
Abstract
A total of 124 fecal specimens were collected from four deer farms in Zhengzhou City, China and examined for Cryptosporidium by Sheather’s sugar flotation technique. Cryptosporidim oocysts were detected in two 1-year-old sika deer, and one of the two specimens was genotyped by sequence and phylogenetic analyses of the small subunit ribosomal RNA (rRNA) (18S rRNA), 70-kDa heat shock protein (HSP70), actin, and Cryptosporidium oocyst wall protein (COWP) genes. Results obtained suggested that the Cryptosporidium studied belonged to Cryptosporidium cervine genotype, although slight sequence differences were noticed at the three loci. The similarities between this isolate and other Cryptosporidium cervine genotype isolates were 99.1–99.8%, 9.8%, 99.7%, and 100% at the 18S rRNA, HSP70, actin, and COWP loci, respectively. This study is the first report of Cryptosporidium infection in sika deer in China.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cryptosporidiosis is a significant cause of diarrheal diseases in human worldwide, usually by routes of waterborne or food-borne transmission. So far, at least seven Cryptosporidium species (C. hominis, C. parvum, C. meleagridis, C. felis, C. canis, C. muris, and C. suis) and one genotype (Cryptosporidium cervine genotype) have been shown to be responsible for human infections (Xiao et al. 2004).
Cryptosporidium cervine genotype has a wide host and geographic range. It has been found in sheep in Australia and USA (Ryan et al. 2005; Santin et al. 2007), an ibex in China (Karanis et al. 2007), one blesbok, mouflon sheep, and nyala each in Czech Republic (Ryan et al. 2003), humans in Canada, England, Slovenia, and USA (Blackburn et al. 2006; Feltus et al. 2006; Leoni et al. 2006; Ong et al. 2002; Soba et al. 2006; Trotz-Williams et al. 2006), wild white-tailed deer, eastern gray squirrels, eastern chipmunk, beaver, red squirrel, oodchuck, deer mice, and raccoon in New York (Feng et al. 2007; Perz and Le Blancq 2001), lemurs in USA (da Silva et al. 2003), and water samples in various geographic locations (Feltus et al. 2006; Jiang et al. 2005; Nichols et al. 2006; Ruecker et al. 2007; Xiao et al. 2000).
There is no report of Cryptosporidium cervine genotype infection in sika deer in China. In the present study, we have characterized a Cryptosporidium cervine genotype isolate from a sika deer (Cervus nippon Temminck) by sequence and phylogenetic analyses of four genes. Results suggest that the Chinese sika deer isolate had minor sequence difference from other known isolates.
Materials and methods
A total of 124 fecal specimens from sika deer (C. nippon Temminck), red deer (C. elaphus) and Pere David’s deer (Elaphurus davidianus) were obtained directly by rectal collection from four deer farms in Zhengzhou City, China between 2002 and 2007 (Table 1). No clinical sign was observed when the fecal specimens were collected. The fecal specimens were concentrated by the Sheather’s sugar flotation technique and examined by bright field microscope under 400× and 1,000×. Two fecal specimens from two 1-year-old sika deer were Cryptosporidium-positive. One hundred twenty-four fecal specimens were all stored in 2.5% potassium dichromate at 4°C. After purifying the oocysts from the two positive specimens by the discontinuous density sucrose gradient centrifugation, genomic DNA was extracted using Mag Extractor-Genome kit (Toyobo, Osaka, Japan). The DNA was kept at −20°C before it was used in molecular analysis.
Primers to amplify 18S ribosomal RNA (rRNA; ∼1,700 bp), HSP70 (∼1,950 bp), actin (∼1,066 bp), and Cryptosporidium oocyst wall protein (COWP; ∼530 bp) genes were adopted from previous studies (Xiao et al. 2004). Polymerase chain reaction (PCR) products of the actin gene were cloned, and four recombinant plasmids were sequenced by TaKaRa Biotechnology Co. Ltd (Dalian, China) with an ABI PRISM™ 3730 XL DNA Analyzer (Applied Biosystems, USA) using the Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). PCR products of the 18S rRNA, HSP70, and COWP genes were sequenced directly without cloning. The sequences were deposited in GenBank under accession numbers DQ898159, DQ898163, EF030519, and EF030518. Phylogenetic analyses were performed using the software PHYLIP version 3.67 (Felsenstein 1989).
Results and discussion
Sequences of the 18S rRNA, HSP70, actin, and COWP genes were obtained from one of the two Cryptosporidium-positive specimens (another specimen failed to amplify the specific fragment of four genes studied). They were aligned with sequences from other known Cryptosporidium species and genotypes.
In the 18S rRNA gene, there were two to seven nucleotide differences out of 667 bp with other Cryptosporidium cervine genotype isolates (EF362480, EF613340, AF262328, DQ640639, EF641017, AY737593, AY737592, DQ19250, EF641018, DQ640640, EF362481, and EF362479). The similarity (99.1%) between this sika deer isolate and the Chinese ibex isolate (EF613340) was the lowest, and the highest similarity (99.8%) was observed with a USA water sample isolate (EF641018). The similarities with other USA water sample, sheep isolates, and Canada human isolates varied from 99.2% to 99.7%. Five copies of the 18S rRNA gene are present in Cryptosporidium genome, and previous studies have suggested that there is slight sequence heterogeneity in some of these copies (Xiao et al. 1999). Therefore, some of the sequence differences could be due to variations among different copies of the rRNA gene.
In the HSP70 gene, comparing to DQ389176, two nucleotide changes of T to C and one change of A to C were observed at nucleotides 594 and 1062 and nucleotide 1809, respectively. The sequence similarity between the two was 98.8%. This might be due to the single nucleotide polymorphy (SNP) of HSP70 gene. For example, three SNPs were present between C. hominis sequences AF221535 and XM_661662. Similarly, in the actin gene, there were two nucleotide changes between the current isolate and DQ389175 at nucleotide 322 and 580, with a sequence similarity of 99.7%. Further comparison was limited because of the lack of more Cryptosporidium cervine genotype actin sequences. On the contrary, no sequence difference was observed in the COWP gene.
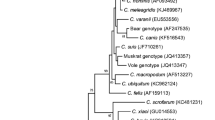
Phylogenetic trees were constructed, and two methods (neighbor-joining and parsimony) produced similar topology for each of the four loci. In each of the analysis, sequence from sika deer grouped together with other Cryptosporidium cervine genotype isolates (a neighbor-joining tree of the 18S rRNA gene is showed in Fig. 1).

Phylogenetic relationship of Cryptosporidium parasites inferred by neighbor-joining analysis of the 18S rRNA gene based on Kimura two-parameter model. Bootstrap values (in percentage) above 50% from 1,000 pseudo-replicates are shown for both the neighbor-joining (the first value) and maximum parsimony analyses (the second value). Low = node with bootstrap values lower than 50%. Scale bar indicates an evolutionary distance of 0.1 substitutions per site in the sequence
Cryptosporidium cervine genotype is the only species/genotype with noticeable broad host range (Tables 2 and 3). Since its initial finding in storm runoff in a wilderness area, it has been found in domestic and wild ruminants (sheep, mouflon sheep, ibex, blesbok, nyala, white-tailed deer, and Pere David’s deer), rodents (squirrels, chipmunks, woodchucks, beavers, and deer mice), carnivores (raccoons), and primates (lemurs and humans; Xiao et al. 2000; Perz and Le Blancq 2001; Ong et al. 2002; da Silva et al. 2003; Ryan et al. 2003, 2005; Blackburn et al. 2006; Feltus et al. 2006; Leoni et al. 2006; Nichols et al. 2006; Soba et al. 2006; Trotz-Williams et al. 2006; Feng et al. 2007; Karanis et al. 2007). The generalist nature of the host specificity of the parasite and habitat sharing are probably responsible for the wide occurrence of the cervine genotype in animals.
According to available data, the cervine genotype has been found in various environmental samples such as source water, stream storm runoff, stream sediment, and raw wastewater in several countries (Table 4). In studies conducted in wilderness areas of the New York City drinking water supply watershed, the cervine genotype was the most common Cryptosporidium species/genotype found in river/stream water (Xiao et al. 2000, 2006b; Jiang et al. 2005) and was the only year-round Cryptosporidium in storm water (Xiao et al. 2000, 2006b; Jiang et al. 2005). The finding of the cervine genotype in these areas suggest the potential for spreading among wildlife species that routinely drink from streams.
An increasing number of human cases have been associated with the Cryptosporidium cervine genotype (Table 2). It has been reported in ten patients in Canada, seven in the UK, three in the USA, and one in Slovenia (Ong et al. 2002; Blackburn et al. 2006; Feltus et al. 2006; Leoni et al. 2006; Nichols et al. 2006; Soba et al. 2006; Trotz-Williams et al. 2006). Likewise, the increasing number of humans infected with the cervine genotype might be related to its wide range of mammalian hosts (Feng et al. 2007). Almost all the human cases are reported in industrialized nations where it ranked number four in Cryptosporidium spp. in humans (more human cases have been attributed to the cervine genotype than C. canis in industrialized nations), indicating that zoonotic transmission is probably responsible for most of the cases with cervine genotype. However, it has been recently seen in one child in a shantytown in Lima, Peru where cryptosporidiosis is known to be transmitted main via the anthroponotic route (Xiao and Cama unpublished).
In this study, four deer farms were screened for Cryptosporidium oocysts for 5 years, but only two fecal specimens were positive, with an average infection rate of 1.61% (2/124). The low infection rate suggests that deer studied here are not very susceptible to Cryptosporidium and are not an important reservoir of cryptosporidiosis. Moreover, the four farms are all far from the drinking water supply system. Therefore, the deer populations on farms may be not a major source of cryptosporidiosis in humans in Zhengzhou region.
This study broadens the host and geographic ranges of the Cryptosporidium cervine genotype and provides new sequence data. In conjunction with previous reports, it is expected that the Cryptosporidium cervine genotype will be named as a new species in future.
References
Blackburn BG, Mazurek JM, Hlavsa M, Park J, Tillapaw M, Parrish M, Salehi E, Franks W, Koch E, Smith F, Xiao L, Arrowood M, Hill V, da Silva A, Johnston S, Jones JL (2006) Cryptosporidiosis associated with ozonated apple cider. Emerg Infect Dis 12:684–686
da Silva AJ, Cacciò S, Williams C, Won KY, Nace EK, Whittier C, Pieniazek NJ, Eberhard ML (2003) Molecular and morphologic characterization of a Cryptosporidium genotype identified in lemurs. Vet Parasitol 111:297–307
Felsenstein J (1989) PHYLIP: phylogeny inference package (version 3.2). Cladistics 5:164–166
Feltus DC, Giddings CW, Schneck BL, Monson T, Warshauer D, McEvoy JM (2006) Evidence supporting zoonotic transmission of Cryptosporidium spp. in Wisconsin. J Clin Microbiol 44:4303–4308
Feng Y, Alderisio KA, Yang W, Blancero LA, Kuhne WG, Nadareski CA, Reid M, Xiao L (2007) Cryptosporidium genotypes in wildlife from a New York watershed. Appl Environ Microbiol 73:6475–6483
Jiang J, Alderisio KA, Xiao L (2005) Distribution of Cryptosporidium genotypes in storm event water samples from three watersheds in New York. Appl Environ Microbio 171:4446–4454
Karanis P, Plutzer J, Halim NA, Igori K, Nagasawa H, Ongerth J, Liqing M (2007) Molecular characterization of Cryptosporidium from animal sources in Qinghai province of China. Parasitol Res 101:1575–1580
Leoni F, Amar C, Nichols G, Pedraza-Diaz S, McLauchlin J (2006) Genetic analysis of Cryptosporidium from 2414 humans with diarrhoea in England between 1985 and 2000. J Med Microbiol 55:703–707
Nichols GL, Chalmers RM, Sopwith W, Regan M, Hunter CA, Grenfell P, Harrison F, Lane C (2006) Cryptosporidiosis: a report on the surveillance and epidemiology of Cryptosporidium infection in England and Wales. Drinking Water Directorate Contract Number DWI 70/2/201. Drinking Water Inspectorate, UK, 142
Ong CS, Eisler DL, Alikhani A, Fung VW, Tomblin J, Bowie WR, Isaac-Renton JL (2002) Novel Cryptosporidium genotypes in sporadic cryptosporidiosis cases: first report of human infections with a cervine genotype. Emerg Infect Dis 8:263–268
Perz JF, Le Blancq SM (2001) Cryptosporidium parvum infection involving novel genotypes in wildlife from lower New York State. Appl Environ Microbiol 67:1154–1162
Ruecker NJ, Braithwaite SL, Topp E, Edge T, Lapen DR, Wilkes G, Robertson W, Medeiros D, Sensen CW, Neumann NF (2007) Tracking host sources of Cryptosporidium spp. in raw water for improved health risk assessment. Appl Environ Microbiol 73:3945–3957
Ryan UM, Xiao L, Read C, Zhou L, Lal AA, Pavlasek I (2003) Identification of novel Cryptosporidium genotypes from the Czech Republic. Appl Environ Microbiol 69:4302–4307
Ryan U, Bath C, Robertson I, Read C, Elliot A, McInnes L, Traub R, Besier B (2005) Sheep may not be an important zoonotic reservoir for Cryptosporidium and Giardia parasites. Appl Environ Microbiol 71:4992–4997
Santin M, Trout JM, Fayer R (2007) Prevalence and molecular characterization of Cryptosporidium and Giardia species and genotypes in sheep in Maryland. Vet Parasitol 146:17–24
Soba B, Petrovec M, Mioc V, Logar J (2006) Molecular characterisation of Cryptosporidium isolates from humans in Slovenia. Clin Microbiol Infect 12:918–921
Trotz-Williams LA, Martin DS, Gatei W, Cama V, Peregrine AS, Martin SW, Nydam DV, Jamieson F, Xiao L (2006) Genotype and subtype analyses of Cryptosporidium isolates from dairy calves and humans in Ontario. Parasitol Res 99:346–352
Xiao L, Escalante L, Yang C, Sulaiman I, Escalante AA, Montali RJ, Fayer R, Lal AA (1999) Phylogenetic analysis of Cryptosporidium parasites based on the small-subunit rRNA gene locus. Appl Environ Microbiol 65:1578–1583
Xiao L, Aldeeisio K, Limor J, Royer M, Lal AA (2000) Identification of species and sources of Cryptopsporidium oocysts in storm waters with a small-subunit rRNA-based diagnostic and genotyping tool. Appl Environ Microbiol 66:5492–5498
Xiao LH, Fayer R, Ryan U, Upton SJ (2004) Cryptosporidium taxonomy: recent advances and implications for public health. Clin Microbiol 17:72–97
Xiao L, Alderisio K, Singh A (2006a) Development and standardization of a Cryptosporidium genotyping tool for water samples. Awwa Research Foundation, Denver, CO
Xiao L, Alderisio KA, Jiang J (2006b) Detection of cryptosporidium oocysts in water: effect of the number of samples and analytic replicates on test results. Appl Environ Microbiol 72:5942–5947
Zhou L, Singh A, Jiang J, Xiao L (2003) Molecular surveillance of Cryptosporidium spp. in raw wastewater in Milwaukee: implications for understanding outbreak occurrence and transmission dynamics. J Clin Microbiol 41:5254–5257
Ziegler PE, Wade SE, Schaaf SL, Chang YF, Mohammed HO (2007) Cryptosporidium spp. from small mammals in the New York city watershed. J Wildl Dis 43:586–596
Acknowledgments
This study was supported in part by Henan Innovation Project for Prominent University Research Talents (number 2004KYCX002).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wang, R., Wang, J., Sun, M. et al. Molecular characterization of the Cryptosporidium cervine genotype from a sika deer (Cervus nippon Temminck) in Zhengzhou, China and literature review. Parasitol Res 103, 865–869 (2008). https://doi.org/10.1007/s00436-008-1069-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-1069-2