
Abstract
A collection of 483 samples representing Fasciola from six naturally infected host species and 16 localities in Spain, previously identified morphologically and genetically as Fasciola hepatica, was characterized by a novel genetic marker, namely sequence-related amplified polymorphism (SRAP), aiming to reveal genetic variability within F. hepatica in Spain. Visualization of amplification fragments was carried out on 6% denaturing polyacrylamide gels, followed by staining with 0.1% AgNO3 solution. Ten SRAP primer combinations were tested—six of them turned out to be polymorphic. Thirty-four representative F. hepatica samples from six host species and 16 geographical localities showed polymorphic banding patterns using SRAP primer combinations and were grouped into four major clusters using the unweighted pair-group method with arithmetic averages, indicating the existence of genetic variability within the examined F. hepatica samples. These four clusters were not related to particular host species and/or geographical origins of the samples. The results of the present study revealed that SRAP markers were useful in revealing sufficient polymorphism in F. hepatica samples from Spain and had implications for studying the population genetic structure of the Spanish F. hepatica. To our knowledge, this is the first application of SRAP marker to study genetic variation in parasites of human and animal health significance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fasciola hepatica is the common liver fluke of a wide range species of animals and has a global geographical distribution (Spithill and Dalton 1998; Mas-Coma et al. 2005). In Spain, F. hepatica occurs in both domestic and wild hosts, including European rabbit (Oryctolagus cuniculus), hare (Lepus granatensis), donkey (Equus asinus), horse (Equus caballus), pig and wild boar (Sus scrofa), fallow deer (Dama dama), cattle (Bos taurus), domestic sheep (Ovis aries), goat (Capra hircus), Iberian ibex (Capra pyrenaica), red deer (Cervus elaphus), European mouflon (Ovis aries), chamois (Rupicapra pyrenaica), roe deer (Capreolus capreolus), and man (Cordero del Campillo et al. 1994; Lavín et al. 1995; Pérez et al. 2006; Alasaad et al. 2007, 2008). The prevalence in domestic animals is high, up to 85.1% and 83.3% in cattle and ovine, respectively, under field conditions from Galicia (Northwest Spain; Sánchez-Andrade et al. 2000; Paz-Silva et al. 2003) but less important in wildlife, 0.7% in Iberian ibex (Alasaad et al. 2008).
Despite the substantial economic losses caused by Fasciola, estimated at US$ 2 billion per year worldwide (Spithill and Dalton 1998), and the vast distribution of this cosmopolitan parasite (Mas-Coma et al. 2003), little attention was given to study the morphometric and/or genetic variation of F. hepatica from different host species and/or geographical localities (Valero et al. 2001a, b; Periago et al. 2006; Alasaad et al. 2007).
Genetic approaches employing a range of molecular makers have proven useful for parasite identification and studies of genetic variability among parasite populations (Gasser 1999; Prichard and Tait 2001; Gasser 2006; Bildfell et al. 2007; Zhu et al. 2007). Previous studies using nuclear ribosomal DNA markers, mitochondrial DNA markers, and isoelectric focusing banding patterns of whole-body protein have demonstrated the existence of genetic variability among different populations of F. hepatica (Lee et al. 1992; Lotfy et al. 2002; Huang et al. 2004; Morozova et al. 2004). A recent study has showed a slight sequence variation in the second internal transcribed spacer (ITS-2; 1/362, 0.3%) among Fasciola samples from different host species and geographical localities in Spain and that Spanish F. hepatica examined in that study differed from Fasciola from elsewhere by two nucleotides in the ITS-2 (Alasaad et al. 2007). Another study concluded that there were some genetic variations in F. hepatica by studying the 28S rDNA (Vara-Del Río et al. 2007).
Sequence-related amplified polymorphism (SRAP) is a novel and efficient genetic marker system, revealing genetic variation in open reading frames among related organisms (Li and Quiros 2001). Because of its simplicity and efficiency, SRAP has been used in the construction of genetic maps (Li and Quiros 2001), comparative genetic studies (Li et al. 2003), and examination of genetic diversities (Riaz et al. 2001; Ferriol et al. 2003) in many plant species. It has also been used to study genetic variations in seaweeds (Qiao et al. 2007). Nevertheless, to our knowledge, there were no reports using this marker in the field of parasitology before the present study. The objective of the present study was to investigate the genetic variability among F. hepatica from different host species and geographical localities in Spain using the novel SRAP marker.
Materials and methods
Parasite collection and identification
Between 2002 and 2006, 483 adult trematodes were collected from the liver of 108 naturally infected animals; all of them were local animals, representing six species and 16 geographical locations in Spain. Individual flukes were washed extensively in physiological saline, identified morphologically as F. hepatica according to existing keys and descriptions (Yamaguti 1958) and then fixed in 70% ethanol until extraction of genomic DNA. The identity of 18 representative flukes was confirmed by sequencing the internal transcribed spacers of nuclear ribosomal DNA (Alasaad et al. 2007). The codes, host species, and geographical origins of the fluke samples used in the present study are listed in Table 1 and Fig. 1.

Spain map showing approximate sites for sample collection. Codes next to the sites represent the sample codes in Table 1
Extraction and purification of genomic DNA
Genomic DNA was extracted from a portion of individual adult trematodes treated with sodium dodecy1-sulfate or proteinase K (Zhu et al. 2002), column-purified using WizardTM DNA Clean-Up System (Promega), and then eluted into 65 μl H2O according to the manufacturer’s recommendations. All the DNA samples were stored at -20°C until further use.
SRAP procedure
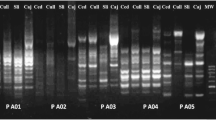
The SRAP technique described by Li and Quiros (2001) was followed with slight modifications. Ten different primer combinations were employed, using five forward primers and five reverse primers. Each 25 μl polymerase chain reaction (PCR) mixture consisted of 1.0 μl genomic DNA, 2.5 μl PCR buffer, 200 μM of dNTPs, 4 mM of MgCl2, 0.5 μM of primer, 1.25 unit of Taq polymerase (TaKaRa), and sterile double-distilled water. Samples were subjected to the following thermal profile for amplification in a thermal cycler (Biometra): 5 min of initial denaturation at 94°C, then five cycles of three steps: 1 min of denaturation at 94°C, 1 min of annealing at 35°C, 1 min of elongation at 72°C, followed by further 35 cycles with annealing temperature being increased to 50°C, with a final elongation step of 5 min at 72°C. Separation of amplified fragments was accomplished on 6% denaturing acrylamide gels [acrylamide-bisacrylamide (19:1), 1 × TBE] at 90 V for 2.5 h. The gel was stained with 0.1% AgNO3 solution and then photographed by digital camera. Primer banding patterns which were difficult to score and those primers that failed to amplify consistently were excluded. Consequently, only six primer combinations out of ten were selected and employed to amplify 483 F. hepatica samples (Table 2, Fig. 2).

Representative gel showing SRAP profiles produced for seven representative Fasciola hepatica samples used to test the amplification efficiency of SRAP primers by primer combinations ME1/EM3, ME2/EM1 and ME3/EM6 (cf. Table 2), respectively. ‘Control’ represents no-DNA control. ‘Marker’ represents a DNA size marker 2000 (ordinate values in bp). Codes in this figure represent the sample codes in Table 1 and Fig. 1
Data analysis
Photographs of denaturing acrylamide gels showing different banding patterns were digitalized and analyzed. SRAP fragments were scored for presence (the presence of the specific allele, coded “1”) or absence (the absence of the specific allele, coded “0”) in each sample. The distance matrix and dendrogram were constructed using the Numerical Taxonomy Multivariate Analysis System (NTSYS-pc) version 2.1 software package. An unweighted pair-group method with arithmetic averages (UPGMA) dendrogram was constructed. FIND module (part of the NTSYS package) was used to identify all trees that could result from different choices of tied similarity of dissimilarity values. To test the robustness of the tree topology, the trees were compiled by Coordinate Open Neural info-Space of European Network e-centers (part of the NTSYS package).
Results and discussion
A total of ten different combinations of SRAP primers, using five forward primers and five reverse primers, were evaluated on seven representative flukes to test the amplification efficiency of each primer pair, and representative results are shown in Fig. 2. Six primer combinations displayed better polymorphisms and, thus, were chosen for the amplification of 483 F. hepatica samples. These six primer combinations were ME1/EM3, ME2/EM1, ME3/EM6, ME4/EM1, ME4/EM6, and ME5/EM4 (Table 2). The number of fragments amplified from F. hepatica samples using these primer combinations ranged from 12 to 20, with an average of 15 polymorphic bands per primer combination, which is consistent with results of previous studies (Li and Quiros 2001; Budak et al. 2004; Ferriol et al. 2004). A total of 51 main polymorphic bands were observed, ranging in size from approximately 100 to 2000 bp. Thirty-four F. hepatica samples, representing the 483 F. hepatica samples from six host species and 16 geographical localities examined in this study, displayed representative polymorphic banding patterns using SRAP primer combinations (Table 2, Fig. 3), and hence were used for cluster analysis.

The UPGMA clustering algorithm grouped the 34 representative F. hepatica samples into four clusters based on SRAP profiles, with the exception of four F. hepatica specimens, namely FhCe1(1), FhBt14(2), FhBt4(2), and FhOa3(1). F. hepatica samples from different host species and/or geographical localities tended to cluster in the same group, and F. hepatica samples from the same host species, e.g., FhOa3 (1) and FhOa3 (2); FhCe1 (1) and FhCe1 (2), and/or from the same geographical location, e.g., FhBt12 (1) and FhBt12 (2); FhEc2 and FhBt2, were grouped into different clusters (Fig. 4).

Dendrogram of 34 representative Fasciola hepatica samples from different host species and geographical localities in Spain constructed on the basis of genetic similarity analysis using six SRAP primer combinations. Codes in this dendrogram represent the sample codes in Table 1 and Fig. 1. The resulting four clusters are labelled as І, ІІ, ІІІ, and ІV
The possible explanation for such a low level of intraspecific variation among F. hepatica samples from different host species and/or geographical localities in Spain may be that the examined sequences belonged to the coding regions of the genomes of F. hepatica samples and these regions did not mutate much due to their functional constraints (Crow and Simmons 1983; Crow 2000). This may be related to the lifecycle of Fasciola, which probably acts against speciation processes, decreasing at the same time their genetic variability and, thus, allowing Fasciola to infect new host species (Alasaad et al. 2007; Vara-Del Río et al. 2007).
As expected, SRAP marker was highly polymorphic and inexpensive technique to establish the genetic variability among Fasciola samples from different host species and geographical localities. Nevertheless, as this novel marker targets the whole genomic DNA and some of the resulting bands are so large in size, working with such marker requires genomic DNA samples of good quality.
In conclusion, the present study demonstrated that genetic variability in the coding regions of the genomes of 483 F. hepatica samples from six host species and 16 geographical locations in Spain was low, indicating that there seemed no genetic association between F. hepatica and their hosts and/or geographical locations in Spain. This study also demonstrated that the SRAP is a useful genetic marker for the studies of genetic variability in parasite populations, and may find broad applications in parasitology.
References
Alasaad S, Huang CQ, Li QY, Granados JE, García-Romero C, Pérez JM, Zhu XQ (2007) Characterization of Fasciola samples from different host species and geographical localities in Spain by sequences of internal transcribed spacers of rDNA. Parasitol Res 101:1245–1250
Alasaad S, Granados JE, Cano-Manuel FJ, Meana A, Zhu XQ, Pérez JM (2008) Epidemiology of fasciolosis affecting Iberian Ibex, Capra pyrenaica, in southern Spain. Parasitol Res (in press)
Bildfell RJ, Whipps CM, Gillin CM, Kent ML (2007) DNA-based identification of a hepatic trematode in an elk calf. J Wildl Dis 43:762–769
Budak H, Shearman RC, Parmaksiz I, Gaussoin RE, Riosdan TP, Dweikat I (2004) Molecular characterization of buffalograss germplasm using sequence-related amplified polymorphism markers. Theor Appl Genet 108:328–334
Cordero del Campillo M, Castañón-Ordóñez L, Reguera-Feo A (1994) Índice-catálogo de zooparásitos ibéricos (2nd ed). Universidad de León, León
Crow JF (2000) The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1:40–47
Crow JF, Simmons MJ (1983) The mutation load in Drosophila. In: Ashburner M, Carson HL, Thompson JN (eds) The genetics and biology of Drosophila. vol. 3C. Academic, London, pp 1–35
Ferriol M, Picó B, Nuez F (2003) Genetic diversity of a germplasm collection of Cucurbita pepo using SRAP and AFLP markers. Theor Appl Genet 107:271–282
Ferriol M, Picó B, Fernandez de Córdova P, Nuez F (2004) Molecular diversity of a germplasm collection of squash (Cucurbita moschata) determined by SRAP and AFLP markers. Crop Sci 44:653–664
Gasser RB (1999) PCR-based technology in veterinary parasitology. Vet Parasitol 84:229–258
Gasser RB (2006) Molecular tools—advances, opportunities and prospects. Vet Parasitol 136:69–89
Huang WY, He B, Wang CR, Zhu XQ (2004) Characterization of Fasciola species from mainland China by ITS-2 ribosomal DNA sequence. Vet Parasitol 120:75–83
Lavín S, Ruiz-Bascarán M, Marco I, Viñas L (1995) Ecopatología del gamo (Dama dama) en el Principado de Asturias. Vet Castilla León 9:14–18
Lee CG, Zimmerman GL, Wee SH (1992) Fasciola hepatica: comparison of fluke from Korea and the United States by isoelectric focusing banding patterns of whole-body protein. Vet Parasitol 42:311–316
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Li G, Gao M, Yang B, Quiros F (2003) Gene for gene alignment between the Brassica and Arabidopsis genomes by direct transcriptome mapping. Theor Appl Genet 107:168–180
Lotfy WM, El-Morshedy HN, Abou El-Hoda M, El-Tawila MM, Omar EA, Farag HF (2002) Identification of the Egyptian species of Fasciola. Vet Parasitol 103:323–332
Mas-Coma S, Bargues MD, Valero MA (2005) Fascioliasis and other plant-borne trematode zoonoses. Int J Parasitol 35:1255–1278
Mas-Coma S, Bargues MD, Valero MA, Fuentes MV (2003) Adaptation capacities of Fasciola hepatica and their relationships with human fasciolosis: from below sea level up to the very high altitude. In: Combes C, Jourdane J (eds) Taxonomy, ecology and evolution of metazoan parasites. Perpignan University Press, Perpignan, pp 81–123
Morozova EV, Khrisanfova GG, Arkhipov IA, Semenova SK (2004) Polymorphism of the ND1 and CO1 mitochondrial genes in populations of liver fluke Fasciola hepatica. Genetika 40:1002–1005
Paz-Silva A, Sánchez-Andrade R, Suárez JL, Pedreira J, Arias M, López C, Panadero R, Díaz P, Díez-Baños P, Morrondo P (2003) Prevalence of natural ovine fasciolosis shown by demonstrating the presence of serum circulating antigens. Parasitol Res 91:328–331
Pérez JM, Meneguz PG, Dematteis A, Rossi L, Serrano E (2006) Parasites and conservation biology: the ‘ibex-ecosystem’. Biodivers Conserv 15:2033–2047
Periago MV, Valero MA, Panova M, Mas-Coma S (2006) Phenotypic comparison of allopatric populations of Fasciola hepatica and Fasciola gigantica from European and African bovines using a computer image analysis system (CIAS). Parasitol Res 99:368–378
Prichard R, Tait A (2001) The role of molecular biology in veterinary parasitology. Vet Parasitol 98:169–194
Qiao LX, Liu HY, Guo BT, Weng ML, Dai JX, Duan DL, Wang B (2007) Molecular identification of 16 Porphyra lines using sequence-related amplified polymorphism markers. Aquat Bot 87:203–208
Riaz A, Li G, Quresh Z, Swati MS, Quiros CF (2001) Genetic diversity of oilseed Brassica napus inbred lines based on sequence-related amplified polymorphism and its relation to hybrid performance. Plant Breed 120:411–415
Sánchez-Andrade R, Paz-Silva A, Suárez J, Panadero R, Díez-Baños P, Morrondo P (2000) Use of a sandwich-enzyme-linked immunosorbent assay (SEA) for the diagnosis of natural Fasciola hepatica infection in cattle from Galicia (NW Spain). Vet Parasitol 93:39–46
Spithill TW, Dalton JP (1998) Progress in development of liver fluke vaccines. Parasitol Today 14:224–228
Valero MA, Panova M, Mas-Coma S (2001a) Developmental differences in the uterus of Fasciola hepatica between livestock liver fluke population from Bolivian highlands and European lowlands. Parasitol Res 87:337–342
Valero MA, Darce NA, Panova M, Mas-Coma S (2001b) Relationships between host species and morphometric patterns in Fasciola hepatica adults and eggs from the Northern Bolivian Altiplano hyperendemic region. Vet Parasitol 102:85–100
Vara-Del Río MP, Villa H, Martinez-Valladares M, Rojo-Vázquez FA (2007) Genetic heterogeneity of Fasciola hepatica isolates in the northwest of Spain. Parasitol Res 101:1003–1006
Yamaguti S (1958) Systema Helminthum, Vol. I. The digenetic trematodes of vertebrates. Interscience, New York
Zhu XQ, D'Amelio S, Palm HW, Paggi L, George-Nascimento M, Gasser RB (2002) SSCP-based identification of members within the Pseudoterranova decipiens complex (Nematoda: Ascaridoidea: Anisakidae) using genetic markers in the internal transcribed spacers of ribosomal DNA. Parasitology 124:615–623
Zhu XQ, Podolska M, Liu JS, Yu HQ, Chen HH, Lin ZX, Luo CB, Song HQ, Lin RQ (2007) Identification of anisakid nematodes with zoonotic potential from Europe and China by single-strand conformation polymorphism analysis of nuclear ribosomal DNA. Parasitol Res 101:1703–1707
Acknowledgements
Project support was provided in part by grants from the Program for Changjiang Scholars and Innovative Research Teams in Chinese Universities (Grant No. IRT0723) to XQZ, and a grant from Euphrates University-Syria to SA. Research activities in Spain were partially supported by the Plan Andaluz de Investigación (group RNM-118). The authors are indebted to Adolfo Paz-Silva (Departamento de Patología Animal, Parasitología y Enfermedades Parasitarias, Facultad de Veterinaria, Universidad de Santiago de Compostela), Ana L. García-Pérez and Ramón A. Juste (Sanidad Animal, Instituto Vasco de Investigación y Desarrollo Agrario, NEIKER), Margarita Buades (Head of the Sección de Mataderos, Islas Baleares) and Santiago Lavín (Universidad Autónoma de Barcelona) for providing F. hepatica samples and for their helpful comments. The experiments comply with the current laws of the countries in which the experiments were performed.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Alasaad and Q. Y. Li contributed equally to this work.
Rights and permissions
About this article
Cite this article
Alasaad, S., Li, Q.Y., Lin, R.Q. et al. Genetic variability among Fasciola hepatica samples from different host species and geographical localities in Spain revealed by the novel SRAP marker. Parasitol Res 103, 181–186 (2008). https://doi.org/10.1007/s00436-008-0952-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-008-0952-1