
Abstract
The protozoan parasite Trypanosoma cruzi has a complex life cycle characterized by intracellular and extracellular forms alternating between invertebrate and mammals. To cope with these changing environments, T. cruzi undergoes rapid changes in gene expression, which are achieved essentially at the posttranscriptional level. At present, expanding families of small RNAs are recognized as key players in novel forms of posttranscriptional gene regulation in most eukaryotes. However, T. cruzi lacks canonical small RNA pathways. In a recent work, we reported the presence of alternate small RNA pathways in T. cruzi mainly represented by a homogeneous population of tRNA-derived small RNAs (tsRNAs). In T. cruzi epimastigotes submitted to nutrient starvation, tsRNAs colocalized with an argonaute protein distinctive of trypanosomatids (TcPIWI-tryp) and were recruited to particular cytoplasmic granules. Using epifluorescence and electronic microscopy, we observed that tsRNAs and the TcPIWI-tryp protein were recruited mainly to reservosomes and other intracellular vesicles including endosome-like vesicles and vesicular structures resembling the Golgi complex. These data suggested that, in T. cruzi, tsRNA biogenesis is probably part of endocytic/exocytic routes. We also demonstrated that epimastigotes submitted to nutrient starvation shed high levels of vesicles to the extracellular medium, which carry small tRNAs and TcPIWI-tryp proteins as cargo. At least a fraction of extracellular vesicle cargo was transferred between parasites and to mammalian susceptible cells. Our data afford experimental evidence, indicating that extracellular vesicles shed by T. cruzi promote not only life cycle transition of epimastigotes to trypomastigote forms but also infection susceptibility of mammalian cells
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trypanosoma cruzi, which is responsible of Chagas’ disease, has a complex life cycle including intracellular and extracellular forms, which alternate between invertebrate insect vectors belonging to the subfamily Triatominae and mammalian hosts including humans (Barrett et al. 2003; de Souza 2008). Life cycle transitions start when the noninfective epimastigote forms proliferate in the midgut of the reduvii bug and advance to the hind gut of the vector. Under these circumstances, epimastigotes suffer a severe nutritional stress, which drives its differentiation into the mammalian-infective metacyclic trypomastigotes (Contreras et al. 1985; Figueiredo et al. 2000). To cope with these changing environments, T. cruzi must undergo rapid and significant changes in gene expression, which are achieved essentially at the posttranscriptional level through modulation of messenger RNA (mRNA) stability and translational control mechanisms (Clayton and Shapira 2007). Nevertheless, the precise mechanisms of gene expression regulation in these parasites remain to be completely elucidated.
Over the last decade, an expanding family of small regulators RNAs (i.e., microRNAs, small interfering RNAs, and Piwi-interacting RNAs) were recognized as key players in novel forms of posttranscriptional gene regulation in most eukaryotes (Ghildiyal and Zamore 2009). Soon after its initial description, the presence and relevance of small RNA pathways in the control of posttranscriptional gene regulation were reported for virus, bacteria, and most eukaryotes. However, the machinery associated with small RNA biogenesis was thought to be either entirely lost or extensively simplified in some unicellular organisms, including T. cruzi, Saccharomyces cerevisiae, Leishmania major, and Plasmodium falciparum (Cerutti and Casas-Mollano 2006).
In a recent work aimed to identify the presence of alternate small RNA pathways that could contribute to the posttranscriptional control in T. cruzi, we reported the presence of a homogeneous population of small RNAs derived from mature transfer RNAs (tRNAs) representing from 25 to 30 % of the small RNA population. The levels of these tRNA-derived halves (tsRNAs) were more pronounced in parasites undergoing nutritional stress and more than 98 % of cloned tsRNAs derived from the 5′ halves of a restricted group of tRNA issoaceptors (Garcia-Silva et al. 2010a). Surprisingly, tsRNAs were recruited to particular cytoplasmic organelles in T. cruzi epimastigotes, which colocalized at least partially with reservosomes. In trypomastigotes, tsRNAs represented about 62 % of small RNAs and were mainly derived from the 3′ arm of precursors tRNAs (Reifur et al. 2012). Of note, small RNAs derived from tRNAs have been recently reported by several groups as a novel population of small regulators RNAs (Cole et al. 2009; Haussecker et al. 2010; Thompson and Parker 2009b), which appear as the most broadly conserved pathway of small RNAs regulating gene expression and frequently initiated after nutritional, biological, or physicochemical stress in prokaryotes as well as in eukaryotes.
The present work was aimed to characterize the cytoplasmic granular structures recruiting tRNA fragments and to gain insights in the biological significance of this small RNA population in the protozoan parasite T. cruzi.
Here, we report that tsRNAs were actively produced in epimastigotes undergoing nutritional stress and recruited to cytoplasmic organelles associated with clathrin and with the T. cruzi argonaute protein TcPIWI-tryp (Garcia-Silva et al. 2010b). Transmission electron microscopy further confirmed that tsRNAs colocalized with TcPIWI-tryp in reservosomes, uncharacterized intracellular vesicles, and Golgi-like vesicular structures. Vesicles carrying small tRNAs and TcPIWI-tryp proteins were secreted to the extracellular medium and acted as vehicles for the transfer of these molecules to other parasites and to mammalian susceptible cells but not to nonsusceptible cells. Our data provide experimental evidence indicating that vesicles shed by T. cruzi were not only associated with life cycle transition of epimastigotes toward the infective trypomastigote forms but associated to infection susceptibility of mammalian cells.
Materials and methods
Parasites and infection assays
Epimastigote forms of the T. cruzi Dm 28c clone (Contreras et al. 1988) were cultured and expanded axenically at 28 °C by weekly transfers in liver infusion tryptose (LIT) medium supplemented with 10 % heat-inactivated fetal bovine serum. Parasites at the late-stationary phase of growth (1 × 109 parasites ml−1) were washed twice and resuspended in serum free or 1 % fetal bovine serum (FBS) RPMI-1640 medium at 5.107 × ml−1 and further cultured for 24 or 48 h. Parasite viability was monitored by light microscopy through the analysis of motility and by flow cytometry with propidium iodide, which showed >98 % viable parasites within over a period of 72 h.
As we have previously reported (Garcia-Silva et al. 2010a), the optimal conditions to induce small tRNA production and vesicle secretion are achieved when parasites were cultured in serum free or up to 1 % FBS RPMI medium. For extended periods of cultures media were supplemented with 1 % FBS in order to assure viability higher than 98 %. Parasites under exponential growing or late-stationary phase do not efficiently produce and secrete adequate amounts of tRNA halves and vesicles. As reported (Hernandez et al. 2012), this nutrient starvation has been recognized as an important condition inducing a strong nutritional stress promoting the emergence of a significant fraction of infective trypomastigotes. The dynamic of cytoplasmic accumulation of tsRNA in unstressed and stressed epimastigotes compared to trypomastigotes was assessed by immunofluorescence (Supplementary Fig. 1). As depicted in Supplementary Fig. 2, epimastigotes become progressively transformed in trypomastigotes over time representing about 30 % of parasites after 48 h, which were infective for the Vero (monkey kidney epithelial cells) and HeLa (human cervical cancer cells) cell lines. Parasites submitted to nutrient starvation for 24 h (sE24) were used mainly for subcellular localization assays (where accumulation of signals for tRNA halves reach to a maximum), and those cultured for 48 h (sE48) were used selectively for infection assays (highly infective with a high ratio of trypomastigotes). Infection assays were performed by incubating sE48 parasites with cells at 1:20 cell/parasite ratio for 4 h at 37 °C in serum-free RPMI and washed twice with phosphate-buffered saline (PBS). For nonsusceptible wild-type or electroporated K-562 cells (electroporation control and treated cells) infection assays were prolonged up to 24 h.
Labeling of parasites by transfection with fluorescent synthetic tsRNAs
Epimastigotes at the late-stationary phase of growth (1 × 109 ml−1) were transfected with synthetic Cy3-labeled oligoribonucleotides (IDT Inc., IA, USA) chemically modified to avoid degradation by ribonucleases (terminal phosphorothioate bonds and 2′ O-methyl ribonucleotides). Probes used were the sense 5′-derived tRNA halves from tRNAAsp GUC (5′ CTC GGT AGT ATA GTG GTA AGT ATA CCC 3′) and from tRNAGlu CUC (5′ CGG TGT GGT ATA GTG GTT AGA ACA AGC GGC T 3′). About 1 × 108 parasites were resuspended in 0.4 ml of Hepes buffer solution (21 mM Hepes, 137 mM NaCl2, 5 mM KCl, and 6 mM glucose, pH 7.4) and electroporated in 0.4-cm cuvettes in the presence of 100 nM of the fluorescent probe using an ECM-600 Electroporation System (BTX CA, USA). Parasites were submitted to two 450 V/500 μF pulses and allowed to recuperate for 24 h in LIT 10 % FBS before use. Nonelectroporated parasites cultured in identical conditions and exposed to equal concentrations of the fluorescent probe were used as controls to exclude the uptake of the free probe (soaking). Nonelectroporated control parasites were followed over a period of 2–24 h without evidences of detectable intracellular fluorescent signals.
Immunofluorescence and fluorescence in situ hybridization
Indirect immunofluorescence microscopy (IIFM) was used to visualize clathrin and TcPIWI-tryp proteins using unconjugated specific antibodies and secondary antibodies conjugated to either FITC or Alexa-Fluor 488 dyes. The monoclonal anticlathrin antibody (kindly gifted by Dr. Maurilio Soares, ICC, Curitiba Brazil) was used at 1:300, and the anti-TcPIWI-tryp antibody was used at 1:200. The rabbit anti-TcPIWI-tryp polyclonal antibodies were produced and characterized elsewhere (Garcia-Silva et al. 2010b). For immunofluorescence, harvested parasites were washed and fixed in 4 % of buffered formaldehyde, freshly prepared from paraformaldehyde, for 10 min at room temperature, washed twice with PBS, and blocked by incubating in 25 mM NH4Cl for 10 min. Parasites were allowed to adhere to 0.1 % poly-l-lysine-coated microscope slides and permeabilized with 0.2 % Triton X-100 in PBS for 5 min. Slides were then incubated with primary antibodies at the indicated dilutions in PBS containing 0.2 % bovine serum albumin (BSA) and 0.1 % Tween 20 (PBS-T) in for 2 h at room temperature. After extensive washing in PBS-T, the slides were further incubated with fluorescent secondary antibodies diluted 1:500 in PBS-T, for 1 h at room temperature. After washing, slides were incubated with 4,6-diamidino-2-phenylindole (DAPI) at 1 mg ml−1 for 5 min at room temperature. Slides were mounted with 5 μl of Fluor Save Reagent (DAKO) or 0.2 M N-propyl-gallate in glycerol-PBS (9:1). IIFM assays were performed alone or coupled to fluorescence in situ hybridization (FISH) to identify tRNAAsp- or tRNAGlu-derived 5′ halves by incubating the respective antisense oligoprobes directly on IIFM slides as previously reported (Garcia-Silva et al. 2010a). Briefly, parasites were fixed in 4 % para-formaldehyde and allowed to adhere to polylysine-coated slides. Samples were further permeabilized with 0.2 % Triton X-100 for 5 min. Slides were then prehybridized at room temperature in 2 % BSA, 5× Denhardt, 4× saline-sodium citrate (SSC), and 35 % deionized formamide for 2 h. Hybridization was performed overnight at room temperature in a humid chamber by the addition of 1 ng ml−1 of the indicated oligonucleotide conjugated to fluorescein isothiocyanate (FITC). After hybridization, slides were washed once in 2× SSC plus 50 % deionized formamide, once in 2× SSC, once in 1× SSC plus 4,6-diamidino-2-phenylindole (DAPI) at 1 mg ml−1, and twice in 0.5× SSC. Merged images were obtained by superimposing the indicated images files. The hybridization probes used were 5′ CGG GTA TAC TTA CCA CTA TAC TAC CGA 3′ (tRNAAsp GUC) and 5′CGC TTG TAG CCA CTA TAC CAC 3′ (tRNAGlu CUC). The respective scramble probes were used as control probes. As previously reported (Garcia-Silva et al. 2010a), the specificity and characterization of oligoprobes used in FISH assays were performed by using scramble and “bridging” control probes (recognizing only the anticodon loop of the respective full-length tRNA) without detectable reactivity with 5′ or 3′ halves. All hybridization was performed in nondenaturating conditions to avoid cross-hybridization with the respective full-length tRNA. FISH assays for TcPIWI mRNA were performed similarly to tRNA halves detection except for the addition of a prehybridization (denaturing step) by heating slides at 75 °C for 15 min. Hybridization probes were antisense (AAG ATT TAC CCC ATG AGC GGG GTC CGT CTC AAA TTT CG) and sense (CGA AAT TTG AGA CGG ACC CCG CTC ATG GGG TAA ATC TTC). Control assays to evaluate the specificity and reactivity of probes used to detect TcPIWI-tryp mRNA are depicted in Supplementary Fig. 3. Analyses were performed in an Olympus IX 81 microscope, ZEISS Axioplan II microscope equipped with a Collor View XS camera, and in a Confocal Leica TCS SP5 AOBS coupled to a Hamamatsu Orca-ER camera (Diagnostic Instruments). Merged images were obtained by superimposing the indicated images files.
Clathrin and TcPIWI-tryp immunoprecipitation: TcPIWI-tryp Western blot
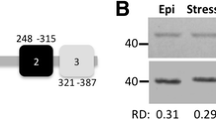
For cell extracts, 1 × 109 sE48 parasites were collected by centrifugation, washed twice with PBS, and resuspended in 1 ml of hypotonic lysis buffer (50 mM Tris at pH 8.0, 100 mM NaCl, 3 mM MgCl2, 0.5 % NP-40, and protease inhibitor cocktail). The lysates were centrifuged at 15,000×g for 15 min at 4 °C to eliminate debris and aggregates. The total protein concentration of supernatants was determined using the bicinchoninic acid protein assay (Sigma-Aldrich Co.) according to the manufacturer’s protocol. Immunoprecipitation was performed by incubating 30 μg of cell lysates with either anticlathrin antibodies diluted 1:100 anti-TcPIWItryp serum diluted 1/100 or an isotype-matched control antibody for 2 h at 4 °C. After addition of secondary antibody-coated magnetic beads (Sigma-Aldrich Co.), samples were further incubated overnight (Sigma) at 4 °C with gentle agitation. The bound fraction was purified by immunomagnetic separation and washed three times in ice-cold hypotonic lysis buffer. The fraction of proteins and total RNA bound to magnetic bead were isolated simultaneously with the Trizol reagent (Sigma-Aldrich Co.) according to the manufacturer’s protocol. The extracted proteins were fractionated by an 8 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (PAGE) and electrophoretically transferred to 0.45-μm nitrocellulose membranes. Western blot was done as previously described (Garcia-Silva et al. 2010b). The specificity of anti-TcPIWI-tryp antibodies used was verified using preimmune sera as negative control and by competition with the recombinant peptide used as immunogen. The absence of reactivity shown by the “unbound fraction” in Fig. 1c further support the anti-TcPIWI-tryp specificity. Additionally, irrelevant primary isotype-matched immunoglobulins were used as controls for anticlathrin and anti-TcPIWI-tryp antibodies (data not shown).

Colocalization of tRNA-derived fragments with clathrin and TcPIWI-tryp proteins. a Upper panel Immunofluorescence coupled to FISH showing colocalization of tRNAAsp GUC 5′ halves (red) and TcPIWI-tryp (green); bottom panel Immunofluorescence coupled to FISH showing colocalization of tRNAAsp GUC 5′ halves (red) with clathrin (green). Merged images were counterstained with DAPI for visualization of nuclei and kinetoplasts. The respective differential interference contrast images are depicted (DIC) in order to identify the parasite cellular body and the respective flagellum origin (scale bars = 5 μm). The parasites used in these experiments were submitted to nutrient starvation for 24 h (sE24) where intracellular accumulation of analyzed signals was optimal. The embedded scatter grams in the bottom right corners of merged images are depicted to estimate the degree of overlapping signals of FITC (green, y-axis) and Cy3 (red, x-axis) channels. Overlapping pixels of yellow color are depicted along the diagonal of the scatter gram. b RT-PCR assays for 5′ and 3′ halves of tRNAAsp and tRNAGlu, respectively, were performed from equivalent amounts of total cellular RNA (total) and clathrin (anti-clathrin) or TcPIWI-tryp (anti-TcPIWI-tryp) immunoprecipitates. Relative levels of amplicons in each immunoprecipitated fraction were quantified by qRT-PCR. The relative abundance of each tsRNA associated to clathrin is expressed as the percentage of the respective total intracellular tsRNAs. c TcPIWI-tryp detection by Western blot in clathrin immunoprecipitates (clathrin fraction) and the respective supernatant (unbound fraction). Total cell extracts were used as controls
RT-PCR of tRNA halves
Twenty micrograms of total RNA obtained after clathrin and TcPIWItryp immunoprecipitation was size-fractionated by denaturating 15 % PAGE, and the fraction from 20 to 60 nt was eluted from the gel and submitted to reverse transcription using the miScript kit (Qiagen) following the manufacturer’s recommendations. Amplification was performed using a universal reverse primer recognizing a 3′ adaptor and specific locked nucleic acid (LNA)-enhanced forward primers (Exiqon) specially designed for tRNA halves. The primers used were (LNA bases represented by uppercase characters): tRNAAsp 5′ ctcGgtaGtataGtg, tRNAAsp 3′ acGcgGgtga, and tRNAGlu 5′ cGgtGtGgtata tRNAGlu 3′ tCacCcGctaga. Amplification was performed with 40 cycles of 15″ at 95 °C and 50″ at 56 °C. Visualization of amplicons was performed after migration in a 15 % PAGE and stained with SYBRGold. In all cases, the identity of amplicons was verified by direct sequencing of purified PCR products.
Exchange of tsRNAs and TcPIWI-tryp protein by extracellular vesicles
These assays were designed to verify the eventual transfer of small vesicle cargo from parasite to parasite and from parasites to susceptible mammalian cells. For parasite-to-parasite exchange 2 × 106 ml−1 unlabeled sE48 parasites were incubated in serum-free RPMI supplemented with 0.24 ng ml−1 of isolated extracellular vesicles (EVs) derived from labeled sE48 parasites for 24 h at 28 °C. The EVs to parasite ratio used was designed in order to add an amount equivalent to that estimated to be eventually produced by the target parasites to keep a range of physiological concentrations. In our experimental conditions, we obtained an equivalent of 0.12 ng of protein in the purified vesicular fraction per million of sE48 parasites. To further validate this transfer and to exclude the eventual presence of unwanted labeled parasites or cellular debris in the purified vesicular fraction a series of transwell experiments were performed. For this, the bottom chambers of transwell plates (0.4 and 1 μm pore size HTS Transwell-24, Corning Inc.) were loaded with 2 × 106 unlabeled sE48 parasites in 1 ml of RPMI-1640 medium supplemented with 1 % of vesicle-depleted FBS, while upper chambers were loaded with 2 × 106 of labeled sE48 in 1 ml of the same medium. The sE48 parasites were labeled with synthetic tRNA 5′ halves from tRNAAsp conjugated to Cy3 as indicated above. Scrambled 5′ halves from tRNAAsp were used as irrelevant control probes. To deplete FBS of all potential vesicular content, FBS was submitted to two round of ultracentrifugation at 110,000×g for 70 min at 4 °C and filtered through 1 μm pore diameter filters to eventually exclude remnant cells or cellular debris. For parasite-to-cell assays, we replaced unlabeled parasites in the bottom chambers by 2 × 105 Vero cells in 1 ml of the same medium. Transwell plates were then incubated at 28 °C for 24 h.
The transfer of labeled tRNA halves from the upper to the bottom chamber was monitored by epifluorescence microscopy of unlabeled parasites or Vero cells. As controls, unlabeled parasites at 2 × 106 ml−1 or Vero cells at 2 × 105 ml−1 were cultured in the presence of free Cy3-labeled tRNA halves at 100 nM in RPMI-1640 medium supplemented with 1 % of vesicle-depleted FBS.
Small vesicle purification
The supernatants of 1 × 1011 sE48 parasites cultured for 24 h were collected and centrifuged at 2,000×g for 15 min to eliminate remnant cells. The 2,000×g supernatants were collected and centrifuged at 15,000×g at 4 °C for 30 min to remove cell debris and eventual apoptotic blebs. The 15,000×g supernatant was ultracentrifuged at 110,000×g 4 °C for 70 min to pellet small vesicles. The pellet was washed once by resuspending it in PBS and further ultracentrifuged at 110,000×g for 1 h. The isolation procedures were evaluated by transmission electron microscopy, and quantification of MVs was done by determining the total protein concentration by the Bradford protein quantification assay (Pierce). By these procedures, the total protein yield of the small vesicular fraction was about 1.2 μg per 1 × 1010 parasites.
Small RNA deep sequencing of secreted vesicles
Total RNAs were extracted from purified EVs obtained in supernantants from 1 × 1011 sE48 parasites using the Trizol reagent (Sigma-Aldrich Co.). Small RNA libraries were prepared using the small RNA sample preparation kit (v1.0, Illumina Inc.), which is specifically designed to isolate small RNAs having 5′ phosphate and 3′ hydroxyl ends. The sequencing was performed using a Genome Analyzer II (Illumina Inc.). Adaptor sequences were identified and trimmed from each read using a customized Python script. Reads in which the adaptor could not be identified, reads shorter than 16 nt long, and homopolymers were discarded. The sequence data were stripped of the “CCA” extension and aligned with the T. cruzi strain CL Brenner genome assembly (Esmeraldo and non-Esmeraldo) (El-Sayed et al. 2005; Weatherly et al. 2009) using Novoalign V2.07.08 (http://www.novocraft.com), which finds global optimum alignments using full Needleman–Wunsch algorithm with affine gap penalties. Novoalign parameters were configured to allow up to three mismatches. Only sequence data showing a single genome location were used for subsequent analysis. Classification of reads in functional classes was realized according to annotation provided in the TriTrypDB database (Aslett et al. 2010). In cases where a small RNAs overlapped protein coding genes, the reads were discarded.
In vitro infection and electroporation of K562 cells with T. cruzi-derived small vesicles
The myelogenous leukemia cell line K562 was artificially fused with labeled vesicles by electropration using an ECM-600 Electroporation System (BTX CA, USA) to transfect 1 × 108 cells in 0.4-cm cuvettes with 1.2 μg of small vesicle preparation (amounts obtained from 10.10 sE48 parasites). Cells were submitted to two 350 V pulses of 130 ms and allowed to recuperate for 24 h in RPMI-1640 supplemented with 20 % FBS before use. Nonelectroporated K562 cells soaked for 24 h in 1.2 μg of small vesicle preparation and K562 cells electroporated in vesicle-depleted media were used as controls using identical experimental conditions. After 24 h K562 cells were coculture with sE48 parasites in a cell/parasite ratio of 1:20 at 37 °C in a 5 % CO2 incubator for the indicated times. Productive infection was assessed at 2, 6, and 12 h postinfection by transmission electron microscopy and after 72 h by fluorescence microscopy.
Reservosomes prelabeling
sE48 parasites were washed three times in PBS (pH 7.2) at room temperature, counted in a Neubauer hemocytometer, and suspended at 5 × 106 cells ml−1 in serum-free RPMI medium. Samples were incubated for 4 h at 28 °C with 10 μg ml−1 of Alexa Fluor 594-labeled human transferrin. Human transferrin samples obtained from Molecular Probes (Invitrogen) were anonymized. Finally, cells were rinsed and processed for immunofluorescence microscopy.
Scanning electron microscopy
Parasites harvested from the culture medium as previously described were washed twice with PBS and fixed for 1 h with 2.5 % glutaraldehyde (GA) in 0.1 M cacodylate buffer (pH 7.2), 5 mM calcium chloride, and 2 % sucrose. The parasites were then washed with the same buffer and adhered to glass coverslips coated with 0.1 % poly-L-lysine (M.W. 70,000, Sigma-Aldrich Co.). After postfixation for 15 min with 1 % osmium tetroxide (OsO4) containing 0.8 % potassium ferrocyanide and 5 mM calcium chloride in cacodylate buffer 0.1 M (pH 7.2), cells were washed, dehydrated in graded ethanol, and then critical point dried with CO2. Samples were adhered to scanning electron microscopy stubs, coated with a 20-nm-thick gold layer in a sputtering device and then observed in a FEI Quanta 250 scanning electron microscope operating at 25 kV.
Transmission electron microscopy
For routine analysis, samples (parasites and host cells) were washed twice in Dulbecco’s PBS and fixed for 1 h at room temperature with 2.5 % GA in 0.1 M cacodylate buffer (pH 7.2), 5 mM calcium chloride, and 2 % sucrose. All samples were postfixed in 1 % osmium tetroxide (OsO4) containing 0.8 % potassium ferrocyanide and 5 mM calcium chloride in 0.1 M cacodylate buffer (pH 7.2) for 1 h at room temperature, dehydrated in graded acetone, embedded in PolyBed812 (Polysciences Inc., Warrington, PA, USA), and then polymerized for 3 days at 60 °C. Ultrathin sections obtained with a Leica (Nussloch, Germany) ultramicrotome were stained with uranyl acetate and lead citrate. For negative staining, purified MVs were absorbed onto formvar/carbon-coated copper grids for 10 min, fixed in 4 % formaldehyde in 0.1 M cacodylate buffer (pH 7.2) for 10 min at room temperature, washed in PBS, and stained with 2 % aqueous uranyl acetate for 30 s. For immunolabeling, grids were blocked after fixation with 50 mM NH4Cl in PBS for 5 min, washed and incubated for 10 min PBS, pH 7.2, 0.2 % gelatin, 0.1 % azide (PGN), and then permeabilized with 0.1 % saponine in PGN for 5 min. Grids were sequentially incubated with anti-TcPIWI-tryp 1:500 or equivalent amounts of rabbit IgGs as controls diluted in PGN for 30 min, washed in PGN, and incubated again with 10 nm gold-labeled antirabbit secondary antibody (Sigma-Aldrich Co.) for 30 min. Samples were washed with PGN. For in situ hybridization, permeabilized samples were washed twice with 2× SSC 50 % formamide and prehybridized for 20 min at room temperature in 2 % BSA, 5× Denhardt, 4× SSC, and 35 % deionized formamide (hybridization solution). Hybridization was performed overnight at room temperature in a humid chamber in the presence of 1 ng ml−1 of the probe conjugated to biotin. At the end of hybridization, grids were washed twice in 2× SSC plus 50 % deionized formamide, once in 2× SSC, once in 1× SSC, once in 0.5× SSC, and once in 0.1× SSC and incubated with 20 nm gold ExtrAvidin (Sigma) for 30 min. After washes with 2× SSC, grids were stained with 2 % aqueous uranyl acetate for 30 s.
Cryoultramicrotomy and immunolabeling
After incubation with specific or irrelevant primary antibodies as described above, parasites were loaded with 1:50 dilution of antirabbit IgG–gold complex 10 nm in diameter (Sigma-Aldrich Co.) in serum-free RPMI and, after washing in PBS, were fixed in 0.1 % glutaraldehyde, 4 % formaldehyde in 0.1 M sodium cacodylate buffer supplemented with 3.7 % sucrose and 5 mM CaCl2 (pH 7.2) for 60 min at room temperature. Parasites were then washed three times in PBS, infiltrated in 10 % gelatin in 0.1 M sodium cacodylate buffer, cut into cubes of 1 mm, and infiltrated with 25 % poly-vinylpyrrolidone in 2.3 M sucrose overnight in a cold room. After that, cells were mounted on cryoultramicrotome stubs flash frozen by immersion in liquid nitrogen. Cryosections were obtained in a temperature range of −70 to −90 using an Ultracut UCT cryo-ultramicrotome (Reichert). The material was collected in 2.3 M sucrose in PBS loop and transferred to Formvar-carbon-coated 400mesh silver grids and stored in PBS, pH 8.0, containing 3 % BSA. For in situ hybridization, samples were washed with 2× SSC 50 % formamide and prehybridized for 20 min at room temperature in hybridization solution. Hybridization was performed with 1 ng ml−1 of the Cy3-conjugated probe in fresh hybridization solution overnight at room temperature in a humid chamber at 37 °C. Then, grids were washed twice in 2× SSC 50 % formamide in a decreasing concentration of this buffer. After hybridization, grids were washed twice in PBS and incubated for 1 h at room temperature with a mouse anti-Cy3 antibody (1:100) in PBS, pH 8.0, containing 1.5 % BSA and 0.01 % Tween20. Samples were then washed and incubated for 1 h at room temperature with an antimouse IgG–gold complex 10 nm in diameter ( Sigma-Aldrich Co., 1:100 dilution) in PBS, pH 8.0, containing 1.5 % BSA and 0.01 % Tween20. At the end of both preparations, samples were washed twice in PBS. Grids were thinly embedded in 3 % polyvinyl alcohol–uranile acetate (9:1). All TEM samples were observed in a FEI Morgagni F268 (Eindhoven, The Netherlands) transmission electron microscope operating at 80 kV.
Dose response curves analyzing the influence of small vesicles on T. cruzi metacyclogenesis in axenic culture
Epimastigotes at the late-stationary phase were harvested and cultured by triplicate in 12-well plates at 4 × 106 ml−1parasites per well in serum-free RPMI. Parasites were exposed to different levels of exogenous EVs for 4 days. Parasites incubated without the addition of exogenous EVs were used as controls. At indicated times, aliquots of cultures were fixed and the absolute number of trypomastigotes was counted by light microscopy.
Results
TcPIWI-tryp and tRNA halves are recruited to reservosomes and intracellular vesicles
In a previous work (Garcia-Silva et al. 2010a), we reported that T. cruzi epimastigotes submitted to nutrient starvation produce an abundant population of tRNA-derived halves, which were recruited to cytoplasmic granular structures. Colocalization studies by epifluorescence microscopy showed that these structures were partially associated with reservosomes and did not significantly associate with other known organelles. Reservosomes are T. cruzi storage organelles where a significant proportion of nutrients from the extracellular media are delivered through endocytic vesicles originated in the cytostome and the flagellar pocket (Cunha-e-Silva et al. 2006; Porto-Carreiro et al. 2000). As in other eukaryotes, trypanosomatids have a clathrin-mediated endocytosis machinery where clathrin-coated vesicles were demonstrated on both early endocytic vesicles and membranes of the trans-Golgi network (Allen et al. 2003; Correa et al. 2007).
To gain insights into the intracellular distribution of tsRNAs and its eventual association with clathrin-coated vesicles, we performed colocalization assays by combining immunofluorescence with specific antibodies recognizing clathrin and TcPIWI-tryp proteins and FISH to detect representative tsRNAs. Results depicted in Fig. 1a showed a colocalization of tsRNAs with the TcPIWI-tryp protein, which significantly overlapped with clathrin-containing intracellular structures. A minor fraction of weak fluorescent signals for TcPWI-tryp and tsRNAs were located at the anterior region of the parasite, which corresponds with the regions of flagellar pocket and cytostome. As depicted in Fig. 1b and c, 5′-derived tRNA halves from tRNAGlu and tRNAAsp (most abundant intracellular fragments) as well as the TcPIWI-tryp protein were enriched in immunoprecipitated clathrin fractions as demonstrated by quantitative reverse transcription PCR (qRT-PCR) and Western blot, respectively. These results suggested that at least half of tsRNA could be directly associated to clathrin or indirectly through intracellular structures containing clathrin. Nevertheless, the fact that only two main tRNA halves were used as representative small tRNAs do not exclude that other tRNA halves could be also associated to clathrin. These data suggest that tRNA halves are not restricted to the clathrin fraction. This eventuality is supported by data indicating that tRNA halves have a wider distribution inside the cell including fractions that do not colocalize with clathrin. Immunoprecipitation assays of TcPIWItryp were also performed to verify its association with representative tsRNAs (Fig. 1b). As assessed by qRT-PCR, a significant fraction of total tsRNAs derived from Asp and Glu including 5′ and 3′ halves were effectively associated with TcPIWItryp.
Further immunogold and gold-labeled streptavidin hybridization assays were performed to detect TcPIWI-tryp protein and tRNA-halves from tRNAGlu and tRNAAsp by transmission electron microscopy. Results clearly demonstrated their localization in intracellular vesicles including principally reservosomes, and Golgi-like vesicular structures and other vesicles dispersed in the cytoplasm (Fig. 2). As depicted in Fig. 2h, the tsRNAs included in intracellular vesicles seem to originate from intracellular complexes or uncharacterized structures carrying tsRNAs apparently by a fusion process with intracellular vesicles. When sE24 parasites were preloaded with transferrin-Alexa Fluor 594 to label the reservosomes (Fig. 2a–d), we could observe a partial but significant colocalization with TcPIWI-tryp. The values of overlapping fluorescent intensities from the green channel over the red channel by confocal microscopy revealed an overlap coefficient (R) of 0.708 indicating that about 70 % of fluorescent signals from TcPIWI-tryp colocalized with transferin.

Subcellular localization of TcPIWI-tryp and tRNAGlu and RNAAsp halves in sE24 parasites. a–d sE24 parasites were preloaded with transferrin-Alexa Fluor 594 to label the reservosomes and later incubated in the presence of anti-TcPIWI-tryp antibodies (b and c, respectively) followed by a secondary goat–antirabbit Alexa Fluor 488. The overlay of both b and c images identified some double-labeled vesicles (arrows in d) located at the posterior region of the parasite. The embedded scatter gram in the bottom left corner of merged images in d is depicted to estimate the degree of overlapping signals of FITC (green, y-axis) and Cy3 (red, x-axis). Overlapping pixels of yellow color are depicted along the diagonal of the scatter gram. e–i In situ hybridization of tRNA-derived halves with 20 nm gold ExtrAvidin; representative micrographs showing recruitment of tRNAAsp GUC halves (arrows) in reservosomes (R), intracellular vesicles (V), and in Golgi-like structures (G). Some uncharacterized cytoplasmic complexes carrying tsRNA are showed in a process of fusion with intracellular vesicles (arrows in h)
Transferrin is a classical cargo entering by endocytosis through the cytostome and early endosomal networks before its storage in reservosomes. These results confirmed that at least in part both molecules were recruited to reservosomes and endosomal networks, but they do not necessarily share the same intracellular fates.
Of note, we could never observe high density of signals within the flagellar pocket and the cytostome that could be the consequence of a very fast clearance in this region. These results represent the first description of recruitment of tRNA-derived halves and the trypanosomatid argonaute protein TcPIWI-tryp protein in intracellular vesicles, suggesting their involvement in endocytic and/or exocytic pathways of T. cruzi.
The argonaute TcPIWI-tryp protein and tRNA-derived halves from T. cruzi are exchanged between parasites through shed vesicles
The recruitment of tsRNAs and TcPIWI-tryp protein in intracellular vesicles suggested to us that these molecules could be part of an endocytic/exocytic pathway in T. cruzi (Allen et al. 2003; Correa et al. 2007; Ungewickell and Hinrichsen 2007). Indeed, a growing body of evidence indicates that secreted vesicles (i.e., exosomes and ectosomes) from mammalian cells serve as a mean of delivering genetic information and proteins to recipient cells playing pivotal roles in cell-to-cell communication (Camussi et al. 2010). To trace small tRNA transfer parasites were labeled by electroporation with synthetic fluorescent 5′ halves from tRNAGlu and tRNAAsp. Electroporation efficiency and intracellular distribution of the fluorescent probe (Fig. 3a) showed that after 2 h the fluorescent signal had a diffuse cytoplasmic pattern in more than 95 % of parasites which adopted the typical granular distribution observed in stressed parasites beyond 24 h. Fluorescent signals remained detectable beyond 72 h of cultures with a slow but progressive decline after 4 days (data not shown). Of note, it was previously reported that T. cruzi are able to endocytose free synthetic probes when exposed for 24 h to concentrations of 20 μM (Malaga and Yoshida 2001). However, in our experimental conditions, nonelectroporated control parasites incubated with 0.1–2.0 μM of the free fluorescent probe for a period of 2–24 h did not incorporate the free probe, indicating that the parasite could not incorporate detectable amounts of free probe when using more physiological concentrations (Fig. 3 and data not shown). We considered that exposition of wild-type parasites to small vesicles secreted by electroporated parasites represented a more natural condition when compared with high nonphysiological concentrations of the free probe.

Exchange of small tRNAs between parasites through shed vesicles. a Epifluorescence microscopy of parasites labeled with fluorescent tRNAAsp halves after soaking for 2 h in the presence of the free probe (left panel) and after 2 (middle panel) or 24 h (right panel) after electroporation. In all cases, only merged images are depicted. b Unlabeled parasites cultured for 24 h in serum-free RPMI depleted from small vesicles (upper panel) and in serum-free RPMI enriched with labeled small vesicles purified from supernatants of labeled parasites (bottom panel). c Experimental evidences for transfer of small RNAs from labeled parasites toward unlabeled parasites in separated chambers of transwells with 0.4 μm of pore diameter. Upper panel Parasites labeled with fluorescent tRNAAsp-derived 5′ halves in the upper chamber; bottom parasites labeled with the respective fluorescent scrambled probe in the upper chamber. Unlabeled parasites were added to bottom chambers and incubated for 24 h at 28 °C
Next, small secreted vesicles from labeled parasites were collected after 48 h of culture in serum-free RPMI by ultracentrifugation and subsequently filtered through 1- and 0.4-μm pore diameter filters to eventually exclude remnant parasites or cellular debris. When unlabeled parasites were incubated in supernatants depleted from labeled small vesicles, they did not incorporate any detectable fluorescent signal (Fig. 3b). In contrast, parasites incubated with serum-free RPMI enriched with purified small vesicles from sE48 rapidly incorporate the fluorescent signals adopting the typical granular distribution in the posterior region of the cell. To exclude an eventual contamination with intact or fragmented cells, labeled as well as unlabeled parasites were cocultivated in separated chambers of transwells with a pore diameter of 0.4 μm (Fig. 3c). A rapid incorporation of fluorescent signals was observed within 2 h in unlabeled parasites located in the bottom chamber of transwells, which adopted the typical granular pattern observed in sE48 parasites (upper panel in Fig. 3c). Of note, identical experiments performed with control fluorescent scrambled probes showed that they were also transferred between parasites, indicating a sequence-independent phenomenon. These data strongly suggested that at least tRNA-derived small RNAs were released to the medium included in vesicles, which deliver their cargo to other parasites in a homotypic manner.
To verify whether sE48 parasites effectively shed small vesicles carrying tRNA-derived small RNAs and TcPIWI-tryp protein, we performed both scanning and transmission electron microscopy of intact parasites and immune detection of TcPIWI-tryp coupled to in situ hybridization for 5′halves derived from tRNAGlu and tRNAAsp on purified extracellular vesicles. Of note, differentiating epimastigotes as well as trypomastigote forms (Fig. 4a and b, respectively) are releasing vesicles from the plasma membrane. Results depicted in Fig. 4a–f revealed the presence of small vesicles being released throughout the cell body as well as from the flagellar pocket and at the posterior end of the parasite. In the specific case of Fig. 4e, we noted that the vesicle attached to the outer side of the parasite cell membrane has a similar size to that found within the reservosome (asterisk), suggesting that these organelles could participate in the generation and release/uptake of vesicles to/from the extracellular compartment. Another important observation is that this reservosome showed a sharp region that seems to touch the cytoplasmic face of the plasma membrane, between the subpellicular microtubules, suggesting that some kind of fusion process could occur at this region. As depicted in Fig. 4h, the diameter of vesicles was heterogeneous and ranged from 20 to 200 nm. Additionally, gold labeling (Fig 4h–j) confirmed that 5′derived small tRNAs, and the TcPIWI-tryp protein were included by sE48 parasites in secreted vesicles.

TcPIWI-tryp and tRNA-derived halves in shed small vesicles from T. cruzi epimastigotes. a, b Scanning electron microscopy of intact sE48 parasites showing a parasite under a process of differentiation (a) and a differentiated trypomastigote (b) with vesicles emerging from the flagellum and the cell body. c–j Transmission electron micrographs: c–f longitudinal sections of SE48 parasites showing small vesicles inside the flagellar pocket (small arrows in c and f), in reservosomes (arrowheads in e) and other smaller vesicles being liberated from the cell body membrane (arrows in d and e). Note that the two vesicles in e (arrowhead and black arrow) present the same size and that the membrane of the reservosome (asterisk) are in close contact with the cell membrane (white arrow). g Purified secreted small vesicles. h In situ hybridization of purified shed vesicles to detect tRNAAsp 5′ halves labeled with 20 nm gold ExtrAvidin streptavidin. i Immuno-electron microscopy with gold-labeled anti-TcPIWI-tryp antibodies (10 nm. j)Double-labeling staining revealing the presence of TcPIWI-tryp protein (arrowheads) and tRNAAsp 5′ halves (arrows) in the same vesicle. F flagellum, K kinetoplast, R reservosome, N nucleus
Small vesicles shed from the surface of many mammalian cells both in vitro and in vivo are a mixed population of vesicles with different origins including exosomes (derived from the endosomal membrane compartments through multivesicular bodies), ectosomes (shedding microvesicles) originated by direct budding from the cell plasma membrane and apoptotic blebs (Camussi et al. 2010; Mathivanan et al. 2010). In this respect, it was recently reported (Bayer-Santos et al. 2013) that T. cruzi epimastigotes as well as the respective trypomastigote forms release two classes of vesicles including large vesicles budding from the plasma membrane resembling ectosomes and a second population of smaller vesicles mainly derived from the exocytic fusion of multivesicular bodies with the flagelar pocket membrane. Thus, in our experimental conditions the secreted vesicular fraction represents a mixed population of both classes of vesicles. For this reason, the vesicular fraction studied in this work will be referred as extracellular vesicles (EVs).
Small RNA cargo from T. cruzi shed vesicles is transferred to infection susceptible Vero cells.
A similar set of experiments were performed to analyze if small vesicles shed by sE48 parasites also deliver their cargo in a heterotypic manner to mammalian cells. As depicted in Fig. 5 (upper panel), Vero cells cultured in the presence of the free tracing probe added to vesicle-depleted supernatants from labeled parasites did not incorporate detectable fluorescent signals for a period of 24 h. When Vero cells were coincubated with labeled parasites in separated chambers of 0.4 μm pore transwells, they showed a clear incorporation of fluorescent signals adopting a perinuclear pattern (Fig. 5, middle panel). As depicted, both the tracing probe (tRNAAsp) and the TcPIWI-tryp argonaute protein were transferred to Vero cells. We speculated about the possibility that the incorporation of TcPIWI-tryp by Vero cells could be the consequence of either the transfer of the protein itself or through a functional mRNA carried out by shed vesicles. As observed in Fig. 5 (middle and bottom panels), when Vero cells were pretreated with cycloheximide to inhibit protein synthesis, the appearance of fluorescent signals remained unchanged. Additionally, the absence of TcPIWI-tryp mRNA inside the treated Vero cells (bottom panel in Fig. 5) further validated the assumption that the TcPIWI-tryp protein was effectively transferred through small vesicles. Taken together, these data indicated that the TcPIWI-tryp protein and specific small tRNAs were effectively delivered into Vero cells by shed small vesicles derived from differentiating forms of T. cruzi. It is noteworthy that, in contrast to their localization inside parasites, the TcPIWI-tryp and tsRNAs do not colocalize inside Vero cells. One can speculate that both species follow different intracellular routes reaching distinct subcellular compartments. However, other experimental approaches are needed to validate this assumption.

Transfer of vesicle cargo from T. cruzi to mammalian susceptible cells. Visualization of fluorescent tRNAAsp 5′ halves secreted from sE48 parasites coupled to immunodetection of TcPIWI-tryp in the infection susceptible Vero cells. Upper panel Control Vero cells cultured for 24 h in the presence of free tRNAAsp 5′ halves and counterstained with DAPI; middle panel untreated Vero cells cocultured for 24 h in the bottom chamber of transwells with sE48 parasites labeled with synthetic fluorescent tRNAAsp 5′ halves in the upper chamber (Vero cells + labeled parasites) compared to the same experiment with cycloheximide pretreated Vero cells for 24 h (Vero cells + labeled parasites + cycloheximide); bottom panel fish assay to detect TcPIWI-tryp mRNA in Vero cells cocultured with labeled parasites
Secreted small vesicles contain a highly represented population of small RNAs derived from tRNAs and rRNAs
In order to know the whole spectrum of small RNAs contained in secreted vesicles, we performed a deep sequencing of an 18–60-nt size-fractionated complementary DNA library of EVs purified from supernatants of starved sE48 parasites (Fig. 6). The sequence data were aligned separately with annotated genomes from the non-Esmeraldo and Esmeraldo haplotypes and additionally with a collection of unassigned contigs from T.cruzi, which represents the sequences that could not be assigned to either haplotype. Sequence analysis resulted in a total of 10,107,119 aligned reads with a fraction of 6,100,251 reads (60 %) with a single alignment location in the genome (single mappers). The length distribution of unique reads (Fig. 6a) was clearly segregated into two main populations with median sequence length of ∼20 and ∼34 nt. Categorization in classes using genome annotations revealed that small RNAs derived from rRNA and tRNA represented about 45.5 % for each (Fig. 6b). Of note, more than 90 % of reads within ∼33 nt population belonged to the tRNA category. Additionally, more than 90 % of tsRNAs derived from a restricted group of six tRNAs: Leu, Thr, Glu, Gly, and Arg (Fig. 5c). The remaining reads were derived from CDS (≈5 %), sno/snRNAs (≈1.5 %), and intergenic regions (≈3 %).

Deep sequencing of small RNAs included in shed small vesicles from T. cruzi. a Graphical representation of length distribution vs number of reads with a single alignment location in the genome. b Categorization of reads into known RNA classes using genome annotations. c Distribution of reads derived from tRNAs according to precursor tRNAs
Electrofusion of vesicles from T. cruzi with infection resistant K562 cells confers susceptibility to infection
In order to gain insight about the biological significance on the transfer of genetic information and proteins to mammalian susceptible cells through secreted vesicles, we performed a set of experiments using as recipient cells the human erythroleukemic K562 cell line, which is known to resist infection by T. cruzi (Magdesian et al. 2001; Ruiz et al. 1998). In contrast to susceptible cells, K562 cells cultured in the presence of labeled sE48-derived vesicles for 24 h did not spontaneously incorporate detectable amounts of labeled tRNA halves (Fig. 7a). Speculating that fusion of T. cruzi-derived EVs with target cells could be required to achieve a productive infection, we performed a series of in vitro infection assays aimed to evaluate the susceptibility of electroporated K562 cells to T. cruzi infection either in the absence or the presence of sE48-derived vesicles. Twenty-four hours after electroporation in vesicle-depleted media, K562 cells do not incorporate detectable fluorescent signals as assessed by fluorescence microscopy (upper panel in Fig. 7b). In contrast, when K562 cells were electroporated in the presence of labeled EVs, they showed a clear incorporation of fluorescent signals, indicating that electroporation was successful to deliver vesicle cargo to K562 cells (bottom panel in Fig. 7b). The ratio of K562 cells to EVs used to obtain this effect was optimal at 1 × 106 cells for 12 ng of vesicle preparation (equivalent to vesicles produced by 1 × 108 parasites cultured for 24 h). To exclude a nonspecific effect of irrelevant vesicles, we performed an additional control experiment by electroporating K562 cells with microvesicles purified from HeLa cells. In these conditions, K562 cells remained resistant to infection by the parasite (data not shown). Fluorescent signals remained virtually unchanged for the first 48–72 h with a slow decline of signals over the following days when the signals became undetectable at day 5 after electroporation.

Assessing infection of vesicle-treated K562 cells by fluorescence microscopy. a Purified small vesicles labeled with Cy3-tRNAAsp 5′ synthetic halves (EVs-Cy3) from sE48 were cocultured with K562 cells for 24 h and stained with the plasma membrane dye CellMask OrangeTM (Invitrogren). b Control experiments showing K562 cells electroporated in the absence of labeled vesicles and further incubated for 24 h (upper panel) and K562 cells 24 h after electroporation in the presence of EVs-Cy3 (bottom panel). c Control K562 cells infected with sE48 parasites 24 h after electroporation in the absence of EVs-Cy3 (upper panel) or infected 24 h after electroporation in the presence of EVs-Cy3 (bottom panel). Images were obtained 3 days after infection
When electroporated K562 cells were infected with sE48 parasites and observed 48–72 h after infection, only k562 cells electroporated with EVs appeared to become susceptible to infection as indicated by the presence of rounded forms of parasites (presumably amastigotes) inside and outside the cell (bottom panel in Fig. 7c). However, in three independent assays, we could not evidence the emergence of trypomastigote forms, suggesting that in spite of become infected T. cruzi could not complete its life cycle in K562 cells. A plausible explanation could be that K562 cells possess a high nucleus/cytoplasm ratio, which limits the extent of amastigote replication inside the cell with the subsequent premature disruption of plasma membrane hampering the emergence of trypomastigote forms. When control K562 cells were incubated with EVs 24 h after electroporation, they did not become infected (upper panel in Fig. 7c), indicating that infection susceptibility was not the consequence of electroporation.
To verify the entry of infective parasites in electroporated K562 cells, we followed the infectious process by transmission electron microscopy in the first 12 h after infection (Fig. 8). These results revealed that, although control K562 cells electroporated in vesicle-depleted supernatants were able to interact with infection-competent parasites by surface-to-surface contact, we never could observe the presence of intracellular parasites in this period. Surprisingly, K562 cells electroporated with small labeled vesicles from sE48 parasites became susceptible to infection as revealed by the presence of several intracellular parasites in the first 12 h after infection. Images from Fig. 8b–d are representatives of the first 2, 6, and 12 h after infection, respectively, depicting a parasite penetrating the cell, and intracellular parasites in a process of differentiation toward round forms characterizing the intracellular replicative phase (amastigotes). These results suggested that fusion of T. cruzi-derived small vesicles with target cells could be a relevant step in the infectious process.

Monitoring infection of EVs-treated K562 cells by transmission electron microscopy. a Representative micrographs of control electroporated K562 cells cocultured with infective parasites (sE48) for 24 h in a 1:20 cell/parasite ratio at 37 °C in serum-free RPMI. Representative micrographs show residual parasites adhered to the plasma membranes (P). b–d K562 cells electroporated with extracellular vesicles (EVs) from sE48 parasites. Representative micrographs showing parasites (P) in early stages of interaction with host cells. b After 2 h of infection. d After 6 h of infection. c After 12 h of infection. The membranes surrounding parasites resembling parasitophorous vacuoles are indicated by small arrows (c and inset of d)
Small vesicles secreted by T. cruzi significantly enhanced metacyclogenesis in axenic cultures
As we have previously reported, both the generation of tsRNAs and the abundance and size of cytoplasmic granules recruiting them are significantly increased after submitting epimastigote forms of T. cruzi to nutrient starvation as differentiating media. Nutritional stress is a biologically relevant condition for the life cycle of this parasite because this condition is specifically sensed by the parasite in the digestive tract of insect vectors. This stress initiates a series of metabolic changes, which, in turn, induce transformation into the infective trypomastigotes. Therefore, we speculated about a relevant participation of shed vesicles from starved parasites in metacyclogenesis. For this, epimastigotes at the late-stationary phase were exposed to increasing levels of purified sE48-derived vesicles, and the total number of trypomastigotes was counted over time by light microscopy. The levels of small vesicles added to cultures were expressed as micrograms of protein per milliliter so that we could assure a significant increase in the exogenous/endogenous small vesicle ratios. As expected, the levels of vesicles added to cultures increased the absolute number of trypomastigotes in a dose-dependent manner (Fig. 9). In our experimental conditions, after 4 days, the higher level of small vesicles added, induced about fivefold increase in trypomastigotes when compared to cultures without addition of vesicles. These trypomastigotes were confirmed to be infection competent in further in vitro assays on Vero cells (data not shown).

Exogenous addition of T. cruzi-derived small vesicles increases metacyclogenesis in axenic cultures. T. cruzi epimastigotes at the late-stationary phase were cultured at 2 × 106/ml in serum-free complete RPMI medium for 4 days and exposed to increasing amounts of purified small vesicles from sE48 parasites expressed as μg of protein in this fraction. At the starting point (day 0), the concentration of parasites was adjusted to 4 × 106 ml−1for all experimental points. The amounts of EVs added to cultures were expressed as microgram of protein in the vesicular preparation. As a reference 0.6 μg correspond to vesicles produced by 5 × 1010 sE48 parasites. Control parasites without addition of exogenous small vesicles were exposed to their own endogenous vesicles. Results are the mean and the standard error of triplicate experiments where the absolute number of trypomastigotes forms per milliliter was obtained by visual observation from a total of 20 microscopic fields
Discussion
As reported, tRNA cleavage appeared as a highly conserved small RNA pathway from prokaryotes to higher eukaryotes, frequently initiated after nutritional, biological, or physicochemical stress (Haiser et al. 2008; Jochl et al. 2008; Kawaji et al. 2008; Lee and Collins 2005; Li et al. 2008; Zhang et al. 2009). Although the biological significance remains to be completely elucidated, recent experimental evidence suggested that tRNA-derived small RNAs emerges as an evolutionary conserved pathway with a putative role in the regulation of gene expression under certain survival pressures (Elbarbary et al. 2009a, b; Haussecker et al. 2010; Li and Zhou 2009; Nakashima et al. 2007).
The stress-induced tRNA cleavage is mediated by anticodon nucleases in bacteria (Ogawa et al. 1999), Rny1p in yeasts (Thompson and Parker 2009a), and angiogenin (ANG) in humans (Fu et al. 2009). These stress-induced tRNA cleavage products are 30–40 nt in length and seem to form related RNA classes, termed generically tsRNAs. An additional class of small RNAs that are ∼20–30 nt long derived from 5′ or 3′ ends of tRNAs was also reported and broadly termed tRNA fragments (Lee et al. 2009).
We found that a significant proportion of tsRNA colocalized with the T. cruzi-specific argonaute protein TcPIWI-tryp (Garcia-Silva et al. 2010b) and with clathrin, suggesting that they could be associated to intracellular vesicles or membranous compartments. Clathrin is a scaffold protein found in different types of coated vesicles in most eukaryotic cells playing an important role in receptor-mediated endocytosis at the plasma membrane when associated with different adaptors proteins (Ungewickell and Hinrichsen 2007). In this respect, a clathrin-mediated membrane budding system was recently demonstrated in T. cruzi playing pivotal roles in endocytic and exocytic pathways (Correa et al. 2007). Clathrin-coated vesicles can also be formed from other membranous compartments such as endosomal membranes (Raiborg et al. 2001), the trans-Golgi network (Deborde et al. 2008), and endosome-like vesicles close to the plasma membrane (Keyel et al. 2004). However, because clathrin-mediated vesicle generation is a transient process where the coat rapidly disassembles allowing clathrin to recycle, we cannot exclude that its colocalization with tsRNAs and TcPIWI-tryp be the consequence of an unspecific recruitment in storage organelles as reservosomes (Soares 1999).
Nevertheless, further analysis by transmission electron microscopy using gold–streptavidin in situ hybridization coupled to immunogold staining showed that tsRNAs and TcPIWI-tryp protein were included in several membrane-bounded organelles including mainly reservosomes, vesicular structures resembling Golgi, and other intracellular vesicles distributed along the cell body. Taken together, these data suggested that tsRNA biogenesis could be part of endocytic/exocytic routes in T. cruzi.
Using immunoprecipitation assays, we obtained experimental evidence showing the association of TcPIWI-tryp with two of the most abundant intracellular tsRNAs (i.e., Asp and Glu). In addition, in a recent work from our lab (submitted manuscript), a deep sequencing of immunoprecipitated TcPIWI-tryp identified abundant small RNAs derived from rRNAs, tRNAs, and snoRNAs ranging from 27 to 31 nt in length. Taken together, these results strongly suggested that at least a fraction of intracellular small tRNAs could be associated with the TcPIWI-tryp argonaute protein from T. cruzi.
The protozoa of the Trypanosomatidae family have a highly polarized endocytic activity that occurs exclusively at the flagellar pocket and the cytostome regions (de Souza et al. 2009). The cytostome is a specialized structure formed by an invagination of a well-defined region of the protozoan surface located at the anterior region and close to the flagellar pocket. Endocytic vesicles formed at the flagellar pocket or the cytostome fuse with a branched vesicular–tubular network that compose the early endosomes. Following this, cargo-containing vesicles deliver nutrients to the T. cruzi storage organelles, the reservosomes (Cunha-e-Silva et al. 2006; de Souza et al. 1978; Porto-Carreiro et al. 2000).
The reported location of tsRNAs and associated proteins in these subcellular compartments suggested to us an endocytic pathway involved in the uptake of small vesicles containing tsRNAs and TcPIWI-tryp as cargo among other biomolecules. Additionally, the presence of these molecules in vesicles underlying the plasma membrane or included in secreted vesicles also suggested their involvement in exocytic pathways. Indeed, we demonstrated that epimastigotes submitted to nutrient starvation as differentiation media (sE48) shed small vesicles ranging from 20 to 200 nm in diameter throughout the cell body including the flagellum with tRNA-derived small RNAs and the argonaute protein TcPIWI-tryp as cargo. Surprisingly, tsRNAs were internalized not only by surrounding parasites but by susceptible mammalian host cells, suggesting that small vesicle could be captured or engulfed by other parasites and mammalian cells. Shedding vesicles of 20–80 nm secreted from T.cruzi trypomastigotes were initially reported by Gonçalves et al. along the whole extent of the plasma membrane, which represented a physiological mechanism to deliver surface antigens to the extracellular medium (Goncalves et al. 1991). More recent experimental evidence suggested that shed vesicles by T. cruzi play key roles in Chagas’ disease pathogenesis by increasing tissue parasitism and inducing strong inflammatory responses (Trocoli Torrecilhas et al. 2009).
Studies over the past few years reported that cell-free mRNA, miRNAs, and other noncharacterized small RNAs are normally secreted from a variety of normal and diseased cells to the extracellular media either through membranous vesicles (Camussi et al. 2010) or included in ribonucleoprotein complexes (Arroyo et al. 2011; Vickers et al. 2011; Wang et al. 2010).
It is now accepted that secreted exosomes and shed microvesicles (ectosomes) (Cocucci and Meldolesi 2011) serve as a mean of delivering genetic information and proteins between cells playing pivotal roles in cell-to-cell communication (Camussi et al. 2010). In an elegant study, Valadi et al. (2007) reported that exosomes from different mammalian cells carry at least 1,300 mRNAs and more than 120 known microRNAs and other noncharacterized small RNAs. Interestingly, these exosomal mRNAs and microRNAs were completely functional in recipient cells.
Overall, these data strongly suggest that shedding of vesicles by T. cruzi represents an integral part of an endo-/exocytic pathway involved in cell-to-cell communication, where tsRNAs and TcPIWI-tryp are at least one of several involved molecules. Other proteins and glycoconjugates were also demonstrated to be included in shed vesicles including members of the gp85/transialidase family, proteases as cruzipain, cytoskeleton proteins, mucin-associated surface proteins, and a variety other proteins with known and unknown functions (Aparicio et al. 2004; Frasch 2000; Tribulatti et al. 2005). A recent proteomic analysis of the two major fractions of extracellular vesicles shed by T. cruzi revealed the presence of more than 300 proteins belonging to relevant pathways, including trafficking and membrane fusion, host–parasite interaction, signaling, and metabolism among others (Bayer-Santos et al. 2013).
Surprisingly, using synthetic small RNAs derived from tRNAGlu and tRNAAsp as tracer molecules, we demonstrated that tsRNAs contained in EVs were efficiently transferred to infection susceptible Vero cells associated to the TcPIWI-tryp argonaute protein but not to the nonsusceptible K562 cells. Therefore, we speculated that vesicles shed by T. cruzi could have a role in facilitating or promoting infection susceptibility of mammalian cells. Indeed, electroporation-mediated fusion of T. cruzi small vesicles with K562 cells turned them susceptible to infection by infective sE48 parasites. However, parasites internalized by K562 cells do not appear to complete their life cycle due to the premature disruption of the plasma membrane with the subsequent release of rounded forms resembling amastigotes. Nevertheless, the amastigote forms have proven to be relevant in the infective cycle of this parasite. In this respect, it is known that extracellular amastigotes are infective for mammalian cells, indicating that they can initiate an alternative subcycle within the life cycle of this parasite in mammalian cells (Ley et al. 1988).
We also describe a cross-kingdom transfer of tRNA-derived small RNAs through secreted vesicles from T. cruzi to mammalian susceptible cells. In this respect, recent evidences demonstrated another type of cross-kingdom transfer of a functional microRNA from plants to humans (Zhang et al. 2012). Some recent reports afforded experimental evidence demonstrating the transfer of transposons across phyla (Gilbert et al. 2010), including the transfer of mitochondrial minicircles from T. cruzi to host cells (Hecht et al. 2010; Teixeira et al. 2011).
The idea that the transfer of T. cruzi small vesicle cargo to mammalian cells could be involved in susceptibility to infection could afford novel avenues to elucidate the molecular mechanisms involved in T. cruzi infection of target cells. In agreement with this idea, it was recently reported that in both phagocytic and nonphagocytic cells, the process of T. cruzi entry into the host cell is drastically diminished when host cells are treated with dynasore, a potent inhibitor of dynamin-dependent endocytosis at the plasma membrane (Barrias et al. 2010). In agreement with this idea, in a recent work, Torrecilhas et al. (2012) demonstrated that shed vesicles from T. cruzi are engulfed by susceptible mammalian cells in the absence of viable parasites. However, the structures or biomolecules (e.g., membranes, lipids, proteins and nucleic acids other than small tRNAs) involved in these processes remain to be elucidated.
Additionally, we afford experimental evidences strongly suggesting that shed vesicles facilitated or significantly enhanced metacyclogenesis. These results suggested that EVs and their cargo represent a route of intercellular communication delivering “molecular messages” to others cells aimed to induce coordinated responses to cope with adverse environmental conditions and thus assuring parasite survival through the emergence of the infective forms. Indeed, recent unpublished results from Torrecillhas et al. (2012) revealed that T. cruzi trypomastigotes invade fivefold as much susceptible cells when cells were preincubated with purified parasite small vesicles. These results suggest that secreted vesicles from T. cruzi and their cargo could act as virulence factors by either promoting metacyclogenesis, enhancing host cell susceptibility or both. However, these assumptions remain to be further validated.
In order to know the total spectrum of small RNA included in shed vesicles, we performed a deep sequencing of short RNAs included in purified small vesicles. Our data revealed that rRNA- and tRNA-derived small RNAs equally contributed to more than 90 % of short RNA cargo of EVs. Of note ∼90 % of tsRNAs were derived from a restricted group of tRNA precursors including tRNALeu, tRNAThr, tRNAGlu, tRNAGly, and tRNAArg, representing about one half of small RNAs included in secreted vesicles where ∼56 % derived from the 3′ arm of precursor tRNAs. The short intracellular noncoding RNAs from the Dm28c clone submitted to nutrient starvation was recently analyzed by our group (Garcia-Silva et al. 2010a) using low-scale sequencing. These results revealed that tsRNAs represented about 25 % of intracellular small RNAs and more than 90 % of them derived from the 5′ arm of tRNAAsp GUC, tRNAGlu CUC, and tRNAGlu UUC. These results indicated that tsRNAs included in small vesicles are not only quantitatively enriched but qualitatively different from its intracellular counterpart, suggesting a selective recruiting process. However, one can speculate that the relative abundance of 5′ halves could not be the consequence of their preferential association with TcPIWI-tryp but the consequence of the higher instability of 3′ halves.
Although small tRNAs derived from tRNAGlu and tRNAAsp were the most abundant intracellular fragments, deep sequencing of EVs revealed that tRNAAsp halves represent a minor fraction of small tRNAs included in extracellular vesicles. Even though this phenomenon could be the consequence of a preferential recruitment of certain tRNA-derived species in EVs, we cannot exclude a bias induced by different sequencing methods used to analyze the pattern of intracellular and secreted tsRNAs. Whatever the case, these eventualities do not invalidate the use of tRNAAsp as a tracer molecule in some experiments. Recent results from our lab (manuscript in preparation) confirmed a selective recruitment of tsRNAs in secreted vesicles. Recently, Franzen et al. (2011) reported the intracellular short non-coding transcriptome of the CL-Brener clone, which was quali- and quantitatively different from that reported for the Dm28c clone. However, we cannot elucidate if these differences are the consequence of species-specific profiles or they reflect differences induced by nutritional stress when compared to normal growing conditions.
These data extended previous results indicating that besides microRNAs and mRNAs, tsRNAs, and other small RNAs are also included in vesicles and secreted to the medium by certain cells. In recent years, various studies have suggested that tRNA-derived small RNAs could act as effector molecules to regulate global gene expression (Li and Zhou 2009; Thompson and Parker 2009b). It is tempting to speculate that similarly to the vesicle-mediated epigenetic reprogramming of cells through either exosome- or ectosome transfer of mRNAs and microRNAs as a novel mechanism of genetic exchange between cells, these tsRNAs could also perform pivotal roles in cell-to-cell communication in T. cruzi (Camussi et al. 2011; Valadi et al. 2007).
The endo-/exocytic pathways involved in small vesicle uptake/release by T. cruzi epimastigotes are presently unknown. However, the presence of tsRNA within vesicular organelles as reservosomes, Golgi-like structures, intracellular vesicles, and plasma membrane strongly suggest that endocytosis of EVs cargo uses the highly polarized endocytic process characteristic of the Trypanosomatidae family (Soares 2006; Souza 2009).
Experimental evidence presented here and in previous reports from our lab (Garcia-Silva et al. 2010a) indicated that a major fraction of tsRNAs are recruited to reservosomes. Reservosomes are big compartments present at the postnuclear region of epimastigote forms of parasites belonging to the genus Trypanosoma, subgenus Schizotrypanum, described initially as multivesicular bodies (de Souza et al. 1978). Reservosomes deserved their name because they store tracer macromolecules ingested by the parasite through an endocytic process (Soares and de Souza 1991), which were reported as late endosomal or endosomal compartments in T. cruzi epimastigotes (Cunha-e-Silva et al. 2006; Souto-Padron et al. 1990). In addition, studies on reservosome biogenesis revealed that vesicles originated from the Golgi complex also fuse with components of the endocytic pathway. Indeed, the cystein protease cruzipain, a major protease synthesized by T. cruzi, is mainly recruited to reservosomes (Souto-Padron et al. 1990). The fact that cruzipain as well as tsRNAs are simultaneously observed in reservosomes and shed vesicles strongly suggested the idea that reservosomes could be organelles with exocytic functions (Aparicio et al. 2004).
Today, it is accepted that reservosomes do not constitute a uniform organelle population. Indeed, different reservosomes might have storing, recycling, or lysosome typical functions (Cunha-e-Silva et al. 2006). Taken into account these experimental evidences, we hypothesized that accumulation of tsRNAs and TcPIWI-tryp protein in reservosomes could result from two different pathways: one representing the contribution of small vesicles through the uptake via endocytic pathways (exogenous origin) and other representing the contribution of vesicles from the trans-Golgi network of the own cell (endogenous origin). In this respect, it was recently reported (Bayer-Santos et al. 2013) that T. cruzi epimastigotes as well as trypomastigotes forms use different populations of extracellular vesicles resembling ecto- and exosomes to excret/secret proteins and eventually deliver cargo into host cells.
Overall, our results afford new relevant pieces of evidence on the role of secreted vesicles in parasite biology especially in life cycle transitions, infectivity, and cell-to-cell communication, which undoubtedly should contribute to gain insights in the molecular mechanisms involved in host–parasite interactions. Additionally, the transfer of proteins and genetic material from protists to mammalian cells is an emerging field, which could conduct us to rethink some concepts in biology.
References
Allen CL, Goulding D, Field MC (2003) Clathrin-mediated endocytosis is essential in Trypanosoma brucei. Embo J 22(19):4991–5002
Aparicio IM, Scharfstein J, Lima AP (2004) A new cruzipain-mediated pathway of human cell invasion by Trypanosoma cruzi requires trypomastigote membranes. Infect Immun 72(10):5892–5902
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, Tait JF, Tewari M (2011) Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A 108(12):5003–5008
Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Logan FJ, Miller JA, Mitra S, Myler PJ, Nayak V, Pennington C, Phan I, Pinney DF, Ramasamy G, Rogers MB, Roos DS, Ross C, Sivam D, Smith DF, Srinivasamoorthy G, Stoeckert CJ Jr, Subramanian S, Thibodeau R, Tivey A, Treatman C, Velarde G, Wang H (2010) TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res 38(Database issue):D457–D462
Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S (2003) The trypanosomiases. Lancet 362(9394):1469–1480
Barrias ES, Reignault LC, De Souza W, Carvalho TM (2010) Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS One 5(1):e7764
Bayer-Santos E, Aguilar-Bonavides C, Rodrigues SP, Cordero EM, Marques AF, Varela-Ramirez A, Choi H, Yoshida N, da Silveira JF, Almeida IC (2013) Proteomic analysis of Trypanosoma cruzi secretome: characterization of two populations of extracellular vesicles and soluble proteins. J Proteome Res 12(2):883–897
Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L (2010) Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int 78(9):838–848
Camussi G, Deregibus MC, Bruno S, Grange C, Fonsato V, Tetta C (2011) Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res 1(1):98–110
Cerutti H, Casas-Mollano JA (2006) On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet 50(2):81–99
Clayton C, Shapira M (2007) Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol Biochem Parasitol 156(2):93–101
Cocucci E, Meldolesi J (2011) Ectosomes. Curr Biol 21(23):R940–R941
Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, Green PJ, Barton GJ, Hutvagner G (2009) Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. Rna 15(12):2147–2160
Contreras VT, Salles JM, Thomas N, Morel CM, Goldenberg S (1985) In vitro differentiation of Trypanosoma cruzi under chemically defined conditions. Mol Biochem Parasitol 16(3):315–327
Contreras VT, Araujo-Jorge TC, Bonaldo MC, Thomaz N, Barbosa HS, Meirelles Mde N, Goldenberg S (1988) Biological aspects of the Dm 28c clone of Trypanosoma cruzi after metacyclogenesis in chemically defined media. Mem Inst Oswaldo Cruz 83(1):123–133
Correa JR, Atella GC, Menna-Barreto RS, Soares MJ (2007) Clathrin in Trypanosoma cruzi: in silico gene identification, isolation, and localization of protein expression sites. J Eukaryot Microbiol 54(3):297–302
Cunha-e-Silva N, Sant’Anna C, Pereira MG, Porto-Carreiro I, Jeovanio AL, de Souza W (2006) Reservosomes: multipurpose organelles? Parasitol Res 99(4):325–327
de Souza W (2008) An introduction to the structural organization of parasitic protozoa. Curr Pharm Des 14(9):822–838
de Souza W, de Carvalho TU, Benchimol M, Chiari E (1978) Trypanosoma cruzi: ultrastructural, cytochemical and freeze-fracture studies of protein uptake. Exp Parasitol 45(1):101–115
de Souza W, Sant’Anna C, Cunha-e-Silva NL (2009) Electron microscopy and cytochemistry analysis of the endocytic pathway of pathogenic protozoa. Prog Histochem Cytochem 44(2):67–124
Deborde S, Perret E, Gravotta D, Deora A, Salvarezza S, Schreiner R, Rodriguez-Boulan E (2008) Clathrin is a key regulator of basolateral polarity. Nature 452(7188):719–723
Elbarbary RA, Takaku H, Uchiumi N, Tamiya H, Abe M, Nishida H, Nashimoto M (2009a) Human cytosolic tRNase ZL can downregulate gene expression through miRNA. FEBS Lett 583(19):3241–3246
Elbarbary RA, Takaku H, Uchiumi N, Tamiya H, Abe M, Takahashi M, Nishida H, Nashimoto M (2009b) Modulation of gene expression by human cytosolic tRNase Z(L) through 5′-half-tRNA. PLoS One 4(6):e5908
El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B (2005) The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309(5733):409–415
Figueiredo RC, Rosa DS, Soares MJ (2000) Differentiation of Trypanosoma cruzi epimastigotes: metacyclogenesis and adhesion to substrate are triggered by nutritional stress. J Parasitol 86(6):1213–1218
Franzen O, Arner E, Ferella M, Nilsson D, Respuela P, Carninci P, Hayashizaki Y, Aslund L, Andersson B, Daub CO (2011) The short non-coding transcriptome of the protozoan parasite Trypanosoma cruzi. PLoS Negl Trop Dis 5(8):e1283
Frasch AC (2000) Functional diversity in the trans-sialidase and mucin families in Trypanosoma cruzi. Parasitol Today 16(7):282–286
Fu H, Feng J, Liu Q, Sun F, Tie Y, Zhu J, Xing R, Sun Z, Zheng X (2009) Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett 583(2):437–442
Garcia-Silva MR, Tosar JP, Frugier M, Pantano S, Bonilla B, Esteban L, Serra E, Rovira C, Robello C, Cayota A (2010a) Cloning, characterization and subcellular localization of a Trypanosoma cruzi argonaute protein defining a new subfamily distinctive of trypanosomatids. Gene 466(1–2):26–35
Garcia-Silva MR, Frugier M, Tosar JP, Correa-Dominguez A, Ronalte-Alves L, Parodi-Talice A, Rovira C, Robello C, Goldenberg S, Cayota A (2010b) A population of tRNA-derived small RNAs is actively produced in Trypanosoma cruzi and recruited to specific cytoplasmic granules. Mol Biochem Parasitol 171(2):64–73
Ghildiyal M, Zamore PD (2009) Small silencing RNAs: an expanding universe. Nat Rev Genet 10(2):94–108
Gilbert C, Schaack S, Pace JK 2nd, Brindley PJ, Feschotte C (2010) A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature 464(7293):1347–1350
Goncalves MF, Umezawa ES, Katzin AM, de Souza W, Alves MJ, Zingales B, Colli W (1991) Trypanosoma cruzi: shedding of surface antigens as membrane vesicles. Exp Parasitol 72(1):43–53
Haiser HJ, Karginov FV, Hannon GJ, Elliot MA (2008) Developmentally regulated cleavage of tRNAs in the bacterium Streptomyces coelicolor. Nucleic Acids Res 36(3):732–741
Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA (2010) Human tRNA-derived small RNAs in the global regulation of RNA silencing. Rna 16(4):673–695
Hecht MM, Nitz N, Araujo PF, Sousa AO, Rosa Ade C, Gomes DA, Leonardecz E, Teixeira AR (2010) Inheritance of DNA transferred from American trypanosomes to human hosts. PLoS One 5(2):e9181
Hernandez R, Cevallos AM, Nepomuceno-Mejia T, Lopez-Villasenor I (2012) Stationary phase in Trypanosoma cruzi epimastigotes as a preadaptive stage for metacyclogenesis. Parasitol Res 111(2):509–514
Jochl C, Rederstorff M, Hertel J, Stadler PF, Hofacker IL, Schrettl M, Haas H, Huttenhofer A (2008) Small ncRNA transcriptome analysis from Aspergillus fumigatus suggests a novel mechanism for regulation of protein synthesis. Nucleic Acids Res 36(8):2677–2689
Kawaji H, Nakamura M, Takahashi Y, Sandelin A, Katayama S, Fukuda S, Daub CO, Kai C, Kawai J, Yasuda J, Carninci P, Hayashizaki Y (2008) Hidden layers of human small RNAs. BMC Genomics 9:157
Keyel PA, Watkins SC, Traub LM (2004) Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J Biol Chem 279(13):13190–13204
Lee SR, Collins K (2005) Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. J Biol Chem 280(52):42744–42749
Lee YS, Shibata Y, Malhotra A, Dutta A (2009) A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 23(22):2639–2649
Ley V, Andrews NW, Robbins ES, Nussenzweig V (1988) Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. J Exp Med 168(2):649–659
Li Y, Zhou H (2009) tRNAs as regulators in gene expression. Sci China C Life Sci 52(3):245–252
Li Y, Luo J, Zhou H, Liao JY, Ma LM, Chen YQ, Qu LH (2008) Stress-induced tRNA-derived RNAs: a novel class of small RNAs in the primitive eukaryote Giardia lamblia. Nucleic Acids Res 36(19):6048–6055
Magdesian MH, Giordano R, Ulrich H, Juliano MA, Juliano L, Schumacher RI, Colli W, Alves MJ (2001) Infection by Trypanosoma cruzi. Identification of a parasite ligand and its host cell receptor. J Biol Chem 276(22):19382–19389
Malaga S, Yoshida N (2001) Targeted reduction in expression of Trypanosoma cruzi surface glycoprotein gp90 increases parasite infectivity. Infect Immun 69(1):353–359
Mathivanan S, Ji H, Simpson RJ (2010) Exosomes: extracellular organelles important in intercellular communication. J Proteomics 73(10):1907–1920
Nakashima A, Takaku H, Shibata HS, Negishi Y, Takagi M, Tamura M, Nashimoto M (2007) Gene silencing by the tRNA maturase tRNase ZL under the direction of small-guide RNA. Gene Ther 14(1):78–85
Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, Masaki H (1999) A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science 283(5410):2097–2100
Porto-Carreiro I, Attias M, Miranda K, De Souza W, Cunha-e-Silva N (2000) Trypanosoma cruzi epimastigote endocytic pathway: cargo enters the cytostome and passes through an early endosomal network before storage in reservosomes. Eur J Cell Biol 79(11):858–869
Raiborg C, Bache KG, Mehlum A, Stang E, Stenmark H (2001) Hrs recruits clathrin to early endosomes. EMBO J 20(17):5008–5021
Reifur L, Garcia-Silva MR, Poubel SB, Alves LR, Arauco P, Buiar DK, Goldenberg S, Cayota A, Dallagiovanna B (2012) Distinct subcellular localization of tRNA-derived fragments in the infective metacyclic forms of Trypanosoma cruzi. Mem Inst Oswaldo Cruz 107(6):816–819
Ruiz RC, Favoreto S Jr, Dorta ML, Oshiro ME, Ferreira AT, Manque PM, Yoshida N (1998) Infectivity of Trypanosoma cruzi strains is associated with differential expression of surface glycoproteins with differential Ca2+ signalling activity. Biochem J 330(Pt 1):505–511
Soares MJ (1999) The reservosome of Trypanosoma cruzi epimastigotes: an organelle of the endocytic pathway with a role on metacyclogenesis. Mem Inst Oswaldo Cruz 94(Suppl 1):139–141
Soares MJ (2006) Endocytic portals in Trypanosoma cruzi epimastigote forms. Parasitol Res 99(4):321–322
Soares MJ, de Souza W (1991) Endocytosis of gold-labeled proteins and LDL by Trypanosoma cruzi. Parasitol Res 77(6):461–468
Souto-Padron T, Campetella OE, Cazzulo JJ, de Souza W (1990) Cysteine proteinase in Trypanosoma cruzi: immunocytochemical localization and involvement in parasite-host cell interaction. J Cell Sci 96(Pt 3):485–490
Souza W (2009) Structural organization of Trypanosoma cruzi. Mem Inst Oswaldo Cruz 104(Suppl 1):89–100
Teixeira AR, Gomes C, Nitz N, Sousa AO, Alves RM, Guimaro MC, Cordeiro C, Bernal FM, Rosa AC, Hejnar J, Leonardecz E, Hecht MM (2011) Trypanosoma cruzi in the chicken model: Chagas-like heart disease in the absence of parasitism. PLoS Negl Trop Dis 5(3):e1000
Thompson DM, Parker R (2009a) The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J Cell Biol 185(1):43–50
Thompson DM, Parker R (2009b) Stressing out over tRNA cleavage. Cell 138(2):215–219
Torrecilhas AC, Schumacher RI, Alves MJ, Colli W (2012) Vesicles as carriers of virulence factors in parasitic protozoan diseases. Microbes Infect 14:1465–1474
Tribulatti MV, Mucci J, Van Rooijen N, Leguizamon MS, Campetella O (2005) The trans-sialidase from Trypanosoma cruzi induces thrombocytopenia during acute Chagas’ disease by reducing the platelet sialic acid contents. Infect Immun 73(1):201–207
Trocoli Torrecilhas AC, Tonelli RR, Pavanelli WR, da Silva JS, Schumacher RI, de Souza W, NC ES, de Almeida Abrahamsohn I, Colli W, Manso Alves MJ (2009) Trypanosoma cruzi: parasite shed vesicles increase heart parasitism and generate an intense inflammatory response. Microbes Infect 11(1):29–39
Ungewickell EJ, Hinrichsen L (2007) Endocytosis: clathrin-mediated membrane budding. Curr Opin Cell Biol 19(4):417–425
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9(6):654–659
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT (2011) MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 13(4):423–433
Wang K, Zhang S, Weber J, Baxter D, Galas DJ (2010) Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res 38(20):7248–7259
Weatherly DB, Boehlke C, Tarleton RL (2009) Chromosome level assembly of the hybrid Trypanosoma cruzi genome. BMC Genomics 10:255
Zhang S, Sun L, Kragler F (2009) The phloem-delivered RNA pool contains small noncoding RNAs and interferes with translation. Plant Physiol 150(1):378–387
Zhang L, Hou D, Chen X, Li D, Zhu L, Zhang Y, Li J, Bian Z, Liang X, Cai X, Yin Y, Wang C, Zhang T, Zhu D, Zhang D, Xu J, Chen Q, Ba Y, Liu J, Wang Q, Chen J, Wang J, Wang M, Zhang Q, Zhang J, Zen K, Zhang CY (2012) Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res 22(1):107–126
Acknowledgments
This work was supported by grants from the National Agency of Research and Innovation (ANII, Uruguay), Institut Pasteur de Montevideo (Uruguay) and CNPq (Prosul), CAPEs and FAPERJ from Brazil.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
Time-course analysis of tRNA halves accumulation in epimastigotes submitted to nutritional stress. Epimastigotes under optimal growth (unstressed), stressed for 48 h (sE48) and trypomastigotes were fixed and hybridized with probes specific for tRNAGlu 5′ halves (red). Samples were counterstained with DAPI 4,6-diamidino-2-phenylindole (DAPI) at 1 mg ml−1 and merged images were obtained by superimposing the indicated images files. (JPEG 88 kb)
Figure S2
T. cruzi epimastigotes submitted to nutrient starvation. Epimastigotes at the late-stationary phase of growth in LIT medium were washed and resuspended in serum-free RPMI-1640 medium and further cultured for 48 h (sE48). a Visualization of sE48 parasites by differential interference contrast microscopy. b HeLa cells after 2 days of infection with sE48 parasites where DAPI stained amastigotes are indicated by arrows. c Percentage of trypomastigotes overtime in different culture conditions. Normal growing parasites in LIT medium (LIT), epimastigotes at the late-stationary phase in LIT medium (stationary phase) and epimastigotes submitted to nutrient starvation in RPMI (RPMI). (JPEG 106 kb)
Figure S3
Reactivity and specificity of TcPIWI-tryp mRNA probes used in FISH assays. Fixed parasites were hybridized for TcPIWI-tryp mRNA with antisense (positive control) or sense (negative control) oligoprobes conjugated to fluorescein isothiocyanate (FITC). Samples were counterstained with DAPI 4,6-diamidino-2-phenylindole (DAPI) at 1 mg ml−1. Merged images were obtained by superimposing the indicated images files. (JPEG 113 kb)
Rights and permissions
About this article
Cite this article
Garcia-Silva, M.R., Cura das Neves, R.F., Cabrera-Cabrera, F. et al. Extracellular vesicles shed by Trypanosoma cruzi are linked to small RNA pathways, life cycle regulation, and susceptibility to infection of mammalian cells. Parasitol Res 113, 285–304 (2014). https://doi.org/10.1007/s00436-013-3655-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-013-3655-1