
Abstract
We developed a real-time LC PCR assay to detect a 152 bp sequence in an uncharacterized region of the Blastocystis genome. The described assay detected 11 of 11 ATCC strains of Blastocystis from subtypes 1, 3, and 4. Three of three stool samples from Oregon and California military personnel that were negative for Blastocystis by an ova and parasite test as well as a conventional PCR assay were positive for Blastocystis using our real-time LC PCR assay. Diagnosis of Blastocystis infections using this sensitive method, including DNA extraction and real-time PCR, only requires 3 h. The lower limit of detection for Blastocystis in stool using the real-time LC PCR assay was calculated to be 760 cells of Blastocystis per 100 mg of stool, an estimated 760 parasites per reaction. The assay did not cross-react with Ruminococcus hansenii, Anarococcus hydrogenalis, Bifidobacterium adolescentis, Fusobacterium prausnitzii, Staphylococcus aureus, Escherichia coli, Enterococcus faecalis, or Lactobacillus acidophilus. Because of the ease of use, sensitivity, specificity, and increase in Blastocystis infections in the USA we believe this assay has the potential to be useful as a clinical diagnosis tool of Blastocystis infection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Blastocystis is a genus of protozoan-like single-celled parasitic organisms that infects the gastrointestinal tracts of humans and other vertebrates. The prevailing view for much of the twentieth century was that these infections were harmless. However, reports associating Blastocystis with pathogenicity emerged in the late 1980s, primarily in travelers returning from less developed countries (Sheehan et al. 1986; O’Gorman et al. 1993). Prevalence of Blastocystis in developing countries is reported to be between 30 and 50% (Stenzel and Boreham 1996). In the USA, prevalence was reported to be 25% (Amin 2002). Current reports suggest that prevalence in Oregon is increasing (Boorom 2006). Physicians in the Willamette Valley of Oregon recognize this as a public health crisis and desire a rapid and cost-effective method to detect Blastocystis (K. Boorom, personal communication).
Early studies associated Blastocystis infections with symptoms such as abdominal pain, diarrhea, constipation, fatigue, headaches, and depression (Qadri et al. 1989). Subsequent reports added skin rash and joint pain to the list (Kruger et al. 1994; Lee et al. 1990; Pasqui et al. 2004; Armentia et al. 1993; Cassano et al. 2005; Biedermann et al. 2002). The United States’ Center for Disease Control states that the symptoms reported to be associated with blastocystosis are diarrhea, watery or loose stools, anal itching, abdominal pain, weight loss, and excess gas (Center for Disease Control Fact Sheet). This wide array of non-specific symptoms has confounded the understanding of the potential pathogenicity of Blastocystis species. As a result, many of these infections likely go undiagnosed.
Detection of Blastocystis can be accomplished by many methods such as microscopy, culture, and formyl acetate concentration technique (FECT). Nevertheless, these methods all have flaws that make them unreliable or time consuming. Since Blastocystis has several morphological forms (vacuolar, cyst, amoeboid, granular, multivacuolar, and avacuolar), microscopy is very difficult (Stenzel and Boreham 1996). In addition, FECT is unreliable because it destroys the multivacuolar, vacuolar, and granular forms of the parasite during stool processing (O’Gorman et al. 1993). Culture is believed to be the current gold standard for detecting Blastocystis, but time to diagnosis is 2–3 days and in some instances allows preferential growth of one subtype over another if more than one subtype is present in the stool (Parkar et al. 2006).
Recently, several studies have described the use of conventional PCR to detect Blastocystis. Parkar et al. (2006) demonstrated that culture prior to PCR is three times more sensitive than culture alone. In contrast, using conventional PCR alone, other studies show the ability to detect Blastocystis at concentrations as low as 13 and 32 parasites per 200 mg of stool (Pasqui et al. 2004, Qadri et al. 1989). Currently, there are no reliable tests readily available to physicians to quickly diagnose blastocystosis. In this study, we developed the first real-time Light Cycler (LC) PCR assay targeting an unknown region of the Blastocystis genome.
Materials and methods
Study subjects
The criteria for inclusion of subjects in the study were a physician’s diagnosis of symptomatic Blastocystis infection or adult onset of unexplained lower gastrointestinal symptoms (diarrhea, constipation, abdominal pain) and subsequent continuance for over a year. Two participants were from a town in Western Oregon, and a third was from a military facility in California. Informed consent and a short medical history was obtained, and each participant and provided 4ccs of stool in empty stool collection vials (Fisher 23-005-31). Vials were preserved with ice by the patient for up to 24 h, then transferred to a holding facility and held at −15°C for varying times, up to 6 weeks. Anonymized vials were packaged with dry ice and shipped to David Grant USAF Medical Center in July 2007.
Specimen processing
Stool specimens were processed in a BSL-2 hood. One hundred milligrams of stool were added to 900 μls of 0.85% saline. Stool was vortexed with glass beads, and then boiled for 2 min. Next, the samples were centrifuged at 1,500×g for 15 min then allowed to settle for 10 min at room temperature. Samples were transferred to a 32-well plate for nucleic acid isolation.
Nucleic acid extraction
Genomic DNA from Blastocystis was extracted from processed stool samples using the MagNA Pure LC DNA Isolation Kit I (Roche, Indianapolis, IN, USA) according to the manufacturers’ recommendations for the MagNA Pure LC automated nucleic acid extraction system.
Blastocystis strains
Blastocystis strains ATCC 50177, ATCC 50587, ATCC 50608, ATCC 50609, ATCC 50610, ATCC 50613, ATCC 50629, ATCC 50751, ATCC 50752, ATCC 50753, and ATCC 50754 were acquired from the American Type Culture Collection (ATCC; Manassas, VA, USA).
Real-time light cycler PCR detection of Blastocystis
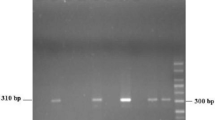
Real-time fluorescent LC PCR was used to detect an unknown gene in the Blastocystis genome. The primers used to detect the 152 bp fragment of Blastocystis strain 50587 (GenBank accession no. M87661) were prMSJ2-F, 5′-CACACTGTGATTCTCGGG-3′ and prMSJ2-R, 5′-GAAATGGAAGATGGAATTGATGAC-3′. The hybridization probes used to detect the unknown Blastocystis gene have the following sequence: prb-blasto3-1, 5′-CACCTCGATCTCGATCTGCTCCA-Fluorescein and prb-blasto3-2, LC Red 640-TTCCGATTCTCTTCACTCATTTGCTCAATCTCAC-Phosphate. For real-time LC PCR, cycling was carried out in a LightCycler 2.0 (Roche, Indianapolis, IN, USA) thermocycler using 1 μl (~2 mg of stool) of extracted RNA in 0.4 μl of LightCycler RT PCR Enzyme Mix, 4 μl of 5× LightCycler RT-PCR Reaction Mix Hyb-Probe (Roche), containing 5 mM MgCl2 and 400 nM forward and reverse primers. Our reaction conditions were as follows: initial denaturation at 95°C for 15 min, followed by 40 cycles of denaturation at 95°C for 1 s, annealing at 60°C for 15 s, and extension at 72°C for 5 s. The progress of real-time fluorescent PCR was monitored at 640 nm. Initial PCR products were resolved on 6.5% acrylamide gel electrophoresis and visualized by SYBR Green staining at 1:10,000 dilution.
Sensitivity and specificity
To determine the range of our real-time LC RT-PCR assay, 76,000, 7,600, 760, 76, and 8 cells of Blastocystis strain 50751 (ATCC) were added to Blastocystis negative stool and assayed according to the real-time LC PCR conditions described above. To determine specificity, nucleic acid extracted via MagNA Pure LC from Ruminococcus hansenii (ATCC 27752), Bifidobacterium adolescentis (ATCC 15703), Fusobacterium prausnitzii (ATCC 27766), Staphylococcus aureus (ATCC 25923), Escherichia coli (ATCC 43895), Enterococcus faecalis (ATCC 51299), Lactobacillus acidophilus (ATCC 11974), and Anarococcus hydrogenalis (ATCC 49630) strains were assayed with the same primer pairs in the LC real-time PCR assay.
Phylogenetic analysis of Blastocystis
Subtype classification was determined phylogenetically and assigned to Blastocystis spp. subtypes as designated by Stensvold et al. (2007). Partial Blastocystis SSU sequences were generated using pr3-3-gdF, GTCAATTCCTTTAAGTTTCAGCC and pr3-3-gdR, TCCAGCTCCAATAGCGT, and placed in an alignment generated by CMW consisting of 62 sequences representative of all Blastocystis species and subtypes (Stensvold et al. 2007; Yoshikawa et al. 2007). Proteromonas lacertae was used as an outgroup. The sequence alignment was manually edited and analyzed in PAUP (Swofford et al. 2001) by parsimony analysis as described in Whipps and Kent (2006). Bayesian analyses were conducted in Mr. Bayes (Ronquist and Huelsenbeck 2003) under a GTR model, with 106 generations, tree sampling every 100 generations, with a burn-in of 100 trees.
Nucleotide sequence accession numbers
Blastocystis sequence has been deposited to GenBank under accession numbers EU290745.
Results
Real-time LC PCR Blastocystis detection assay
The results of our real-time LC PCR assay are in Tables 1 and 2. Our real-time LC PCR assay, which detects a 152-bp region in an unknown gene of the Blastocystis genome, detected 11 of 11 Blastocystis strains (100%) from the ATCC. We also detected Blastocystis in three out of three clinical stool samples (100%) from patients who presented with fatigue, depression, skin rash, joint pain, constipation, abdominal pain, and diarrhea (Table 2) in 3 hours. In contrast, the clinical stool samples were negative using the traditional ova and parasite (O&P) test as well as a conventional PCR assay developed by Stensvold et al (2006). The Blastocystis genome has not been sequenced, therefore it was not possible to relate the copy number of our marker directly to a number of cells.
Sensitivity of assay
To determine the sensitivity of our assay in clinical samples, we spiked Blastocystis negative stool with serial dilutions of Blastocystis (76,000, 7,600, 760, 76, and 8 cells), then used our real-time LC PCR assay to detect the pathogen. The lower limit of detection for Blastocystis in stool was calculated to be 760 parasites per 100 mg of stool, an estimated 760 parasites per reaction (Table 3).
Specificity of assay
Because Blastocystis is an enteric pathogen (Ronquist and Huelsenbeck 2003), we wanted to determine whether or not our assay was able to detect other pathogens commonly found in the gastrointestinal tract of humans. Using our real-time LC PCR assay, we were unable to amplify our 152 bp amplicon from genomic DNA extracted from Ruminococcus hansenii, Anarococcus hydrogenalis, Bifidobacterium adolescentis, Fusobacterium prausnitzii, Staphylococcus aureus, Escherichia coli, Enterococcus faecalis, and Lactobacillus acidophilus (Table 4), all of which are bacteria commonly found in the intestinal tract of humans. These results suggested that our assay was specific to Blastocystis.
Subtype specificity
Phylogenetic analysis of Blastocystis spp. SSU by either Bayesian or parsimony analysis yielded several well-defined clades corresponding to the various subtypes designated by Stensvold et al. (2007; Fig 1). American type culture collection isolates were categorized as subtypes 1, 3, or 4 (Fig 1; Table 5), representative of multiple Blastocystis subtype lineages. Tree topology generated from Bayesian and parsimony analyses were almost identical with the exception of the placement of Blastocystis cycluri. Bayesian analysis placed B. cycluri as a sister taxon to subtype 3, whereas parsimony analysis placed it basal to subtypes 3, 4, and 8 (not shown). Two isolates, ATCC 50588 and 50613, were excluded from the phylogentetic analyses because they appeared to consist of more than one subtype as evidenced by multiple peaks in our DNA sequence output (data not shown). Regardless, our real-time LC PCR assay was able to detect all ATCC strains (Table 1).

Relationships among Blastocystis sp. subtypes
Discussion
This is the first study demonstrating a sensitive and specific real-time LC PCR assay that detects Blastocystis in clinical stool samples. Our assay detected a 152-bp amplicon in an unknown region of the Blastocystis genome in stool samples from patients who tested negative for Blastocystis via O&P.
Detection of Blastocystis is very important public health information. Currently, there is no FDA approved nucleic acid-based test to detect Blastocystis. In the Willamette Valley of Oregon, the number of people with symptoms suggesting Blastocystis infections has increased at such an alarming rate that physicians in that state have ardently requested that the organism be a reportable infection (Boorom 2006). We believe that the Blastocystis detection assay described in this study has the potential to be a helpful tool in blastocystosis diagnosis.
To date, there are several methods to detect Blastocystis (Parkar et al. 2006; Pasqui et al. 2004; Qadri et al. 1989; Suresh and Smith 2004; Swofford et al. 2001; Termmathurapoj et al. 2004). Culture has been regarded as the standard for Blastocystis detection. However, culturing Blastocystis is time consuming and can bias subtyping results due to preferential growth of one subtype versus another (Parkar et al. 2006). O&P is currently used in many public health labs, yet this technique has been deemed to be poor in studies from Denmark (Qadri et al. 1989), the UK (Swofford et al 2001) and Thailand (Termmathurapoj et al. 2004). Recently, detection of Blastocystis directly from stool via conventional PCR has been reported by three different research groups (Parkar et al. 2006; Pasqui et al. 2004; Qadri et al. 1989). Stensvold et al. reported a lower limit of detection of 32 parasites per 200 mg of fecal sample, and Menounos et al. (2008) were able to detect 13 parasites per 200 mg of fecal sample (Pasqui et al. 2004; Qadri et al. 1989). Interestingly, when we used the assay by Stensvold et al. we were unable to detect Blastocystis in three clinical samples (Table 2). The lower limit of detection for our real-time LC PCR assay was 760 parasites per 100 mg of stool (Table 3). The clinically relevant question is, “How much Blastocystis is present in the stool of a symptomatic patient?” Unfortunately, this has not been elucidated.
The reliability of Blastocystis testing is problematic because shedding is irregular, with patients shedding the pathogen on some days yet not on others (Vennila et al. 1999). For nucleic acid testing to be reliable in a clinical setting, we suggest that it be performed on stools from two consecutive days, thus increasing the probability that the pathogen will be detected.
An advantage of the described assay is that it detects 11 of 11 strains from the ATCC as well as strains found in clinical stool samples (Tables 1 and 2). Another advantage is that we were able to detect Blastocystis infections in clinical samples in 3 h. A disadvantage is that it detects a region with unknown function which does not discriminate between pathogenic and non-pathogenic strains of Blastocystis. Currently, there is no way to differentiate between non-pathogenic and pathogenic strains of Blastocystis. Past studies demonstrate that Blastocystis infection is increasing in the USA (Boorom 2006; Wang et al. 2002). Thus, it is imperative that the non-pathogenic and pathogenic Blastocystis genomes are sequenced.
The ATCC strains of Blastocystis were classified as subtypes 1, 3, or 4 (Fig 1; Table 5). Although we were unable to evaluate the ability of our assay to detect other Blastocystis subtypes, we are confident that it would be effective for other lineages as types 1, 3, 4 are representative of the phylogenetic diversity found within this group. ATCC 50588 and 50613 yielded mixed results in our DNA sequencing analyses, suggestive of multiple subtypes within the sample or multiple ribosomal DNA alleles within those isolates. Hoevers et al. (2000) noted multiple SSU alleles in ATCC 50177 using universal eukaryotic primers. These data highlight the importance of using PCR primers that are inclusive of all Blastocystis subtypes: a challenging task given the diversity within this taxon.
While we did not determine whether our assay cross-reacts with every known pathogen, we demonstrated that it was unable to detect pathogens commonly found in the gastrointestinal tract of humans (Table 4). If our assay detected other pathogens in the stool that we did not test for, we would have been unable to generate a standard curve when we spiked Blastocystis negative stool data not shown. Therefore, we conclude that our assay is specific and useful for detecting Blastocystis in clinical stool samples.
The real-time LC PCR assay that we developed is sensitive and specific for Blastocystis. Because of the increase in Blastocystis infections in North America (Boorom 2006; Wang et al. 2002), we believe this assay has the potential to be useful as a clinical diagnosis tool of Blastocystis infection.
References
Amin OM (2002) Seasonal prevalence of intestinal parasites in the United States during. Am J Trop Med Hyg 66:799–803
rmentia A et al (1993) Urticaria by Blastocystis hominis. Successful treatment with paromomycin {paromycin}. Allergol Immunopathol (Madr) 21:149–151
Biedermann T, Hartmann K, Sing A, Przybilla B (2002) Hypersensitivity to non-steroidal anti-inflammatory drugs and chronic urticaria cured by treatment of Blastocystis hominis infection. Br J Dermatol 146:1113–1114
Boorom KF (2006) Commensal and pathogenic Blastocystis with case studies from Oregon’s Willamette Valley. BRF Press, Corvallis OR USA
Cassano N, Scoppio BM, Loviglio MC, Vena GA (2005) Remission of delayed pressure urticaria after eradication of Blastocystis hominis. Acta Derm Venereol 85:357–358
Center for Disease Control Fact Sheet, Blastocystis hominis, available online
Hoevers J, Holman P, Logan K, Hommel M, Ashford R, Snowden K (2000) Restriction-fragment-length polymorphism analysis of small-subunit rRNA genes of Blastocystis hominis isolates from geographically diverse human hosts. Parasitol Res 86:57–61
Kruger K, Kamilli I, Schattenkirchner M (1994) Blastocystis hominis as a rare arthritogenic pathogen. A case report. Z Rheumatol 53:83–85
Lee MG, Rawlins SC, Didier M, DeCeulaer K (1990) Infective arthritis due to Blastocystis hominis. Ann Rheum Dis 49:192–193
Menounos PG, Spanakos G, Tegos N, Vassalos CM, Papadopoulou C, Vakalis NC (2008) Direct detection of Blastocystis sp. in human faecal samples and subtype assignment using single strand conformational polymorphism and sequencing. Mol Cell Probes 22:24–29
O’Gorman MA, Orenstein SR, Proujansky R, Wadowsky RM, Putnam PE, Kocoshis SA (1993) Prevalence and characteristics of Blastocystis hominis infection in children. Clin Pediatr (Phila) 32:91–96
Parkar U, Traub RJ, Kumar S, Mungthin M, Vitali S, Leelayoova S, Morris K, Thompson RC (2006) Direct characterization of Blastocystis from faeces by PCR and evidence of zoonotic potential. Parasitology 19:1–9
Pasqui AL, Savini E, Saletti M, Guzzo C, Puccetti L, Auteri A (2004) Chronic urticaria and Blastocystis hominis infection: a case report. Eur Rev Med Pharmacol Sci 8:117–120
Qadri SM, al-Okaili GA, al-Dayel F (1989) Clinical significance of Blastocystis hominis. J Clin Microbiol 27:2407–2409
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Sheehan DJ, Raucher BG, McKitrick JC (1986) Association of Blastocystis hominis with signs and symptoms of human disease. J Clin Microbiol 24:548–550
Stensvold CR, Suresh GK, Tan KS, Thompson RC, Traub RJ, Viscogliosi E, Yoshikawa H, Clark CG (2007) Terminology for Blastocystis subtypes–a consensus. Trends Parasitol 23:93–96
Stensvold R, Brillowska-Dabrowska A, Nielsen HV, Arendrup C (2006) Detection of Blastocystis hominis in unpreserved stool specimens by using polymerase chain reaction. J Parasitol 92:1081–1087
Stenzel DJ, Boreham PF (1996) Blastocystis hominis revisited. Clin Microbiol Rev 9:563–584
Suresh K, Smith H (2004) Comparison of methods for detecting Blastocystis hominis. Eur J Clin Microbiol Infect Dis 23:509–511
Swofford DL, Waddell PJ, Huelsenbeck JP, Foster PG, Lewis PO, Rogers JS (2001) Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Syst Biol 50:525–539
Termmathurapoj S et al (2004) The usefulness of Short-Term In-Vitro cultivation for the detection and molecular study of Blastocystis hominis in stool specimens. Parasitol Res 93:445–447
Vennila GD, Suresh Kumar G, Khairul Anuar A, Rajah S, Saminathan R, Sivanandan S, Ramakrishnan K (1999) Irregular shedding of Blastocystis hominis. Parasitol Res 85:162–164
Wang KX, Li CP, Wang J, Cui YB (2002) Epidemiological survey of Blastocystis hominis in Huainan City, Anhui Province, China. World J Gastroenterol 8:928–932
Whipps CM, Kent ML (2006) Phylogeography of the cosmopolitan marine parasite Kudoa thyrsites (Myxozoa: Myxosporea). J Eukaryot Microbiol 53:364–373
Yoshikawa H, Wu Z, Howe J, Hashimoto T, Geok-Choo N, Tan KS (2007) Ultrastructural and phylogenetic studies on Blastocystis isolates from cockroaches. J Eukaryot Microbiol 54:33–37
Acknowledgments
The work reported herein was performed under United States Air Force Surgeon General-approved Clinical Investigation No. FDG20070009N, FDG20070010N. The experiments comply with the current laws of the country in which they were performed. The views expressed in this material are those of the authors, and do not reflect the official policy or position of the U.S. Government, the Department of Defense, or the Department of the Air Force.
Author information
Authors and Affiliations
Corresponding author
Additional information
The work reported herein was performed under United States Air Force Surgeon General-approved Clinical Investigation No. FDG20070009N, FDG20070010N.
Rights and permissions
About this article
Cite this article
Jones II, M.S., Ganac, R.D., Hiser, G. et al. Detection of Blastocystis from stool samples using real-time PCR. Parasitol Res 103, 551–557 (2008). https://doi.org/10.1007/s00436-008-1006-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-1006-4