
Abstract
Sarcocystis cameli was first described in one-humped camels (Camelus dromedarius), and it is the only species which have so far reported in camels. Although more than 150 species of Sarcocystis were described in various animals, only a few data on camel Sarcocystis ultrastructure were published, and this report is the first for molecular information (DNA sequence and RLFP digestion pattern). The main objective of the present work is to characterize Sarcocystis isolated from camels by electron microscopy and PCR-RFLP methods. Muscle samples were taken from the fresh esophagus, diaphragm, skeletal muscles, and heart of one-humped camels (C. dromedarius) slaughtered in abattoirs of Tehran and Ghazvin provinces, Iran. The dissection and trypsin digestion techniques were applied for the detection of the cysts. The infected samples were fixed in glutaraldehyde and/or frozen at −20°C until use for ultrastructural and molecular studies, respectively. The ultrastructural and molecular studies were carried out contemporaneously. The 18S rRNA gene of the parasites was amplified by PCR. The PCR products were cloned into a pTZ57R/T and sequenced. In addition, the PCR products were digested separately with each of the four restriction enzymes for RFLP. Our results indicated that only microcysts were observed in muscle samples. The microcysts were white, elongated, spindled, and a few spiral-shaped, with mean size 260 × 75 μm which are identical with S. cameli. The ultrastructure of microcyst wall had many non-branched finger-like protrusions irregularly folded. There was a 600-bp specific band amplified after PCR with specific primers. The molecular data for camel Sarcocystis is reported for the first time in Iran and the world.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sarcocystosis is caused by different species of Sarcocystis, an intracellular protozoan parasite belonging to phylum Apicomplexa. More than 150 species of Sarcosystis are known as parasites of domestic and wild animals. The genus is worldwide distributed. These parasites have an indirect life cycle, between a definitive and an intermediate host. Intestinal infection occurs in the definitive hosts, and tissue invasion is seen in the intermediate hosts. Some species are pathogenic organisms dangerous to humans and livestock (Butkauskas et al. 2007; Fayer 2004). Sarcocystis is a known parasite of considerable veterinary economic and public health importance. Intermediate hosts become infected when they ingest oocysts or sporocysts (Al-Goraishi et al. 2004). The parasite produces tissue cyst in muscles of intermediate host. The muscle cysts may be macroscopic or microscopic in size.
Sarcocystis cameli was first described in one-humped camels (Camelus dromedarius) in Egypt by Mason (1910), and it is the only species which have so far been reported in camels. There are only three reports on the prevalence of sarcocysts infection in camels from Tehran, Esfahan, and Khorasan Provinces in the central and northeastern area of Iran (Rahbari et al. 1981; Shekarforoush et al. 2006; Valinezhad et al. 2008). Rahbari et al. (1981) and Shekarforoush et al. (2006) examined the camel muscles by impression smear method, and they reported infection rates of 52.6% and 52.3%, respectively. Valinezhad et al. (2008) examined the muscles by histopathological method and reported infection rates of 83.6%. They found only microcysts in camel muscles.
Ultrastructure characteristic of sarcocysts is one of the most important criteria for the specification of Sarcocystis species (Mehlhorn and Heydorn 1978, 1979; Mehlhorn et al. 1976; Melhorn 2008). Ultrastructural study on cysts wall has been used for identification of Sarcocystis species isolated from different animals by many workers (Abdel-Ghaffar et al., 2009; Al-Goraishi et al. 2004; Dalimi et al. 1999; Dubey et al. 1989a; Dubey et al. 1989b; Ghaffar et al. 1989; Mehlhorn and Heydorn 1978, 1979; Mehlhorn et al. 1976). Since studies on Sarcocystis taxonomy in camel are scarce and there is a lack of comprehensive information on species infecting this animal as intermediate host (Shekarforoush, et al. 2006), conducting investigation on species characterization of the parasite is so much necessary in our country. In addition, in recent years, molecular technique has been applied for Sarcocystis characterization isolated from different animals by some investigators (Butkauskas et al. 2007; Costa da Silva et al. 2009; Fischer and Odening 1998; Holmdahl et al. 1999; Li et al. 2002; Yang and Zuo 2000; Yang et al. 2000; Yang et al. 2001a; Yang et al. 2001b; Yang et al. 2002). As a noticeable point, there is no molecular report conducting camel Sarcocystis in the world.
The aim of the present study was to investigate a full description and distinguish the Sarcocystis species isolated from camel by studying the ultrastructure of the cyst wall by electron microscopy and combining these data with information on DNA sequence and restriction fragment length polymorphism (PCR-RFLP) characters.
Materials and methods
Sample collecting
One-humped camels (C. dromedarius) are raised in the semi-arid regions in Iran, mostly for transportation and meat consumption purposes. Muscle samples were taken from the fresh esophagus, diaphragm, skeletal muscles, and heart of one-humped camels slaughtered in abattoirs of Tehran and Ghazvin provinces, Iran. The specimens were tagged according to each organ. Collected samples of each organ were examined by the naked eye for macrocyst. For detection of microscopic sarcocysts in muscles, the surface of the muscles was cut serially and searched with a sterio-microscope at ×12 to ×25 magnification. Cysts were isolated from dissections using sterile needles. Fresh smear preparations by squash and digestive techniques were applied for the detection and verification of microscopic sarcocysts and existence of bradyzoites (Dubey et al. 1989a, b; Odening et al. 1996).
Squash technique
The technique was used for detection of bradyzoite, briefly; a piece of muscle sample was squashed between a cover-glass and slide and then examined unstained using a light microscope (Singh et al. 1990; Dubey et al. 2000).
Pepsin-hydrochloric acid digest
Infected organs (20 g) were incubated (20 min at 40°C) in 50 ml of acid-pepsin (2.6 g pepsin, 5 g NaCl, 7 ml HCl 1 M, 993 ml distilled water). This suspension was filtered through a 38-μm sieve, centrifuged at 2,000×g for 5 min, and sediment-suspended in 0.5 ml of distilled water. A drop of this solution was examined for the presence of bradyzoites with light microscope (Barham et al. 2005; Fischer and Odening 1998).
Electron microscopy
Small pieces of infected organs were washed in 0.9% NaCl, then fixed in the buffered glutaraldehyde (4%) and prepared to examine with transmission electron microscope. The samples were washed in phosphate buffer (0.2 M, pH = 7.2) three times, then post fixed in osmium tetra-oxide (2% w/v, for 2 h), dehydrated in ethanol (30–100%), transferred to propylene oxide (1 h), and embedded in Epoxi Resin (EPON812), and after polymerization (72 h at 60°C), semi-thin and ultra-thin sections were cut with a Om U3 (C. Reichert, Austria) microtome. Semi-thin sections were stained with toluidine blue and examined with light microscope. Ultra-thin sections were stained with uranyl acetate and lead citrate, then examined with (Philips TEM400) transmission electron microscope (Dalimi et al. 1999).
Molecular study
Infected muscles were minced and incubated in pepsin-hydrochloric acid digest solution, and then the bradyzoites were separated from host debris by centrifugation. The sediment was washed three times with buffer saline, and cleaned zoites were pelleted in small conical tubes and held frozen at −20°C until use (Costa da Silva et al. 2009).
Total DNA was extracted from positive samples using DNA Purification Kit (DNP™KIT, CinnaGen Inc., Iran) according to the manufacturer's instruction. The DNA concentration and purity was measured with a Nano drop ND 1000 spectrophotometer. The extracted DNA was kept at −20°C until used. Polymerase chain reaction (PCR) was used to amplify partial sequence of small subunit ribosomal RNA (18S rRNA) gene sequences of camel Sarcocystis. Forward and reverse primers were selected from domestic animals Sarcocystis published sequences in the gene bank. The nucleotides of primers are as:
-
F-5′ GCA CTT GAT GAA TTC TGG CA 3′
-
R-5′ CAC CAC CCA TAG AAT CAA G 3′
The PCR was performed (30 μl reactions) using 1 μl (10 pM) of each primer, 0.5 μl Taq polymerase, 0.5 μl dNTP, 2 μl MgCl2, 10 μl DNA, 3 μl buffer 10× and 12 μl distilled water. Reactions were carried out on an Eppendorf Master Cycler Gradient thermal cycler. The PCR required a preliminary denaturation at 94°C for 5 min. The remaining PCR steps were 35 cycles at 94°C (2 min), 57°C (30 s), 72°C (2 min), with a single terminal step at 72°C (5 min). The PCR products were analyzed using agarose gel electrophoresis and purified by the PCR Purification Kit (Roche), following the manufacturer's instructions. Cloning were carried out by Plasmid Mini Extractin Kit (Bioneer, Republic of Korea) and Ins TAclone™ PCR Cloning Kit (Fermentas), respectively, following the manufacturer's instructions. The PCR products were cloned into a pTZ57R/T and sequenced. The comparison of the obtained sequences with the GenBank was performed using Blast program. After sequencing the 18S rRNA, the PCR products were digested separately with each of the four restriction enzymes (Xba1, Mbo1, EcoR1, Ava11). Each 30 μl restriction digest contained 20 μl of PCR product and 2 μl of the appropriate restriction enzyme and 3 μl associated buffer and 5 μl of distilled water. Incubation times and temperatures were 12 h and 37°C, respectively. The digests were terminated by addition of 4 μl loading mixture (50% glycerol, 0.1 M ethylene diamine tetra-acetic acid, 0.01% bromo-phenol blue, pH = 8). Digestion products were electrophoressed on 2% agarose gel at 60 V (120 min). Gel was stained with ethidium bromide, and the fragments visualized with UV transilluminator. Digestion reactions containing PCR product but lacking restriction enzyme were used as negative controls. A DNA size marker was run on each gel for estimation of fragment size (Yang et al. 2002; Fischer and Odening 1998).
Results
No macroscopic sarcocysts were found in any of the samples obtained from camels (C. dromedarius) and only microcystic species were observed. The microcysts were elongated, spindle, and a few spiral-shaped, with a mean size 250 × 75 μm (305–130 × 105–45). In squash preparations, the bradyzoites were ovoid with mean size of 7 × 3 μm.
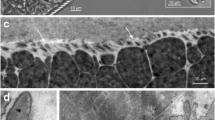
The ultastructure of the primary cyst wall revealed the presence of irregular folded non-branch finger-like villar protrusions that are characterized by the presence of internal fibrillar elements (Fig. 1). The ground substance is located directly under the primary cyst wall and extend into the protrusions and consist mainly fine dense homogenous granules and fibrillar elements which are characteristic for S. cameli (Figs. 2 and 3).

Ultrastructure of primary cell wall of microcyst in S. cameli (magnification ×4,200)

Ultrastructure of primary cell wall of microcyst in S. cameli (magnification ×11,220)

Ultrastructure of fine dense homogenous granules and internal fibrillar elements of primary cell wall of microcyst in camel (magnification ×27,800)
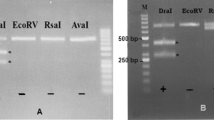
The PCR yielded an amplicon of approximate length of 539 bp for all samples (Fig. 4). The PCR products were cloned in a pTZ57R/T successfully and sequenced. Sequence of PCR product of partial small subunit ribosomal RNA (18S rRNA) gene of S. cameli is shown in Fig. 5, and restriction map of sequence of PCR product is shown in Fig. 6. The PCR amplicon was submitted in GenBank with GU074011 accession number.

PCR products of partial small subunit ribosomal RNA (18S rRNA) gene of Sarcocystis cameli (539 bp) as visualized on 2% agarose gels (stained with ethidium bromide) in UV light: lane 1 DNA size marker (100 bp ladder), lane 2 uninfected tissue (negative control), lane 3 positive control (sheep sarcocyst), lane 4 (negative control), lane 5 camel sarcocyst1, lane 6 camel sarcocyst2, lane 7 camel sarcocyst3, lane 8 Toxoplasma tachyzoite

Sequence of PCR product of partial small subunit ribosomal RNA (18S rRNA) gene of Sarcocystis cameli (539 bp)

Restriction map of sequence of PCR product of partial small subunit ribosomal RNA (18S rRNA) gene of Sarcocystis cameli (539 bp)
Figure 7 shows the RFLP patterns for the samples after digestion with four restriction enzymes (Xba1, Mbo1, EcoR1, Ava11). EcoR1 does not show any restriction sites, but the other three enzymes demonstrate different size of fragments. Xba1 enzyme shows two fragments of 429 and 110 bp, Mbo1 illustrates 274 and 265 bp fragments, and Ava11 reveals 480 and 59 bp fragments. Xba1 electomorph pattern is specific for Sarcocystis in camel and are suitable for differentiate S. cameli from the rest. Thus, the PCR products of all collected samples were digested with this enzyme. Digestion with Xba1 yielded the same electomorph pattern for all samples, which means that the whole samples are synonym species (Fig. 8).

RFLP pattern of PCR product of partial small subunit ribosomal RNA (18S rRNA) gene of Sarcocystis cameli (539 bp) with different restriction enzymes as visualized on 2% agarose gels (stained with ethidium bromide) in UV light: lane 1 DNA size marker (100 bp ladder), lane 2 undigested product (positive control), lane 3 Ava11 enzyme digestion pattern (480 bp, 59 bp), lane 4 EcoR1 enzyme digestion pattern (negative), lane 5 Mbo1 enzyme digestion pattern (274 bp,265 bp), lane 6 Xba1 enzyme digestion pattern (429 bp,110 bp), lane 7 negative control

Xba1 digest pattern of PCR product of partial small subunit ribosomal RNA (18S rRNA) gene of Sarcocystis cameli (539 bp) as visualized on 2% agarose gels (stained with ethidium bromide) in UV light: lane 1 DNA size marker (100 bp ladder), lane 2 undigested product (positive control), lanes 3, 4, 5 Xba1 digestion pattern (429 bp, 110 bp), lanes 6, 7 negative control
Discussion
Up to now, various characters such as host specificity, morphology of cyst, ultra structure of cyst wall, as well as biochemical and molecular characteristics were applied for description of Sarcocystis species. In fact, the cysts have distinctive physical features that aid in species identification such as shape, overall size, and presence or absence of septa. But these features may vary with the age of sarcocyst, the host cell type, and the method of fixation. Therefore, taxonomy based on shape and size of cyst is not reliable. Classification on the basis of the microscopic feature of cyst walls also have often failed to provide reliable characters because of unsuitable way of cyst isolation and effect of fixation substances, but the ultrastructural characters of cyst wall was found a good indication for identification of the parasite species. In addition, in recent years, molecular technique has been applied with high efficiency for species characterization of Sarcocystis isolated from different animals.
Although there are some reports about Sarcocystis species in camel, information on the developmental stages and the details of cyst wall are scarce, and only few data on camel sarcocyst ultrastructure were known. It is obvious that the Sarcocystis that infect camel cause microcysts which are hidden within the muscles of animal and can only be observed with armed eye. The present study showed that the examined camels have infected only with microscopic form of Sarcocystis. The ultrastructural observations of the cyst cell wall showed that all cysts resemble S. cameli. The ground substances located directly under the primary cyst wall and extend into the protrusions and consist of mainly fine dense homogenous granules and fibrillar elements are specific characters for S. cameli that was reported before generally. The present observation is similar to the reports of Al-Goraishi et al. (2004).
The variable regions of the 18S rRNA gene have been shown to be good genetic markers for distinguishing certain species of Sarcocystis (Fischer and Odening 1998; Yang and Zuo 2000). Although it is possible to study variation in this gene directly by DNA sequencing, as done by Fischer and Odening (1998); Holmdahl et al. (1999); Mugridge et al. (1999, 2000); and Yang et al. (2000, 2001a, b, 2002). On the other hand, PCR-RFLP technique have been shown to give the same result as detailed morphological studies or DNA sequence-based identifications but in a rapid and more cost-effective manner (Yang et al. 2002). Comparing the sequencing of our result with other sequences in the GenBank showed that it belonged to genus Sarcocystis sequences of other animals with 96–97% identity. Moreover, DNA sequencing and PCR-RFLP characters were found unique for Sarcocystis of camel.
In summary, the present work has demonstrated that both ultrastructural study and RFLP-based techniques may be used to distinguish camel species of Sarcocystis from other species. Xba1 is found to be an appropriate restriction enzyme to differentiate Sarcocystis of camel from other Sarcocystis in goat, sheep, and cattle. The PCR-RFLP technique is cost-effective and rapid in comparison with ultrastructural study or DNA sequencing procedures. In addition, our results indicated that only one Sarcocystis species (S. cameli) was identified in our country.
References
Abdel-Ghaffar F, Mehlhorn H, Bashtar AR, Al-Rasheid K, Sakran T, El-Fayoumi H (2009) Life cycle of Sarcocystis camelicanis infecting the camel (Camelus dromedarius) and the dog (Canis familiaris), light and electron microscopic study. Parasitol Res 106:189–195
Al-Goraishi SAR, Bashtar AR, Al-Rasheid KAS, Abdel-Ghaffar FA (2004) Prevalence and ultrastructure of Sarcocystis species infecting camels (Camelus dromedarius) slaughtered in Riyadh city Saudi Arabia. Saudi J Biol Sci 11(2):135–141
Barham M, Stutzer H, Karanis P, Latif BM, Neiss WF (2005) Seasonal variation in Sarcocystis species infections in goats in northern Iraq. Parasitology 130(2):151–156
Butkauskas D, Sruoga A, Kutkiene L, Prakas P (2007) Investication of the phylogenic relationships of Sarcocystis spp. from Greylag (Anser anser) and white-fronted (Anser aalbifrons) geese to other cyst forming coccidian using 18S rRNA gene sequences. Acta zool 17(2):124–128
Costa da Silva R, Chunlei Su, Langoni H (2009) First identification of Sarcocystis tenella (Railliet, 1886), Moule 1886 (Protozoa: Apicomplexa) by PCR in naturally infected sheep from Brazil. Vet Parasitol 165:332–336
Dalimi A, Khodashenas M, Nouri A, Morovati M (1999) Ultrastructural study of Sarcocystis isolated from water buffalo (Bubalus bubalis) in Khozestan province in Iran. Pajouhesh Sazandegi 43:47–49 (In Persian)
Dubey JP, Speer CA, Charleston WAG (1989a) Ultrastructural differentiation between Sarcocystis of Sarcocystis hirsute and S. hominis. Vet Parasitol 34:153–157
Dubey JP, Speer CA, Shah HL (1989b) Ultrastructural of Sarcocystis from water buffalo in India. Vet Parasitol 34:149–152
Dubey JP, Saville WJA, Lindsay DS, Stich RW, Stanek JF, Speer CA, Rosenthal BM, Njoku CJ, Kwok OCH, Shen SK, Reed SM (2000) Completion of life cycle of Sarcocystis neurona. J Parasitol 86:1276–1280
Fayer R (2004) Sarcocystis spp in human infection. Clin Microbiol 17(4):894–902
Fischer S, Odening K (1998) Characterization of bovine Sarcocystis species by analysis of their 18S ribosomal DNA sequences. J Parasitol 84(1):50–54
Ghaffar FA, Heydorn AO, Mehlhorn H (1989) The fine structure of cysts of Sarcocystis moulei from goats. Parasitol Res 75:416–418
Holmdahl OJM, Morrison DA, Ellis JT, Huong LTT (1999) Evolution of ruminant Sarcocystis (Sporozoa) parasites based on small subunit rDNA sequences. Mol Phylo Evol 11:27–37
Li QQ, Yang ZQ, Zuo YX, Attwood SW, Chen XW, Zhang YP (2002) A PCR-based RLFP analysis of Sarcocystis cruzi (Protozoa: Sarcocystidae) in Yunnan Province, PR China, reveals the water buffalo (Bubalus bubalis) as a natural intermediate host. J Parasitol 88(6):1259–1261
Mason FP (1910) Sarcocystis in the camel in Egypt. J Comp Pathol Therap 23:168–176
Melhorn H (2008) Encyclopedia of parasitology, 3rd edn. Springer, Berlin
Mehlhorn H, Heydorn AO (1978) The Sarcosporidia (Protozoa, Sporozoa): life cycle and fine structure. Adv Parasitol 16:43–93
Mehlhorn H, Heydorn AO (1979) Electron microscopical study on gamogony of Sarcocystis suihominis in human tissue cultures. Z Parasitenkd (Parasitol Res) 58:97–113
Mehlhorn H, Hartley WJ, Heydorn AO (1976) A comparative study of the cyst wall of 13 Sarcocystis species. Protistologica 12:451–467
Mugridge NB, Morrison DA, Heckeroth AR, Johnson AM, Tenter AM (1999) Phylogenetic analysis based on full-length large subunit ribosomal RNA gene sequence comparison reveals that Neospora caninum is more closely related to Hammondia hedorni than to Toxoplasma gondii. Inter J Parasitol 29:1545–1556
Mugridge NB, Morrison DA, Jekel T, Heckeroth AR, Tenter AM, Johnson AM (2000) Effect of sequence alignment for the Protozoa family Sarcocystidae. Soci Mol Biol Evol 17:1842–1853
Odening K, Stolte M, Bockhardt I (1996) On the diagnostics of Sarcocystis in cattle: sarcocysts of a species unusual for Bos taurus in a dwarf zebu. Vet Parasitol 66:19–24
Rahbari S, Bazargani TT, Rak H (1981) Sarcocystosis in the camel in Iran. J Fac Vet Med Univ Tehran 37:1–10
Shekarforoush SS, Shakerian A, Hassanpoor MM (2006) Prevalence of Sarcocystis in slaughtered one-humped camels (Camelus dromedarius) in Iran. Trop Anim Health Prod 38:301–303
Singh KP, Agrawal MC, Shah HL (1990) Prevalence of Sarcocysts of Sarcocystis capracanis in oesophagus and tail muscles of naturally infected goats. J Vet Parasitol 36:153–155
Valinezhad A, Oryan A, Ahmadi N (2008) Sarcocystis and its complications in camels (Camelus dromedarius) of Eastern provinces of Iran. Korean J Parasitol 46(4):229–234
Yang ZQ, Zuo YX (2000) The new views of the researchers on cyst forming coccidia species including Sarcocystis by using the molecular biological techniques. Chines J Parasitol Parasit Dis 18:120–126
Yang ZQ, Zuo YX, Ding B, Chen XW, Luo J, Zhang YP (2000) 18S rRNAgene of Sarcocystis hominis cyst from water buffalo and cattle. Zool Res 21:133–138
Yang ZQ, Zuo YX, Ding B, Chen XW, Luo J, Zhang YP (2001a) Identification of Sarcocystis hominis-like (Protozoa: Sarcocystidae) cyst in water buffalo (Bubalus bubalis) based on 18S rRNA gene sequences. J Parasitol 87:934–937
Yang ZQ, Zuo YX, Yao YG, Chen XW, Yang GC, Zhang YP (2001b) Analysis of the 18S rRNA genes of Sarcocystis species suggests that the morphologically similar organ from cattle and water buffalo should be considered the same species. Mol Biochem Parasitol 115:283–288
Yang ZQ, Li QQ, Zuo YX, Chen XW, Chen YJ, Nie L, Wei CG, Zen JS, Attwood SW, Zhang XZ, Zhang YP (2002) Characterization of Sarcocystis species in domestic animal using a PCR-RLFP analysis of variation in the 18S rRNA gene: a cost effective and simple technique for routine species identification. Exp Parasitol 102:212–217
Acknowledgements
This research has been supported financially by Razi Vaccine and Serum Institute and Tarbiat Modares University. The authors would like to thank the staff of Parasitology, Electron Microscopy and Biotechnology Departments of Razi Institute for their kind cooperation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Motamedi, G.R., Dalimi, A., Nouri, A. et al. Ultrastructural and molecular characterization of Sarcocystis isolated from camel (Camelus dromedarius) in Iran. Parasitol Res 108, 949–954 (2011). https://doi.org/10.1007/s00436-010-2137-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-010-2137-y