia circumcincta (Nematoda) morphs equally able to survive under anthelmintic treatment in sheep on pastures? Parasitol Res 87:687–692
Lichtenfels JR, Hoberg EP (1993) The systematics of nematodes that cause ostertagiasis in domestic and wild ruminants in North America: an update and a key to species. Vet Parasitol 46:33–53
Lv J et al (2014) Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasite Vector 7:93. doi:10.1186/1756-3305-7-93
Maia VH, Mata CS, Franco LO, Cardoso MA, Cardoso SR, Hemerly AS, Ferreira PC (2012) DNA barcoding Bromeliaceae: achievements and pitfalls. PLoS One 7, e29877. doi:10.1371/journal.pone.0029877
Nejsum P, Frydenberg J, Roepstorff A, Parker ED Jr (2005) Population structure in Ascaris suum (Nematoda) among domestic swine in Denmark as measured by whole genome DNA fingerprinting. Hereditas 142:7–14. doi:10.1111/j.1601-5223.2005.01864.x
Nylander JAA (2005) MrModeltest v2. Computer program distributed by the author. Evolutionary Biology Centre, Uppsala University, Uppsala
Prosser SWJ, Velarde‐Aguilar MG, Hebert PDN (2013) Advancing nematode barcoding: a primer cocktail for the cytochrome c oxidase subunit I gene from vertebrate parasitic nematodes. Mol Ecol Resour 13:1108–1115
Ratnasingham S, Hebert PD (2007) Bold: the barcode of life data system (http://www.barcodinglife.org). Mol Ecol Notes 7:355–364 doi:10.1111/j.1471-8286.2007.01678.x
Riggio V et al (2014) A joint analysis to identify loci underlying variation in nematode resistance in three European sheep populations. J Anim Breed Genet 131:426–436. doi:10.1111/jbg.12071
Ronquist F et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. doi:10.1093/sysbio/sys029
Ross HA, Murugan S, Li WL (2008) Testing the reliability of genetic methods of species identification via simulation. Syst Biol 57:216–230. doi:10.1080/10635150802032990
Schindel DE, Miller SE (2005) DNA barcoding a useful tool for taxonomists. Nature 435:17. doi:10.1038/435017b
Shapiro-Ilan DI, Gaugler R (2002) Production technology for entomopathogenic nematodes and their bacterial symbionts. J Ind Microbiol Biot 28:137–146
Snounou G, Viriyakosol S, Jarra W, Thaithong S, Brown KN (1993) Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasit 58:283–292
Tamura K (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28
Theodoridis S, Stefanaki A, Tezcan M, Aki C, Kokkini S, Vlachonasios KE (2012) DNA barcoding in native plants of the Labiatae (Lamiaceae) family from Chios Island (Greece) and the adjacent Cesme-Karaburun Peninsula (Turkey). Mol Ecol Resour 12:620–633. doi:10.1111/j.1755-0998.2012.03129.x
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Whitworth TL, Dawson RD, Magalon H, Baudry E (2007) DNA barcoding cannot reliably identify species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Pro Biol Sci 274:1731–1739. doi:10.1098/rspb.2007.0062
Wijova M, Moravec F, Horak A, Modry D, Lukes J (2005) Phylogenetic position of Dracunculus medinensis and some related nematodes inferred from 18S rRNA. Parasitol Res 96:133–135. doi:10.1007/s00436-005-1330-x
Wu S, Zhang L, Wang F (2001) Fauna Sinica: Nematoda Rhabditida: Strongylata (I). Science Press, Beijing
Zarlenga DS, Hoberg EP, Stringfellow F, Lichtenfels JR (1998) Comparisons of two polymorphic species of Ostertagia and phylogenetic relationships within the Ostertagiinae (Nematoda: Trichostrongyloidea) inferred from ribosomal DNA repeat and mitochondrial DNA sequences. J Parasitol 84:806–812
Zhang CY, Wang FY, Yan HF, Hao G, Hu CM, Ge XJ (2012) Testing DNA barcoding in closely related groups of Lysimachia L. (Myrsinaceae). Mol Ecol Resour 12:98–108. doi:10.1111/j.1755-0998.2011.03076.x
Acknowledgments
We thank Dr. Zhang Luping (Hebei Normal University) and Dr. Yang Xiaoye (Inner Mongolia Agricultural University) for their kind assistance in morphological identification of Ostertagiinae specimens. We also thank Dr. Ji Xincheng (Xinjiang Entry-Exit Inspection and Quarantine Bureau) and Dr. Zhao Qing (Qinghai Entry-Exit Inspection and Quarantine Bureau) for their kind assistance in collection of Ostertagiinae specimens. This research was supported by the National Science and Technology Support Programs of the Ministry of Science and Technology of China (Grant Nos: 2012BAK11B04, 2013BAD12B01).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Shaoqiang Wu and Xiangmei Lin contributed equally to this work.
Rights and permissions
About this article

Cite this article
Lv, J., Zhang, Y., Feng, C. et al. Species discrimination in the subfamily Ostertagiinae of Northern China: assessment of DNA barcode in a taxonomically challenging group. Parasitol Res 115, 987–996 (2016). https://doi.org/10.1007/s00436-015-4826-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-015-4826-z