
Abstract
Fasciola hepatica is a trematode leading to heavy economic setbacks to the livestock sector globally. The population’s genetic information and intimate kinship level are frequently assessed using analysis of mitochondrial DNA. In this analysis, we retrieved cox1 (n = 247) and nad1 (n = 357) sequences of F. hepatica from the NCBI GenBank database and aligned the sequences with the respective reference sequences using MEGA software. The median joining network was drawn using PopArt software while neutrality and diversity indices were estimated with the help of DnaSp software. Neighbor-joining phylogenetic tree was constructed using the MEGA software package. A total of 46 and 98 distinctive haplotypes were observed for cox1 and nad1 genes, respectively. Diversity indices indicated high haplotype and nucleotide diversities in both genes. Positive Tajima’s D and Fu’s Fs values were found for the entire population of both the genes under study. The cox1 and nad1 gene segments in this study showed high Tajima’s D values, suggesting a low likelihood of future population growth. The Tajima’s D value of the nad1 gene sequence is lower (2.14910) than that of the cox1 gene sequence (3.40314), which suggests that the former is growing at a slower rate. However, the region-wise analysis revealed that both the cox1 and nad1 genes showed deviation from neutrality suggesting a recent population expansion as a result of an excess of low-frequency polymorphism. Furthermore, the overall host-wise analysis showed positive and significant Tajima’s D values for the cox1 and nad1 gene sequences. To the best of our knowledge, this is the first attempt to provide insights into genetic variations and population structure of F. hepatica at a global scale using cox1 and nad1 genes. Our findings suggest the existence of specific variants of F. hepatica in different parts of the world and provide information on the molecular ecology of F. hepatica. The results of this study also mark a critical development in upcoming epidemiological investigations on F. hepatica and will also contribute to understanding the global molecular epidemiology and population structure of F. hepatica.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trematodes of the genus Fasciola cause fasciolosis which is often known as liver fluke infection. The parasite F. hepatica infects animals with one of the widest regional distributions and causes significant losses to the global livestock industry (Charlier et al. 2020). It is listed as one of the most important neglected zoonotic diseases which affect both humans and domesticated animals (Mas-Coma et al. 2014). Fasciola hepatica causes a common foodborne illness in individuals that are linked to eating raw vegetables such as aquatic plants or alfalfa and drinking water that has been tainted with infective metacercaria (Cabada et al. 2018; Sabourin et al. 2018). Similar to humans, animals can get fasciolosis by consuming polluted food and water, which has a negative impact on their health and reduces animal output and, consequently, farm economic growth (Beesley et al. 2018).
There are many zoonotic trematodiasis that have been estimated in the past to cause 665,352 disability-adjusted life years (DALYs) throughout the world (Fürst et al. 2012). Fasciolosis, a type of trematode infection that spreads through contaminated food, is found all around the world. Records show that Fasciola infection has been identified in 81 different countries (Caravedo and Cabada 2020). Fasciola hepatica is commonly present in tropical and subtropical regions such as those in the Middle East (Egypt and Iran), South America (Bolivia, Ecuador, and Peru), and Asia (Mas-Coma et al. 2009). Infection with Fasciola spp. increases the risk of co-infections, decreases production and fertility, and causes considerable morbidity and mortality in livestock. These factors add up to approximately €2.5 billion in annual economic losses worldwide (Mazeri et al. 2017). When animals consume infected encysted metacercariae that have erupted from the intermediate host and settled on the grass, they become infected. Following excystment in the gut, the parasite pierces the intestinal wall and migrates to the liver, where it causes two clinical phases of the disease to develop (Corrales et al. 2021). Acute fasciolosis sets in due to tissue destruction and hemorrhages brought on by the parasites tunneling and feeding activity in the liver tissue, as well as immunopathology brought on by the host’s immune responses. In the chronic phase, the flukes move into the bile ducts, where they mature and lay eggs. Generally, the severity of infection is more in sheep than in cattle because they are less resistant to parasites. The most prevalent type of fasciolosis in sheep farms is subclinical and has minimal liver fluke loads, but it can still affect the wool quality and weight gain (Hayward et al. 2021) and make sheep more vulnerable to other diseases (Munita et al. 2019).
In addition to serving as a source of protein, livestock is seen as a direct source of revenue and employment (Mehmood et al. 2017). Thus, the food security and finance of the affected countries are threatened by this parasitosis (Webb and Cabada 2018). It is considered the primary contributor to financial losses in the cattle sector, particularly through liver damage, high morbidity and mortality, decreased output of meat and milk, and increased costs for anthelmintics and veterinary care (El-Tahawy et al. 2017; Fanke et al. 2017).
The occurrence and spread of the disease are also being impacted by climate change (Mas-Coma et al. 2019) and the trading in live animals is accelerating the spread of novel species to new areas as well as encouraging the development of hybrid forms (Agatsuma et al. 2000). Fasciola distribution causes controversy over species identification in several countries where morphological criteria are applied in research investigations. Presently, the GenBank database has a significant number of incomplete nad1 and cox1 gene sequences of Fasciola species isolated from different host species (Reaghi et al. 2016). Indicators like genetic diversity and population enable us to identify parasite adaptation and fitness in specific habitats (Rouhani et al. 2017).
Molecular studies play a significant role in the detection of dissemination of Fasciola hepatica due to host spectrum and ecological diversity (Thang et al. 2019). Diversity indices advocate that using the main population in defined ecosystems assists in better comprehending the biological perspectives and forecasting antigenic and phenotypic differences (Ai et al. 2011).
The genetic diversity of the F. hepatica population is also influenced by the intermediate hosts where clonal expansion occurs (Beesley et al. 2017). The potential of the intermediate host to spread metacercariae was confirmed by Vilas et al. (2012). Furthermore, the same multilocus genotype was found to be shared among cattle and sheep. The non-significant difference in genetic diversity of F. hepatica isolates from domesticated animals was observed by Beesley et al. (2017); however, a wide spectrum of wild animals being infected by F. hepatica is supposed to be responsible for maintaining genetic diversity. The available literature indicates a high genetic diversity level in F. hepatica populations, which could facilitate adaptation to environmental selection pressure through clonal reproduction in snails, allowing for the rapid spread of resistant populations (Elliott et al. 2014).
Molecular studies that employ both nuclear and mitochondrial DNA have identified diverse fluke populations across the globe. The nuclear DNA markers, ribosomal internal transcribed spacer (ITS), phosphoenolpyruvate carboxykinase (pepck), DNA polymerase delta (pold), cytochrome c oxidase I (cox1), and NADH dehydrogenase (nad1) genes are some of the markers that can be used for genetic characterization of Fasciola spp., as described by Sarkari et al. (2017) and Chougar et al. (2019).
Mitochondrial DNA (mt-DNA), has been used as a marker for population diversity. Mitochondrial cox1 and nad1 genes are frequently used as DNA markers to investigate the genetic variability of Fasciola spp. These markers are of high importance to study biogeography and population structure of F. hepatica (Itagaki et al. 2005). Furthermore, microsatellite DNA simple sequence repeat markers have also been studied to reveal the population structure of various parasites (Yin et al. 2016; Moendeg et al. 2017). The genetic information of the population and the level of intimate kinship are frequently assessed using analysis of mt-DNA sequences because of the absence of recombination, maternal inheritance, conserved structure, higher mutation rate and a relatively higher evolutionary rate (Jia et al. 2012). To identify genetic variations within Fasciola species, incomplete sequences of cox1 and nad1 genes have been effectively employed (Bowles and McManus 1993). To understand the population dynamics, it is essential to assess the genetic diversity in F. hepatica isolates globally. This study analyzed the cox1 and nad1 gene sequences available in the GenBank repository to describe genetic variation, population structure, and phylogeny of F. hepatica.
Methods
Data Collection
A total of 604 gene sequences of F. hepatica were retrieved and a dataset was produced after selecting the cox1 (n = 247) and nad1 (n = 357) genes, submitted to the National Center for Biotechnology Information, USA (NCBI), database until 9 February 2023.
Alignment and Phylogenetic Analysis
By using Molecular Evolutionary Genetics Analysis Version 11 (MEGA 11), all gene sequences were assembled into it in FASTA format. The reference sequences cox1 (accession no. LC273025) and nad1 (accession no. MK838688) were used to align all of the sequences after they had been cut off at both ends. Short gene sequences were eliminated leaving 604 gene sequences of 387 bp cox1 (n = 247) and 689 bp nad1 (n = 357) for bioinformatic analysis. Phylogenetic trees were created from the sequences of both gene regions using the neighbor-joining (NJ) model and the Jukes–Cantor nucleotide distance measure. The 1000 bootstrap replicates were used to obtain statistical support for the branch specificity. To determine the strength of the relationships, the sequence of Schistosoma japonicum was used as an outgroup.
Haplotype analysis and networking
The sequences in FASTA format were examined during the haplotype analysis by using the tool DnaSP 6 (Rozas et al. 2017). Calculations were made to estimate the genetic composition of both the genes using the haplotype and nucleotide change values, nucleotide and haplotype numbers, and the neutrality indices. For a visual depiction of the associations between haplotypes, the sequences were converted to Nexus format (Maddison et al. 1997) and a haplotype network was created using the PopArt (Population Analysis with Reticulate Trees) application (Leigh and Bryant 2015).
Results
A total of 604 gene sequences from F. hepatica isolates were analyzed in this study, comprising 247 cox1 sequences obtained from 9 countries and 357 nad1 sequences obtained from 11 countries (as shown in Table 1). All of these sequences were sourced from the NCBI database.
Polymorphism and haplotype analysis
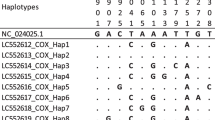
Mutations were detected at 154 and 275 different points within the cox1 and nad1 gene sequences. Forty-six distinctive haplotypes were identified by the analysis of 247 cox1 gene sequences (Table 2). The dominating haplotype among these was Hap01, which had 64 genetic sequences. By evaluating 357 nad1 genetic sequences, 98 haplotypes were recognized (Table 3). Out of these, Hap11 was the dominant haplotype and consisted of 113 gene sequences.
Haplotype network
There were 46 haplotypes in the cox1 haplotype network (Fig. 1). The major haplotype in the network was Hap01, which contributed 25.91% (64/247) and was followed by Hap15 with 18.21% (45/247). A single distinct haplotype mainly composed 65.21% (30/46) of the total haplotype network. Individual haplotypes came from Algeria (n = 13), Iran (n = 7), Ecuador (n = 3), Iraq (n = 3), Spain (n = 3), and Italy (n = 1).

Appearance of cox1 (387 bp) haplotypes F. hepatica sequences. The number of mutations that distinguish haplotypes is indicated by screening marks. The geographical distribution of haplotypes is shown in different colors. The size of the circles is related to the haplotype frequency
There were 98 haplotypes in the nad1 haplotype network (Fig. 2). The major haplotype in the network was Hap11, which contributed 31.65% (113/357) and was followed by Hap40 with 16.52% (59/357). A single distinct haplotype mainly composed 77.55% (76/98) of the total haplotype network. Individual haplotypes came from Spain (n = 24), Brazil (n = 14), Armenia (n = 12), Ecuador (n = 8), Afghanistan (n = 7), Algeria (n = 7), and Iran (n = 4).

Appearance of nad1 (689 bp) haplotypes F. hepatica sequences. The number of mutations that distinguish haplotypes is indicated by screening marks. The geographical distribution of haplotypes is shown in different colors. The size of the circles is related to the haplotype frequency.
Phylogenetic tree
The findings of the phylogenetic analysis were reliable with the haplotype network. The cox1 and nad1 gene sequences were aligned to create the phylogenetic tree as mentioned in Figs. 3 and 4, respectively. Schistosoma japonicum was added as outgroup in both phylogenetic trees. The evolutionary history was inferred using the neighbor-joining method (Saitou and Nei 1987). The optimal trees are shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test are shown next to the branches (Felsenstein 1985). The evolutionary distances were computed using the maximum composite likelihood method (Tamura et al. 2004) and are in the units of the number of base substitutions per site. This analysis involved 52 and 103 nucleotide sequences for cox1 and nad1 trees, respectively. Codon positions included were 1st+2nd+3rd+Noncoding. All ambiguous positions were removed for each sequence pair (pairwise deletion option). Evolutionary analyses were conducted in MEGA11 (Tamura et al. 2021).

Phylogenetic tree view of F. hepatica sequences using cox1 gene (387 bp)

Phylogenetic tree view of F. hepatica sequences using nad1 gene (689 bp)
Gene flow, diversity, and neutrality analysis
In Table 4, the diversity and neutrality indices are given for the cox1 and nad1 genes. To ascertain whether populations were under selection pressure, the values of Tajima’s D and Fu’s Fs were determined.
Diversity and neutrality indices for F. hepatica isolates from various geographical regions
To provide insights into the future population growth in each region, the sequences used in the study were further analyzed to compute diversity and neutrality indices. Diversity and neutrality indices of cox1 and nad1 genes are shown in Tables 5 and 6, respectively.
For cox1 gene sequences from Ecuador, Iran, Italy, and Spain, the negative but insignificant values of Tajima’s D were observed while this parameter was statistically significant and negative for the Iraqian isolates. This indicated deviation from neutrality suggesting a recent population expansion as a result of an excess of low-frequency polymorphism. A positively significant value of Tajima’s D was observed for the isolates from Algeria and the value was also positive but insignificant for the isolates from South Africa. Diversity indices were not calculated for the isolates from Australia and Egypt as four or more sequences are needed to compute Tajima’s D value. Fu’s Fs was negative for the entire population indicating an excess number of alleles, as would be expected during genetic hitchhiking or a recent population expansion except for the Algerian and South African isolates which were positive but insignificant.
Regarding nad1 gene sequences, the negatively significant values of Tajima’s D were observed for the isolates from Algeria, Armenia, Brazil, Ecuador, and Spain. Like cox1 gene sequences, this also indicated deviation from neutrality suggesting a recent population expansion. Diversity indices were not calculated for the isolates from Australia and Uruguay as four or more sequences are needed to compute Tajima’s D value. Fu’s Fs was negative for the entire population indicating an excess number of alleles, except for the Argentinian isolates which were positive but insignificant. For nad1 gene analysis, Japan was excluded in indices calculations as only one sequence was included from Japan in this study.
Host-based diversity and neutrality indices for F. hepatica isolates
In order to provide a better comprehensive understanding of the genetic dynamics of the parasite in various hosts and its circulation in the environment, the sequences used in the study were further analyzed to compute diversity and neutrality indices based on their host origin. A total of 12 cox1 and 20 nad1 sequences available in the NCBI GenBank database were without any information about their host. Thus, only 235 cox1 and 337 nad1 gene sequences were included in the host-wise analysis of data. Diversity and neutrality indices of cox1 and nad1 genes are shown in Tables 7 and 8, respectively.
Regarding the cox1 gene sequences, the values of Tajima’s D were negative but insignificant for the isolates of goat and human origin. Overall, positive and significant Tajima’s D values were observed. A similar pattern of Fu’s Fs values was observed.
Regarding the nad1 gene, the negative and statistically significant value of Tajima’s D indicated events of recent population expansion while this value was positive for the isolates of cattle origin. Fu’s Fs was negative for the isolates of sheep origin while the overall value was insignificant and positive.
The cox1 and nad1 gene sequences were aligned to create the phylogenetic tree (Figs. 5 and 6) to describe host-wise diversity. Schistosoma japonicum was added as an outgroup in both phylogenetic trees. Representative haplotypes were included to construct the phylogenetic trees. For nad1, 52 cattle and 60 sheep isolates were included out of a dataset of 337 sequences while for the cox1 gene, 35 cattle, 15 sheep, 5 goat, and 4 human samples were included out of a dataset of 235 sequences.

Evolutionary analysis of host wise investigation of cox1 gene sequences inferred by maximum likelihood method

Evolutionary analysis of host wise investigation of nad1 gene sequences inferred by maximum likelihood method
Discussion
Infectious diseases including parasitic infestations are important health problems in both animals and human beings (Mo’awad et al. 2022; Mahmoud et al. 2022). Parasites, in addition to causing diseases in animals, indirectly affect the economics of the farmer by decreasing the productivity potential of animals (Mahmood et al. 2022; Alberfkani et al. 2022). Due to the growing infection risks, fasciolosis, a recurrent ignored tropical disease, is becoming a significant issue for both food quality and human safety. The current study offers significant findings about the genetic analysis and global diversity of F. hepatica.
High levels of rainfall, the optimal combination of temperature and humidity, public eating habits, ecological factors of Fasciola transmission, free ruminant grazing, and the presence of an intermediate host favor the propagation of fasciolosis in an area (Akhlaghi et al. 2017). It is widely known that maximum genetic flow, which increases the actual population size across a range of geographical zones where the features associated with heterogeneity may be prevalent, can frequently result in the genetic variation of parasites. Due to the variety of hosts and ecological circumstances, molecular epidemiology investigations are essential for identifying Fasciola dissemination (Thang et al. 2019).
High variability has also been observed in other research that has used haplotype diversity to investigate F. hepatica. For instance, research employing the nad1 gene identified 24 haplotypes from 79 Brazilian individuals and 37 haplotypes from 130 Iranian individuals (Bozorgomid et al. 2020). Eight haplotypes were found within 90 F. hepatica samples (based on a 535 bp nad1 fragment) and six haplotypes were found within 30 F. hepatica samples (based on a 745 cox1-nad1 concatenated fragment) in previous investigations from Iran and Armenia, respectively (Reaghi et al. 2016; Semyenova et al. 2006). In a Peruvian investigation, the nad1 component from 78 distinct parasites was examined. Eight haplotypes were discovered (Hd = 0.685 and π = 0.00175) (Ichikawa-Seki et al. 2016). Seven cox1 gene haplotypes were found in an Argentine investigation that included 22 participants. When nad4 and nad5, two more mitochondrial genes, were examined, four and three haplotypes were found, correspondingly (Carnevale et al. 2017). Only six cox1 haplotypes (Hd = 0.482 and π = 0.003) and 18 nad1 haplotypes (Hd = 0.832 and π = 0.005) were found in an analysis of 208 samples conducted by Elliott et al. (2014) in Australia. Ichikawa-Seki et al. (2017) discovered that in 211 lamellipodia from different geographic regions of China, 11 haplotypes belonged to an F. hepatica population and 18 haplotypes belonged to an F. gigantica population.
The existence of F. hepatica in Peru, Argentina, Australia, and Southern Brazil with a very limited number of individuals, each from a much larger parental population, producing a Founder’s effect, may be one explanation for both high haplotype diversity and low nucleotide diversity. We propose that F. hepatica was introduced into Brazil in a number of distinct waves of human and cattle immigration in order to more adequately explain the large number of haplotypes that were observed.
In the present study, the genetic variation and population dynamics of F. hepatica were evaluated. This was accomplished by utilizing sequenced information from the cox1 and nad1 gene sequences that were obtained from GenBank and are frequently utilized for Fasciola species differentiation. As a result of this work, knowledge about global gene flow and population dynamics in F. hepatica infections was gained. For this in silico analysis, we used a total of 247 cox1 (387 bp) and 357 nad1 (689 bp) gene sequences of F. hepatica isolates that were already registered in the NCBI database to assess the genetic diversity and variation of the F. hepatica. The results of the current study show an extremely high global haplotype diversity within both cox1 and nad1 genes. The 604 samples analyzed represented a total of 46 haplotypes for the cox1 and 98 for nad1 genes.
Nucleotide diversity and population expansion were assessed by using neutrality indices including Tajima D, Fu’s Fs, and Fu’s LD (Ramos-Onsins and Rozas 2002). A positive Tajima D value indicates heterozygosity, which is described as having a selective advantage, whereas negative values show that one allele has a selective advantage over the other allele. The Tajima D test measures how much populations deviate from the traditional neutral model. A negative score also denotes a rapid population increase (Vamathevan et al. 2008; Stephens et al. 2001). Both the cox1 and nad1 gene segments in our study had high Tajima’s D values, which suggested a low likelihood of future population growth. The Tajima’s D value of the nad1 gene sequence is lower (2.14910) than that of the cox1 gene sequence (3.40314), which suggests that the former is growing at a slower rate. With a significantly negative value (p < 0.05), Fu’s Fs serves as a marker of population growth sensitivity, showing that the populations share the same gene pool and exhibit similar growth tendencies (Fu 1997; Li et al. 2009). In both the cox1 and nad1 haplotype groupings, our study produced highly positive and statistically non-significant Fu’s Fs values, indicating that these populations have not spread internationally.
The level of polymorphism in the population was found by evaluating nucleotide diversity. We found that the mean nucleotide difference between the nad1 (0.21029) and cox1 (0.17426) gene sequences was larger. In order to determine the uniqueness of haplotypes within the population, haplotype diversity was also evaluated. The values of the nad1 (0.860) and cox1 (0.869) gene sequences were extremely comparable in our investigation.
Our investigation of the cox1 gene sequences resulted in the discovery of 46 haplotypes in total. The network consisted of 30 individual haplotypes, with the dominant haplotype accounting for 25.91% of the whole. In our examination of the nad1 gene sequence, 98 distinct haplotypes were found. There were 76 individual haplotypes, and the predominant haplotype made up 31.65% of the entire network. The major haplotypes all share a common ancestry.
The 387 bp cox1 gene sequences had 154 unique mutations found throughout them, whereas the 689 bp nad1 sequences had 275 unique mutations found. The longer and more complicated evolutionary history of F. hepatica may be reflected in the higher mutation rates. Worldwide, there is a very high level of genetic variation within the F. hepatica species, and the complicated phylogeographic patterns that have been identified by phylogenetic and geographic analyses imply that the intensive animal trade has influenced the species’ current distribution. Several South American, North African, Asian, and Middle Eastern countries (Ecuador, Brazil, Algeria, Iran, Afghanistan, etc.) had a high number of haplotypes detected in this study, suggesting that F. hepatica has been present in these regions for a longer period of time than in some western nations (Italy and Spain).
Region-wise analysis revealed that both the cox1 and nad1 genes showed deviation from neutrality suggesting a recent population expansion as a result of an excess of low-frequency polymorphism. Furthermore, overall host-wise analysis showed positive and significant Tajima’s D values for both the cox1 and nad1 gene sequences. To the best of our knowledge, this is the first attempt to provide insights into genetic variations and population structure of F. hepatica at a global scale using cox1 and nad1 genes. Our findings suggest the existence of specific variants of F. hepatica in different parts of the world and provide information on the molecular ecology of F. hepatica. In this analysis, 387 bp cox1 and 689 bp nad1 gene consensus sequences were investigated to describe genetic diversity. In the future, we recommend the utilization of longer, rather complete, gene sequences to provide a broader picture of genetic variations among F. hepatica isolates. It is further suggested to study additional mitochondrial markers as the discriminatory power of a set of mitochondrial gene targets is superior to describing genetic diversity.
Conclusion
Communities with a high prevalence of intermediate hosts face a serious threat from F. hepatica. This study is the first bioinformatics investigation to assess the genetic structure F. hepatica gathered globally, despite the fact that numerous molecular studies have been carried out to date. All of the sequencing information from F. hepatica isolates relevant to cattle and humans was presented in this study. We assume that this research can close any knowledge gaps in the area. Our findings also mark a critical development in upcoming epidemiological and bioecological research that may result in effective therapies for specific species or strains.
References
Agatsuma T, Arakawa Y, Iwagami M (2000) Molecular evidence of natural hybridization between Fasciola hepatica and F. gigantica. Parasitol Int 49:231–238
Ai L, Chen MX, Alasaad S, Elsheikha HM, Li J, Li HL, Lin RQ, Zou FC, Zhu XQ, Chen JX (2011) Genetic characterization, species differentiation and detection of Fasciola spp. by molecular approaches. Parasit Vector 4:101
Akhlaghi E, Mohammadi MA, Ziaali N, Baneshi MR, Nasibi S, Kamyabi H, Rostami S, Harandi MF (2017) Morphometric and molecular study of Fasciola isolates from ruminants in Iran. Turkiye Parazitol Derg 41:192
Alberfkani MI, Albarwary AJS, Jaafar GM, Zubair AI, Abdullah RY (2022) Molecular characterization and phylogenetic analysis of cox1 and ITS-1 gene fragments of Moniezia species isolated from sheep. Pak Vet J 42:566–570
Beesley NJ, Williams DJL, Paterson S, Hodgkinson JE (2017) Fasciola hepatica demonstrates high levels of genetic diversity, a lack of population structure and high gene flow, possible implications for drug resistance. Int J Parasitol 47:11–20
Beesley NJ, Caminade C, Charlier J, Flynn RJ, Hodgkinson JE, Martinez-Moreno A, Martinez-Valladares M, Perez J, Rinaldi L, Williams DJL (2018) Fasciola and fasciolosis in ruminants in Europe: identifying research needs. Transbound Emerg Dis 65:199–216
Bowles J, McManus DP (1993) NADH dehydrogenase 1 gene sequences compared for species and strains of the genus Echinococcus. Int J Parasitol 23:969–972
Bozorgomid A, Rouhani S, Harandi M, Ichikawa-Seki M, Raeghi S (2020) Genetic diversity and distribution of Fasciola hepatica haplotypes in Iran: molecular and phylogenetic studies. Vet Parasitol 19:100359
Cabada MM, Morales ML, Webb CM, Yang L, Bravenec CA, Lopez M, Bascope R, White AC, Gotuzzo E (2018) Socioeconomic factors associated with Fasciola hepatica infection among children from 26 communities of the Cusco Region of Peru. Am J Trop Med Hyg 99:1180–1185
Caravedo MA, Cabada MM (2020) Human fascioliasis: current epidemiological status and strategies for diagnosis, treatment, and control. Res Rep Trop Med 11:149–158
Carnevale S, Malandrini JB, Pantano ML, Soria CC, Rodrigues-Silva R, Machado-Silva JR, Velásquez JN, Kamenetzky L (2017) First genetic characterization of Fasciola hepatica in Argentina by nuclear and mitochondrial gene markers. Vet Parasitol 245:34–38
Charlier J, Rinaldi L, Musella V, Ploeger HW, Chartier C, Vineer HR, Hinney B, von Samson-Himmelstjerna G, Bacescu B, Mickiewicz M, Mateus TL, Martinez-Valladares M, Quealy S, Azaizeh H, Sekovska B, Akkari H, Petkevicius S, Hektoen L, Hoglund J et al (2020) Initial assessment of the economic burden of major parasitic helminth infections to the ruminant livestock industry in Europe. Prev Vet Med 182:105103
Chougar L, Amor N, Farjallah S (2019) New insight into genetic variation and haplotype diversity of Fasciola hepatica from Algeria. Parasitol Res 118:1179–1192
Corrales JL, Cwiklinski K, Verissimo CD, Dorey A, Lalor R, Jewhurst H, McEvoy A, Diskin M, Duffy C, Cosby SL, Keane OM (2021) Diagnosis of sheep fasciolosis caused by Fasciola hepatica using cathepsin L enzyme-linked immunosorbent assays (ELISA). Vet Parasitol 298:109517
Elliott T, Muller A, Brockwell Y, Murphy N, Grillo V, Toet HM, Anderson G, Sangster N, Spithill TW (2014) Evidence for high genetic diversity of NAD1 and COX1 mitochondrial haplotypes among triclabendazole resistant and susceptible populations and field isolates of Fasciola hepatica (liver fluke) in Australia. Vet Parasitol 200:90–96
El-Tahawy AS, Bazh EK, Khalafalla RE (2017) Epidemiology of bovine fascioliasis in the Nile Delta region of Egypt: its prevalence, evaluation of risk factors, and its economic significance. Vet World 10:1241–1249
Fanke J, Charlier J, Steppin T, von Samson-Himmelstjerna G, Vercruysse J, Demeler J (2017) Economic assessment of Ostertagia ostertagi and Fasciola hepatica infections in dairy cattle herds in Germany. Vet Parasitol 240:39–48
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evol 39:783–791
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and back ground selection. Genet 147:915–925
Fürst T, Keiser J, Utzinger J (2012) Global burden of human food-borne trematodiasis: a systematic review and meta-analysis. Lancet Infect Dis 12:210–221
Hayward AD, Skuce PJ, McNeilly TN (2021) The influence of liver fluke infection on production in sheep and cattle: a meta-analysis. Int J Parasitol 21:142–149
Ichikawa-Seki M, Ortiz P, Cabrera M, Hobán C, Itagaki T (2016) Molecular characterization and phylogenetic analysis of Fasciola hepatica from Peru. Parasitol Int 65:171–174
Ichikawa-Seki M, Peng M, Hayashi K, Shoriki T, Mohanta UK, Shibahara T, Itagaki T (2017) Nuclear and mitochondrial DNA analysis reveals that hybridization between Fasciola hepatica and Fasciola gigantica occurred in China. Parasitol 144:206–213
Itagaki T, Kikawa M, Sakaguchi K, Shimo J, Terasaki K, Shibahara T, Fukuda K (2005) Genetic characterization of parthenogenic Fasciola sp. in Japan on the basis of the sequences of ribosomal and mitochondrial DNA. Parasitology 131:679–685
Jia W, Yan H, Lou Z, Ni X, Dyachenko V, Li H, Littlewood DTJ (2012) Mitochondrial genes and genomes support a cryptic species of tapeworm within Taenia taeniaeformis. Acta Trop 123:154–163
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Li YL, Kong XY, Yu ZN, Kong J, Ma S, Chen LM (2009) Genetic diversity and historical demography of Chinese shrimp Feneropenaeus chinensis in Yellow Sea and Bohai Sea based on mitochondrial DNA analysis. Afr J Biotechnol 8:1193–1202
Maddison DR, Swofford DL, Maddison WP (1997) NEXUS: an extensible file format for systematic information. Syst Biol 46:590–621
Mahmood Q, Younus M, Sadiq S, Iqbal S, Idress S, Khan S, Zia UR (2022) Prevalence and associated risk factors of cystic echinococcosis in food animals – a neglected and prevailing zoonosis. Pak Vet J 42:59–64
Mahmoud HYAH, Ali AAA, Khalil AM, Amin YA, Ali AO (2022) The infection rate of Fasciola and Anaplasma in cattle and buffaloes in Qena Egypt. Int J Vet Sci 11:308–314
Mas-Coma S, Valero MA, Bargues MD (2009) Chapter 2. Fasciola, lymnaeids and human fascioliasis, with a global overview on disease transmission, epidemiology, evolutionary genetics, molecular epidemiology and control. Adv Parasitol 69:41–146
Mas-Coma S, Valero MA, Bargues MD (2014) Fascioliasis. Adv Exp Med Biol 766:77–114
Mas-Coma S, Valero MA, Bargues MD (2019) Fascioliasis. Adv Exp Med Biol 1154:71–103
Mazeri S, Rydevik G, Handel I (2017) Estimation of the impact of Fasciola hepatica infection on time taken for UK beef cattle to reach slaughter weight. Sci Rep 7:7319
Mehmood K, Zhang H, Sabir AJ, Abbas RZ, Ijaz M, Durrani AZ, Saleem MH, Ur-Rehman M, Iqbal MK, Wang Y, Ahmad HI, Abbas T, Hussain R, Ghori MT, Ali S, Khan AU, Li J (2017) A review on epidemiology, global prevalence and economical losses of fasciolosis in ruminants. Microb Pathog 109:253–262
Mo’awad HFM, Sobhy MM, Ismail TF, Enbaawy M (2022) Seroprevalence of Coxiella burnetti (Q fever) in cows and buffaloes in Egypt. Int J Vet Sci 11:16–22
Moendeg KJ, Angeles JMM, Nakao R, Leonardo LR, Fontanilla IKC, Goto Y, Kirinoki M, Villacorte EA, Rivera PT, Inoue N, Chigusa Y, Kawazu SI (2017) Geographic strain differentiation of Schistosoma japonicum in the Philippines using microsatellite markers. PLoS Negl Trop Dis 11:1–14
Munita MP, Rea R, Martinez-Ibeas AM, Byrne N, McGrath G, Munita-Corbalan LE, Sekiya M, Mulcahy G, Sayers RG (2019) Liver fluke in Irish sheep: prevalence and associations with management practices and co-infection with rumen fluke. Parasit Vectors 12:525
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Reaghi S, Haghighi A, Harandi MF, Spotin A, Arzamani K, Rouhani S (2016) Molecular characterization of Fasciola hepatica and phylogenetic analysis based on mitochondrial (nicotiamide adenine dinucleotide dehydrogenase subunit I and cytochrome oxidase subunit I) genes from the North-East of Iran. Vet World 9:1034–1038
Rouhani S, Raeghi S, Spotin A (2017) Spermatogenic and phylo-molecular characterizations of isolated Fasciola spp. from cattle, north West Iran. Pak J Biol Sci 20:204–209
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Sabourin E, Alda P, Vázquez A, Hurtrez-Boussès S, Vittecoq M (2018) Impact of human activities on Fasciolosis transmission. Trends Parasitol 34:891–903
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sarkari B, Parhoode M, Abdolahi Khabisi S, Shafiei R, Mohammadi-Ghalehbin B (2017) Genetic diversity of Fasciola spp. isolates from northern part of Iran: comparison with southwestern isolates. J Parasit Dis 41:768–772
Semyenova SK, Morozova EV, Chrisanfova GG, Gorokhov VV, Arkhipov IA, Moskvin AS, Movsessyan SO, Ryskov AP (2006) Genetic differentiation in Eastern European and Western Asian populations of the liver fluke, Fasciola hepatica, as revealed by mitochondrial nad1 and cox1 genes. J Parasitol 92:525–530
Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, Messer CJ, Chew A, Han JH (2001) Haplotype variation and linkage disequilibrium in 313 human genes. Sci 293:489–493
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc National Acad Sci 101:11030–11035
Tamura K, Stecher G, Kumar S (2021) MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. https://doi.org/10.1093/molbev/msab120
Thang TN, Hakim H, Rahimi RR, Ichikawa-Seki M (2019) Molecular analysis reveals expansion of Fasciola hepatica distribution from Afghanistan to China. Parasitol Int 72:101930
Vamathevan JJ, Hasan S, Emes RD, Amrine-Madsen H, Rajagopalan D, Topp SD, Kumar V, Word M, Simmons MD, Foord SM (2008) The role of positive selection in determining the molecular cause of species differences in disease. BMC Evol Biol 8:1–14
Vilas R, Vázquez-Prieto S, Paniagua E (2012) Contrasting patterns of population genetic structure of Fasciola hepatica from cattle and sheep: implications for the evolution of anthelmintic resistance. Infect Genet Evol 12:45–52
Webb CM, Cabada MM (2018) Recent developments in the epidemiology, diagnosis, and treatment of Fasciola infection. Curr Opin Infect Dis 31:409–414
Yin F, Gasser RB, Li F, Bao M, Huang W, Zou F, Zhao G, Wang C, Yang X, Zhou Y, Zhao J, Fang R, Hu M (2016) Population structure of Haemonchus contortus from seven geographical regions in China, determined on the basis of microsatellite markers. Parasit Vectors 9:586
Data availability statement
All data supporting the conclusions of this article are included in the article.
Funding
We acknowledge the funding received from the National Key Research and Development Program (2022YFC2304000; 2022YFD1302101), NBCITS (CARS-37) and Central Public-Interest Scientific Institution Basal Research Fund (Y2022GH13; 1610312020016).
Author information
Authors and Affiliations
Contributions
MAA, AK, LL, and HBY conceptualized the study. The methodology was designed by MAA, MS, RMAA, and BA. Formal analysis was carried out by AK, MAA, RMAA, WQ, and MS. Writing of the original draft was done by MAA and AK while WZJ, HBY, and BQF edited the draft. WZJ supervised the project.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Section Editor: Xing-Quan Zhu
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alvi, M.A., Khalid, A., Ali, R.M.A. et al. Genetic variation and population structure of Fasciola hepatica: an in silico analysis. Parasitol Res 122, 2155–2173 (2023). https://doi.org/10.1007/s00436-023-07917-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-023-07917-0