
Abstract
In the present study, the morphological and molecular characterization of Lecithochirium grandiporum, a digenetic trematode infecting the European eel Anguilla anguilla (Family (F): Anguillidae), were described for the first time from Burullus Lake, Kafr El-Sheikh Governorate, Egypt. Twenty-five out of 60 specimens (infection rate of 41.66 %) were found to be naturally infected. Infection was recorded as small worms attached to the inner wall of the intestine of host fish. Adult worms measured 1.59 ± 0.20 (1.3–1.85) mm long and 0.3 ± 0.02 (0.29–0.48) mm wide for everted specimens with a smaller oral sucker measuring 0.15 ± 0.02 (0.13–0.18) mm, and a larger ventral sucker which was 0.16 ± 0.02 (0.14–0.25) mm. Our results recorded morphological differences as smaller dimensions of different body parts and the smaller oral/ventral sucker ratio between Lecithochirium fusiforme and L. grandiporum. Also, the phylogenetic position of the worm was determined by molecular characterization of their 18 SSU rDNA. Results were compared with those of previously recorded species on the Gene Bank. It was found that the present species coincide with those belonging to genus Lecithochirium. Comparison of the nucleotide sequences and divergence showed that the SSU rDNA gene of this Lecithochirium species revealed 92 % sequence identity with L. fusiforme (accession no. DQ413192) differing in 26 nucleotides with lower divergence value. According to these results, this study indicated that the present species is recorded as L. grandiporum with accession no. KC166146 as a parasite with new host and locality records in Egypt.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The potential risk for transmission of zoonotic diseases through consumption of parasitized fish could cause public health problems (Williams and Jones 1994). Marine fish may play an important role as intermediate or definitive hosts for a number of helminthic parasites which have been reported from their digestive tract (Shih et al. 2004). The European eel, Anguilla anguilla Linnaeus 1758 (Actinopterygii: Anguilliformes), is a benthic marine carnivorous predator and feeds on small fish, crustacean, and planktonic invertebrates.
The digenetic trematodes belonging to Family Hemiuridae (Looss 1899) represent a large group of parasitic helminthes that includes numerous subfamilies parasitizing fish and inhibiting mainly the stomach of marine teleosts (Pankov et al. 2006). The major diagnostic feature of this family is the presence of protrusible ecsoma that lies on the posterior region of the body representing the feeding organ and assists in the attachment of worms to the intestinal wall (Gibson and Bray 1979, 1986; Pankov et al. 2006).
The most common genus of this family is Lecithochirium (Lűhe 1901). The systematics of this genus is highly controversial. Identification and taxonomy of the different species in this genus are difficult due to the high level of intraspecific morphological variation and the lack of species-specific morphological characters as a result of the anatomical simplicity and morphological plasticity of these organisms (Vilas et al. 2002; Al-Zubaidy 2010). During the past two centuries, five species of this genus have been described from European benthic ichthyophagous marine fishes, mainly the Anguilliformes. These were the following: Lecithochirium rufoviride (Rudolphi 1819), Lecithochirium fusiforme (Lűhe 1901), Lecithochirium musculus (Looss 1907), Lecithochirium grandiporum (Rudolphi 1819), and Lecithochirium furcolabiatum (Jones 1933). Two pairs of these species appear very closely related: L. rufoviride and L. furcolabiatum, and L. grandiporum and L. fusiforme (Gibson and Bray 1986).
The incomplete description of the different recorded species of this genus provides the need of this group to be revised taxonomically (Bray 1991). Multidisciplinary approach including both morphological and molecular analyses should provide a more reliable means of identification (Casanova et al. 2001; Vilas et al. 2002; Carreras-Aubets et al. 2012). Blair and Barker (1993) concluded that a variable domain V4 region of 18 SSU rDNA gene was useful for phylogenetic studies in trematodes. Recently, molecular studies had characterized some genetically distinct but morphologically very similar species (Criscione and Blouin 2004; Testini et al. 2011).
In the present study, the natural prevalence, morphological, as well as molecular analyses of the 18 SSU rDNA of L. grandiporum infecting the European eel A. anguilla were carried out to determine the exact taxonomy and phylogenetic position of this species.
Materials and methods
Sample collection and parasitological study
During the period from January to November 2012, 60 specimens of the European eel A. anguilla (F: Anguillidae) were collected alive from fishermen at boat landing sites along Burullus Lake at Kafr El-Sheikh Governorate and transported to Parasitology Laboratory, Zoology Department, Faculty of Science, Cairo University, Egypt. The collected fish (small to medium size) reaching an average of 40 cm in length were identified according to Randall (1992). Fish were examined externally to detect any visible lesions. After dissection, internal organs were carefully examined for any helminth infection.
Parasites were recovered and washed in isotonic saline solution (0.65 % NaCl). Some of the recovered parasites were fixed in buffered formalin solution (10 %) after flattening by repression between two slides. For permanent whole mount preparation, fixed worms were stained by iron acetocarmine (Pankov et al. 2006), then dehydrated through an ascending alcohol series, followed by clearing in xylene and mounted on Canada balsam. Stained specimens were examined and photographed using Zeiss photo research microscope supplied by a Canon digital camera. Measurements were taken in millimeter as a mean ± SD followed by a range in parentheses.
For scanning electron microscopy, specimens were fixed in 3 % cold buffered glutaraldehyde (pH, 7.2) for 4 h, washed in sodium cacodylate buffer, and post-fixed in osmium tetroxide for 2 h. Rewashing of fixed specimens in cacodylate buffer and immersing into 2 % tannic acid for 8 h were done (Murakami 1977). The samples were dehydrated in a graded series of ethanol, infiltrated with amyl acetate. After critical drying, specimens were mounted on stubs and coated with gold (Lee 1993). The samples were examined and photographed with high-resolution scanning electron microscope JOEL 6100.
Molecular analysis
DNA extraction, PCR amplification, and sequencing
Parasite specimens for DNA extraction were fixed alive in 70 % ethanol. DNA was extracted using phenol–chloroform method (Sambrook et al. 1989). The portion of SSU rDNA gene that includes the V4 region was selected as a target for molecular analysis and amplified by PCR. All PCR reactions were carried out in a volume of 25 μl reaction mixture comprising of 0.625 unit Taq polymerase, 2 μl 103 PCR buffer, 1.5 μl 25 mM MgCl2, 1.25 μl 4 mM of each dNTP, 10 mol each primer, 100 ng template DNA, completed to 25 μl with distilled water. The forward primer used was SB8 (GGGTGGATTTATTAGAACAG) and the reverse one was PB (CCGTCAATT CMTTTRAGTTT). PCR fragments were generated in capillary thermal cycler by 25 cycles of the following program: 10 s at 95 °C, 10 s at 55 °C, and 120 s at 72 °C. PCR fragments were sequenced directly using 48 capillary ABI PRISM 310 Automatic DNA Sequencer (Applied Biosystems) using the Big-Dye Terminator v3.1 Cycle Sequencing Ready Reaction Kit (Applied Biosystems). The forward primer SB3 (GGAGGGCAAGTCTGGTGC) and the reverse one SB9 (TTTCACCTCTAACACCGC) and A27 (CCATACAAATGCCCCCGTCTG) were used for sequencing. DNA fragments were sequenced in both directions two times at least to ensure accuracy. Despite this, some bases remained unresolved and were shown as “N” in the alignment. Neither unresolved bases nor alignment gaps were taken into account in calculating the number of sites that were variable or phylogenetically informative for parsimony analyses.
Sequence alignment and phylogenetic analysis
To evaluate the relationship of the present studied species, a homology search was performed using NCBI/BLAST database (www.ncbi.nlm.nih.gov/blast) on Gene Bank (Altschul et al. 1997). For phylogenetic analysis, sequences of SSU rDNA for the present species were aligned and compared with those of eight digenean species recovered from Gene bank. Sequences were truncated for homology and sequence identities (percent similarity) between the present and comparable species (Table 1). Sequences were submitted to the Gene Bank database and assigned with accession number KC166146.
Alignments of the newly obtained sequence with other sequences from Gene Bank were performed using CLUSTAL-X v1.83 (Thompson et al. 1997). The data set for the alignment was chosen on the basis of the results on BLAST searches and morphological findings. The alignment was then manually corrected using the alignment editor of the software BioEdit 4.8.9 (Hall 1999) to eliminate minor inconsistencies between different taxa. The resulting sequence fragments were assembled into a single contiguous sequence using the multiple-alignment algorithm in Megalign (DNASTAR, Window version 3.12e). Trees were constructed using the neighbor-joining method. When doing bootstrap resampling, 1,000 resampled data sets were evaluated.
Results
Twenty-five (41.66 %) out of 60 collected and examined specimens of A. anguilla were generally found to be naturally infected with L. grandiporum. The infection was increased during winter to 31.66 % (19 out of 30) and fall to 9.99 % (6 out of 30) in summer.
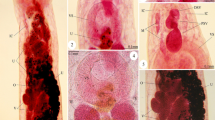
Light and electron microscopic examination of fresh and fixed preparations of the parasites revealed that the adult worm possessed an elongated body, pointed anteriorly, but truncated posteriorly (Figs. 1 and 2). Body measurements were 1.59 ± 0.20 (1.3–1.85) mm long and 0.3 ± 0.02 (0.29–0.48) mm wide. The fore body carried a subterminal oral sucker (Figs. 1 and 2) measuring 0.15 ± 0.02 (0.13–0.18) mm in diameter, which is smaller than the ventral sucker that measured 0.16 ± 0.02 (0.14–0.25) mm in diameter (Figs. 1 and 2). Pharynx is subspherical, well developed, and measured 0.07 ± 0.02 (0.04–0.08) mm in diameter followed by a very short esophagus. The ovary is subspherical, equatorial, post-testicular, and widely separated from the testes by uterine loops. Uterine seminal receptacle is well developed; post-ovarian uterine has numerous coils and fills much of the somatic hind body reaching back to the level of cecal extremities (Fig. 1). Intestinal bifurcation was located slightly anterior to the mid-fore body. Ceca are often inflated with anterior region transversely striated, pass posteriorly in dorsolateral fields and blindly close to the base of ecsoma (Fig. 1). Two rounded testes are located at the ventral field of the body in tandem. Posterior ecsoma is well developed, usually withdrawn (Fig. 3), and sometimes is everted (Figs. 1 and 2). Excretory pore is located at the posterior extremity of the ecsoma (Figs. 1, 2, and 3).

1 Photomicrograph of the adult digenetic trematode L. grandiporum showing the subterminal oral sucker (OS), muscular pharynx (PH), ventral sucker (VS) which extends a small distance to outside, intestinal ceca (IC), a large coiled uterus (U) filled with eggs, and the invaginated ecsoma (EC) with a terminal excretory pore (EP). Scanning electron micrographs showing: 2 the whole body of the adult worm. Observe the subterminal oral sucker (OS) and the large ventral sucker (VS). 3 High magnification of the posterior end of the worm with the retracted ecsoma (EC) terminated at the excretory pore (EP)
Taxonomic summary
Parasite: Lecithochirium grandiporum (Rudolphi 1819) belonging to Family Hemiuridae (Looss 1899)
Type host: European eel Anguilla anguilla (Linnaeus 1758) (F: Anguillidae)
Site of infection: Intestine
Locality: Burullus Lake, Kafr El-Sheikh Governorate, Egypt
Prevalence: Twenty-five (41.66 %) out of 60 specimens of the examined fish were infected
Etymology: The specific name of the parasite (grandiporum) was given because it possesses a ventral sucker with large aperture.
Molecular analysis
Molecular analysis based on 18 SSU rDNA was performed to determine the phylogenetic position of the described species. The amplified and sequenced variable region (V4) of SSU rDNA for the present species was 971 nt and obtained using primers after trimming the 3′ end. Before phylogenetic analysis, only those sites which could be unambiguously aligned among Hemiuridae were used. The GC content of the sequenced gene was 48.09 %. The sequence was deposited in the Gene Bank under accession number KC166146. Submission to the BLAST server showed that eight SSU rDNA sequences including those with the highest BLAST scores were aligned and compared with L. grandiporum, their accession numbers were given in Table 1. Calculation of the percentage of identity (no. of base differences / total number of bases) between this novel sequence and a range of other Hemiuridae predominantly from other hosts demonstrated a high degree of similarity (>82 %) with other Hemiuridae species (Table 1). Comparison of the nucleotide sequences and divergence showed that SSU rDNA of this species revealed 92 % sequence identity with L. fusiforme (accession no. DQ413192) differing in 26 nucleotides with a lower divergence value; 91 % with Lecithochirium caesionis (accession no. AY222200) differing in 31 nucleotides; 90 % with Lecithochirium kawakawa (accession no. AF029800) differing in 40 nucleotides; and 90 % with Lecithochirium genypteri (accession no. AF029799) differing in 46 nucleotides, which represent as the highest four BLAST scores that were aligned with CLUSTAL-X (Fig. 4).

Sequence alignment of L. grandiporum (present study) with those of the most related species. Note: Only variable sites are shown. Dots represented bases identical to those of the first sequences and dashes indicated gaps
Based on SSU rDNA sequence data, the constructed dendrogram splits into two lineages (one major and one minor clade) as shown in Fig. 5. Sequence alignment resulted that the major clade clustering all Hemiuridae species with sequence similarity between 92 and 82 % (Fig. 4). The minor clade containing out-group of SSU rDNA sequence for Lobatostoma manteri (accession no. AY157177) with a high divergence value. The sequence divergences detected between closely related Hemiuridae species varied from low value as 5.2 % (L. grandiporum vs. L. fusiforme) to a high value as 14.5 % (L. grandiporum vs. Lecithaster gibbosus). Phylogenetic relationship between L. grandiporum (present study) and other Hemiuridae species recorded from the Gene Bank which was studied using maximum likelihood, maximum parsimony, and neighbor-joining methods showed that L. grandiporum is deeply embedded in the genus Lecithochirium with close relationship with L. fusiforme and L. caesionis as a more related sister taxons with strong bootstrap values.

A dendrogram based on 18 SSU rDNA gene sequence showing the phylogenetic relationship between the present L. grandiporum and other species belonging to family Hemiuridae recovered from Gene Bank. The 18 SSU rDNA gene sequence of L. manteri was used as out-group
Discussion
Fish parasites have been used for almost a century as biological indicators, markers, or tags to provide information on various aspects of host biology (Williams et al. 1992). Family Hemiuridae comprised the most common parasitic digenean flukes inhabiting the digestive tract of marine fish (Marianne 1990; Shih et al. 2004; Bartoli et al. 2005; Bullard et al. 2011).
Lecithochirium is the most common genus within this family (Shih et al. 2004). This genus now includes at least more than 100 described species (Surekha and Lakshmi 2005). Their host specificity was not distinguished since one species L. fusiforme (Lűhe 1901) was harvested from three different fish species, and another one L. trichiuri (Gu and Shen 1981) occurred in two fish species. L. grandiporum has been reliably reported on seven occasions along the Red Sea (Rudolphi 1819; Looss 1907, 1908; Mola 1928; Sey 1970; Gibson and Bray 1986; Bartoli et al. 2005; Morsy et al. 2012). The same species L. grandiporum was isolated from three different European host species by Bartoli and Gibson (2007) which were Lophius piscatorius, Conger conger, and Muraena helena with all of the fundamental anatomical characters being very similar for example to the digitiform lobes of the vitellarium and the presence of large ejaculatory glands on either side of the sinus sac. It was observed that dimensions of the different body parts detected for the present studied L. grandiporum were smaller than those of the comparable species from the different host species (Table 2).
Gibson and Bray (1986) thought that L. grandiporum is a senior synonym of L. fusiforme and there was no significant morphological difference between the two species. However, both helminthes have different definitive host specificity. Bartoli and Gibson (2007) redescribed L. grandiporum from M. helena which was in a close agreement with that of L. fusiforme from C. conger as provided by Gibson and Bray (1986). All fundamental anatomical characters being very similar. Also, morphologically measurements of the present L. grandiporum were slightly smaller than those of L. fusiforme with a larger sucker ratio, but not significantly different and fall within, or they were close to the range given by Bartoli and Gibson (2007) for L. grandiporum. The ultrastructural morphology in addition to phylogenetic analysis using 18 SSU rDNA genes has become significantly an enhanced tool for the differentiation between species. It is now become an essential criteria for identification of new species and/or redescription of an inadequately described species (Maddison and Maddison 2002; Blair et al. 2005; Testini et al. 2011; Carreras-Aubets et al. 2012). These genes are particularly useful for elucidating relationships in this group because it is highly variable between very closely related species (Bullard 2010).
The general structure of the phylogram obtained in the present study is consistent with previous analyses by Blair et al. (1998), which was constructed using maximum likelihood and maximum parsimony that revealed the same gross topology and showed that L. grandiporum revealed a separate line, which was easily distinguishable to be deeply embedded in the genus Lecithochirium with strongly supported molecular data. Some clades strongly supported by molecular data lack corresponding morphological synapomorphies. The most notable of these is the branch leading to the base of the clade containing the species from related families. This leads us to believe that both kinds of data are valuable in inferring relationships among the Digenea (Testini et al. 2011).
Blair et al. (1998) and Cribb et al. (2001) stated that the Hemiuridae genus Lecithochirium is monophyletic and its division into major clades is consistent with our results. The clustering of the Lecithochirium species is independent of the host species/family and shows no relation to the geographical origin. However, clustering according to host tissue localization can be observed in some species, as L. fusiforme and L. caesionis which infect the host intestine as a result L. grandiporum clustered with them in one clade which having the same host tissue localization.
The close morphological characteristics between the species described here and those of the same genus indicated the urgent need to apply molecular investigation either to confirm or to deny this similarity.
The present molecular investigations revealed 92 % similarity between L. grandiporum and L. fusiforme showing that they are clearly separate species differing in 26 nucleotide positions in its SSU rDNA sequence. Previous molecular phylogenetic studies have demonstrated a high degree of sequence similarity between a subset of Lecithochirium species (Cribb et al. 2001; Vilas et al. 2005; Bullard 2010; Bullard et al. 2011; Carreras-Aubets et al. 2012). The present investigation also observed that all Lecithochirium showed at least 90 % similarity to present sequence. Parasites from other clades showed only 88–82 % similarities.
The addition of new sequences from this study identifies the ancestral marine origin of the present Lecithochirium species and it strongly aids to understand the cladistic arrangement within the more recent clade due to the addition of new species belonging to the previous genera.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402, http://www.ncbi.nlm.nih.gov/blast
Al-Zubaidy A (2010) First record of Lecithochirium sp. (Digenea: Hemiuridae) in the marine fish Carangoides bajad from the Red Sea, coast of Yemen. J KAU Mar Sci 21:85–94
Bartoli P, Gibson DI (2007) The status of Lecithochirium grandiporum (Rudolphi, 1819) (Digenea: Hemiuridae), a rarely reported and poorly known species from the Mediterranean moray eel Muraena helena L. in the Western Mediterranean. Syst Parasitol 68:183–194
Bartoli P, Gibson DI, Bray RA (2005) Digenean species diversity in teleost fish from a nature reserve off Corsica, France (Western Mediterranean), and a comparison with other Mediterranean regions. J Nat Hist 39:47–70
Blair D, Barker SC (1993) Affinities of the Gyliauchenidae: utility of the 18S rRNA gene for phylogenetic inference in the Digenea (Platyhelminthes). Int J Parasitol 23:527–532
Blair D, Bray RA, Barker SC (1998) Molecules and morphology in phylogenetic studies of the Hemiuroidea (Digenea: Trematoda: Platyhelminthes). Mol Phylo Evol 9:15–25
Blair D, Chang Z, Chen M, Cui A, Wu B, Agatsuma T, Iwagami M, Corlis D, Fu C, Zhan X (2005) Paragonimus skrjabini Chen, 1959 (Digenea: Paragonimidae) and related species in eastern Asia: a combined molecular and morphological approach to identification and taxonomy. Syst Parasitol 60:1–21
Bray RA (1991) Hemiuridae (Digenea) from marine fishes of the southern Indian Ocean: Genus Lecithochirium Lűhe, 1901 (Lecithochirriinae). Syst Parasitol 18:193–219
Bullard SA (2010) Littorellicola billhawkinsi n. gen., n. sp. (Digenea: Aporocotylidae) from the myocardial lacunae of Florida pompano, Trachinotus carolinus (Carangidae) in the Gulf of Mexico; with a comment on the interrelationships and functional morphology of intertrabecular aporocotylids. Parasitol Int 59:587–598
Bullard SA, Barse AM, Curran SS, Morris JA (2011) First record of a Digenean from invasive lionfish, Pterois cf. Volitans, (Scorpaeniformes: Scorpaenidae) in the Northwestern Atlantic Ocean. J Parasitol 97(5):833–837
Carreras-Aubets M, Montero F, Kostadinova A, Gibson D, Carrasson M (2012) Redescriptions of two frequently recorded but poorly known hemiurid digeneans, Lecithochirium musculus (Looss, 1907) (Lecithochiriinae) and Ectenurus lepidus Looss, 1907 (Dinurinae), based on material from the western Mediterranean. Syst Parasitol 82:185–199
Casanova JC, Santalla F, Durand P, Vaucher C, Feliu C, Renaud F (2001) Morphological and genetic differentiation of Rodentolepis straminea (Goeze, 1752) and Rodentolepis microstoma (Dujardin, 1845) (Hymenolepididae). Parasitol Res 87:439–444
Cribb TH, Bray RA, Littlewood DTJ, Pichelin SP, Herniou EA (2001) The Digenea. In: Littlewood DTJ, Bray RA (eds) Interrelationships of the Platyhelminthes. Taylor and Francis, London, pp 168–185
Criscione CD, Blouin MS (2004) Life cycles shape parasite evolution: comparative population genetics of salmon trematodes. Evolution 58:198–202
Gibson DI, Bray RA (1979) The Hemiuroidea: terminology, systematics and evolution. Bull Br Mus Nat Hist Zool Ser 36:35–146
Gibson DI, Bray RA (1986) The Hemiuridae (Digenea) of fishes from the north-east Atlantic. Bull Br Mus Nat Hist Zool Ser 51:1–125
Gu CD, Shen JW (1981) Digenetic trematodes of ribbonfish, Trichiurus haumela (Forska) and their distribution in the fishing grounds of China Seas. Act Zool Sin 27:53–63
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nuc A Symp Ser 41:95–98
Jones EI (1933) Ceratotrema furcolabiata n.g. et n. sp and Hemipera sharpei n. sp, two new digenetic trematodes of British marine fishes. Parasitology 25:248–254
Lee RE (1993) Scanning electron microscopy and X-ray microanalysis. PTR Prentice Hall, Englewood Cliffs, p 07632
Looss A (1907) Zur Kenntnis der Distomenfamilie Hemiuridae. Zool Anz 31:585–620
Looss A (1908) Beiträge zur Systematik der Distomen. Zur Kenntnis der Distomenfamilie Hemiuridae. Zool Jahrb Syst 26:63–180
Lűhe M (1901) Über Hemiuriden (Ein Beitrag zur Systematik der digenetischen Trematoden). Zool Anz 24:473–488
Maddison DR, Maddison WP (2002) McClade 4: analysis of phylogeny and character evolution. Sinauer, Sunderland
Marianne K (1990) On the morphology and life-history of Hemiurus luehei Odhner, 1905 (Digenea: Hemiuridae). J Parasitol 64:193–202
Mola P (1928) Vermi parassiti del’ittiofauna Italiana. Boll Pesca Piscicoltura edi Idriobiologica 4:395–443
Morsy K, Bashtar A, Abdel-Ghaffar F, Baksh W (2012) First record of Lecithochirium grandiporum (Digenea: Hemiuridae) infecting the lizard fish Saurida tumbil from the Red Sea. Parasitol Res 111(6):2339–2344
Murakami J (1977) Modified TOA. Murakami procedure. Arch Histo 40:119
Pankov P, Webster BL, Blasco-Costa I, Gibson DI, Littlewood DT, Balbuena JA, kostadinova A (2006) Robinia aurata n. g., n. sp. (Digenea: Hemiuridae) from the mugilid Liza aurata with a molecular confirmation of its position within the Hemiuroidea. Parasitology 133:217–227
Randall J (1992) Red Sea reef fishes. Immel, London, p 192
Rudolphi CA (1819) Entozoorum synopsis, cui accident mantissa duplex et indices locupletissimi. Sumtibus Augusti Rűcker, Berolini, p 811
Sambrook J, Fritsch EF, Maniatis T (1989) Chapter 14: in vitro amplification of DNA by the polymerase chain reaction. In: molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, New York
Sey O (1970) Parasitic helminths occurring in Adriatic fishes. Part II (flukes and tapeworms). Acta Adriat 13(6):3–15
Shih HH, Liu W, Zhao ZQ (2004) Digenean fauna in marine fishes from Taiwanese water with the description of a new species Lecithochirium tetraorchis sp. nov. Zool Stud 43(4):671–676
Surekha P, Lakshmi CV (2005) Lecithochirium testelobatus n. sp. (Digenea: Hemiuridae) from the lizard fish, Saurida undosquamis from Andhrapradesh Coast. J Parasitol Dis 29(2):143–146
Testini G, Papini R, Lia RP, Parisi A, Dantas-Torres F, Traversa D, Otranto D (2011) New insights into the morphology, molecular characterization and identification of Baylisascaris transfuga (Ascaridida, Ascarididae). Vet Parasitol 175:97–102
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL-X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nuc A Res 25:4876–4882
Vilas R, Paniagua E, Outeiral S, Sanmartin ML (2002) Electrophoretic and morphological differentiation of three sympatric species of the genus Lecithochirium (Trematoda: Hemiuridae), parasites of marine fishes. Parasitol Res 88:1055–1060
Vilas R, Criscione CD, Blouin MS (2005) A comparison between mitochondrial DNA and the ribosomal internal transcribed regions in prospecting for cryptic species of Platyhelminthes parasites. Parasitology 131:839–846
Williams H, Jones A (1994) Parasitic worms of fishes. Taylor and Francis, London, p 593
Williams HH, Mackenzie K, McCarthy AM (1992) Parasites as biological indicators of the population biology, migrations, diet and phylogenetics of fish. Rev Fish Biol Fish 2:144–176
Acknowledgments
This work was supported by the Faculty of Science, Cairo University, Egypt.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdel-Ghaffar, F., Bashtar, AR., Mehlhorn, H. et al. Morphological and molecular characterization of Lecithochirium grandiporum (Digenea: Hemiuridae) infecting the European eel Anguilla anguilla as a new host record in Egypt. Parasitol Res 112, 3243–3250 (2013). https://doi.org/10.1007/s00436-013-3502-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-013-3502-4