
Abstract
The complete internal transcribed spacer 1 (ITS1), 5.8S ribosomal DNA, and ITS2 region of the ribosomal DNA from 60 specimens belonging to two closely related bucephalid digeneans (Dollfustrema vaneyi and Dollfustrema hefeiensis) from different localities, hosts, and microhabitat sites were cloned to examine the level of sequence variation and the taxonomic levels to show utility in species identification and phylogeny estimation. Our data show that these molecular markers can help to discriminate the two species, which are morphologically very close and difficult to separate by classical methods. We found 21 haplotypes defined by 44 polymorphic positions in 38 individuals of D. vaneyi, and 16 haplotypes defined by 43 polymorphic positions in 22 individuals of D. hefeiensis. There is no shared haplotypes between the two species. Haplotype rather than nucleotide diversity is similar between the two species. Phylogenetic analyses reveal two robustly supported clades, one corresponding to D. vaneyi and the other corresponding to D. hefeiensis. However, the population structures between the two species seem to be incongruent and show no geographic and host-specific structure among them, further indicating that the two species may have had a more complex evolutionary history than expected.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Members of the family Bucephalidae Poche, 1907, known as gasterostomes, represent a special group of digeneans. After Wang and Wang (1998a), this family consists of 7 subfamilies and 24 genera. Dollfustrema Eckmann, 1934, is a genus belonging to the subfamily Bucephalinae. To date, the taxonomy of this genus is totally dependent on phenetic classification, but the paucity of morphological characters causes misidentifications, particularly in larvae and immature adults.
At present, different views still exist over the taxonomic status in the genus Dollfustrema. One view is that there are five species found in China: D. vaneyi Tseng, 1930, D. sinica Gu et Shen, 1976, D. foochowensis Tang et Tang, 1963 (Syn. D. sinipercae Wang, 1985), D. cociellae Gu et Shen, 1976 (Syn. Telorhynchus cociellae Gu et Shen, 1976), and D. hippocampi Shen, 1982 (Syn. T. hippocampi Shen, 1982) (Wang and Wang 1998b). An alternative view is that, in addition to D. vaneyi, D. sinica, and D. foochowensis, D. sinipercae is considered as a valid species, and another one is described in Zhang et al. (1999), named as D. hefeiensis. But T. cociellae and T. hippocampi were not classified into the genus Dollfustrema in Zhang et al. (1999). Our particular interest is the fact that D. vaneyi and D. hefeiensis are regarded as sibling species, which are nearly indistinguishable morphologically, especially before treated with dyestuff. The key distinguishing feature between the two species is the shape and number of thorns on the anterior sucker. D. vaneyi has three circlewise interleaved thorns, with the middle one being the longest and the rest two being identical in size, while D. hefeiensis has two circlewise interleaved thorns, being different in length (Zhang et al. 1999). However, these characters are often mixed, leading to the confusion between the two species. In fact, identification of species between closely related species is difficult at all stages in the life cycles. This is due in part to the phenotypic plasticity of the organisms themselves, the paucity of morphological features in life-cycle stages, and host-induced variation, artifacts produced during fixation, and the extensive overlap in morphological characteristics that occurs among species (e.g., Niewiadomska and Laskowski 2002). Consequently, whether D. hefeiensis represents a valid species discrete from D. vaneyi is not indisputable.
Wang and Wang (2000) determined that two intermediate hosts (Limnoperna lacustris, small carps and catfishes) and one final host (mandarin fish, i.e., Siniperca chuatsi) are required to maintain the life cycle of D. vaneyi. However, recent surveys have shown that some other sinipercid fishes exclusive of S. chuatsi also harbor D. vaneyi and that the geographical distribution of this bucephalid is still increasing, implying that the parasite may have a high potential to colonize both new definitive hosts and new localities. It would, therefore, be interesting to use modern molecular tools to investigate the evolutionary variation in this parasite during colonization and to elucidate the relationships of D. vaneyi parasitizing sinipercid fishes.
DNA-based approaches provide an independent method of distinguishing between species when morphological criteria are equivocal or are subject to variation (McManus and Bowles 1996). Sequence data of the ribosomal RNA (rRNA) gene, in particular the two highly variable internal transcribed spacer regions (ITS1 and ITS2), have been successfully used to resolve taxonomic questions and to determine phylogenetic affinities between closely related digenean species (e.g., Anderson and Barker 1998; Bell et al. 2001; Galazzo et al. 2002).
Thus, the starting point of our work is to obtain a better understanding of the status of D. hefeiensis and D. vaneyi by DNA-based taxonomy. The ITS1–5.8S–ITS2 sequences were cloned to analyze the genetic structure and phylogenetic relationships of 60 individuals of the two closely related bucephalids from different fish host species and different localities. The aims of the present study are (1) to determine the level of variation among ITS1–5.8S–ITS2 sequences of two closely related bucephalids, (2) to test whether the two ‘recognized’ species correspond to diagnosable genetic disjunctions, and (3) to infer the evolutionary and intraspecific relationships of the two species. In addition, such data will enable the future confident identification of all life-cycle stages of the two parasites, a task that is especially difficult for the larvae, which lack distinctive morphological features and which share common primary intermediate host species (Zhang et al. 1999). This work represents the first molecular characterization of any bucephalid species belonging to the genus Dollfustrema.
Materials and methods
Biological material
A total of 60 specimens of bucephalids were collected from the intestine, ceca, or gill of several fishes, which were indicated in Table 1. Individual bucephalids were washed in 0.85% NaCl solution before being preserved in 95% ethanol. Species identification was made on the basis of morphological description according to Zhang et al. (1999).
DNA extraction, PCR amplification, and sequencing
Total genomic DNA of the parasite was extracted using a standard sodium dodecyl sulfate-proteinase K procedure, as described by Sambrook et al. (1989). Polymerase chain reaction (PCR) was used to generate a fragment spanning ITS1–5.8S–ITS2 ribosomal DNA (rDNA) between the forward primer BD1 (5′-GTC GTA ACA ACG TTT CCG TA-3′) and the reverse primer BD2 (5′-TAT GCT TAA (G/A) TT CAG CGG GT-3′), as employed by Luton et al. (1992). The PCR protocols were 94°C for 3 min followed by 30 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 1 min and then a final elongation step at 72°C for 10 min. The amplified products were purified on a 1.0% agarose gel stained with ethidium bromide, using a commercial DNA purification kit following the manufacturer’s protocol. The purified PCR product was cloned into pMD18-T vector and sequenced with the universal primers M13. The DNA sequences of each individual and each species were deposited in the GenBank database under accession numbers EF198179 to EF198238.
Sequence alignments and analyses
Sequences were aligned using Clustal X (Thompson et al. 1997) with default settings and refined manually. DNAsp version 4.0 (Rozas et al. 2003) was used to define the haplotypes. The ITS1–5.8S–ITS2 fragment of Bucephalus polymorphus (GenBank accession number AY289239, Stunzenas et al. 2004) is included as outgroup. The boundaries between the ITS1 and ITS2 regions and the rRNA coding regions 18S, 5.8S, and 28S were determined by comparing with the ITS1–5.8S–ITS2 sequences of a pseudophyllidean cestode Bothriocephalus acheilognathi (Luo et al. 2002). The alignment is available from the corresponding author upon request.
Base compositional frequencies and nucleotide substitutions between pairwise distances were determined using PAUP* 4.0b10 (Swofford 2002). The base frequency stationarity were evaluated using chi-square (χ 2) tests implemented in PAUP*. The p-distance matrix were computed with MEGA 3.1 (Kumar et al. 2004). We used DNAsp version 4.0 (Rozas et al. 2003) to calculate nucleotide diversity (π), haplotypic diversity (h), and mean number of pairwise differences (κ). To investigate host species’ genetic structure, we constructed unrooted parsimony networks of haplotypes for each species using TCS version 1.18 (Clement et al. 2000).
Phylogenetic analyses
Phylogenetic analyses were conducted on the aligned sequences of ITS1–5.8S–ITS2 rDNA .We performed a wide array of phylogenetic analyses using different methods to gauge the robustness of our resulting hypotheses. These methods were neighbor joining (NJ) with maximum likelihood distance, maximum parsimony (MP) as implemented in PAUP*, and maximum likelihood (ML) as implemented in PhyML 2.4.4 (Guindon and Gascuel 2003), The MP method was performed using heuristic searches with ten random-addition sequence replicates and tree bisection-reconnection branch swapping. Appropriate models of sequence evolution for each data partition were determined using the Bayesian information criterion (Schwarz 1978) as implemented Modeltest 3.7 (Posada and Crandall 1998). Statistical support for the internodes in phylogenetic tree was tested by bootstrap percentages (BP) with 1,000 replicates (Felsensten 1985). Phylogenetic trees were rooted using B. polymorphus. Furthermore, we used partitioned Bayesian analyses as carried out with MrBayes 3.1 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003), which has facilitated the exploration of partition-specific evolutionary models and should reduce systematic error, thus, resulting in more accurate posterior probability estimates (Nylander et al. 2004; Brandley et al. 2005).
In Bayesian analyses, we choose the partitioning strategy according to its genomic assignment. We set the parameters for partitioned likelihood analysis in MrBayes for ITS1, 5.8S, and ITS2 as the Modeltest result suggested. Each analysis consisted of 2 × 106 generations with a random starting tree, default priors, the same set of branch lengths for each partition, and four Markov chains (with default heating values) sampled every 100 generations. To ensure the Bayesian analyses were not trapped on local optima, two separate analyses were performed. We discarded the first 3,000 trees as part of a burn-in procedure and combined the postburn-in trees (whose log-likelihoods converged to stable values) for the two analyses to construct a 50% majority rule consensus tree. The frequency of a particular clade occurred within the collection of trees after the burn-in was interpreted as a measure of clade support.
Testing alternative phylogenetic hypotheses
Because Bayesian inference generates a distribution of trees given the data, prior probabilities and model of evolution, commonly used statistical methods in topological comparisons of phylogenies, such as the approximately unbiased test (Shimodaria 2002), are not plausible. Following the recommendation of Brandley et al. (2005), we employed a Bayesian hypothesis testing and built 95% credible sets of topologies (sampled at stationarity) by using the sumt command in MrBayes. All of the trees of the 95% credible set were imported into PAUP* and filtered with the alternative phylogenetic hypothesis. If the alternative phylogenetic hypothesis was absent, it could be rejected statistically (Buckley 2002).
Results
Sequence variations
The complete ITS1–5.8S–ITS2 fragment, including portions of the 3′ end of the 18S and 5′ start of the 28S, were sequenced for the species and populations considered. The ITS1–5.8S–ITS2 region ranged from 1,067 to1,074 bp in D. vaneyi; while ranged from 1,075 to 1,111 bp in D. hefeiensis. The alignment of the ITS1–5.8S–ITS2 region sequences from the two bucephalids and the outgroup resulted in a total of 1,271 characters, including gaps. The average GC content of the sequences was 0.53, and a χ 2 test at the 5% level of significance for differences in base frequencies showed that there was no base compositional heterogeneity among sequences (χ 2 = 5.05, df = 114, P = 1.00), which is known to adversely affect phylogenetic inference (Jermiin et al. 2004). Based on the alignment of the ITS1–5.8S–ITS2 region sequences from the two bucephalids, there were 1,145 character sites in the matrix, with 153 variable sites and 62 phylogeny-informative sites.
The gene 5.8S was identical among most of specimens. The length of the ITS1 and ITS2 region differed in the two species, ranging from 562 to 591 bp and from 342 to 355 bp, respectively. Genetic distances between the haplotypes in the two species varied from 3.94 to 6.17% (mean = 4.66%). In the haplotypes of D. vaneyi and D. hefeiensis, genetic distance ranged from 0.09 to 1.68% (mean = 0.92%) and from 0.09 to 4.07% (mean = 0.62%), respectively. Comparisons between species and different populations level are shown in Table 2.
Phylogenetic relationships
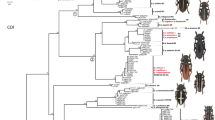
Tree topologies generated by the different building methods were similar and supported by high posterior probability or bootstrap values at main nodes. Two distinct clades (Clade A, B) were obtained using NJ, ML, MP, and Bayesian method (Fig. 1). All major clusters were supported by bootstrap values of more than 50% or posterior probabilities of more than 0.9. Clade A included only the haplotypes of D. hefeiensis (BP = 100% for NJ, MP, and ML; PP = 1.0 for partitioned Bayesian analysis), while Clade B included only the haplotypes of D. vaneyi (BP = 92, 89, and 92% for NJ, MP, and ML, respectively; PP = 0.95 for partitioned Bayesian analysis). As for the intra-relationships of the two clades, there is a slight difference among the four competitive topologies. Yet, based on the Bayesian hypothesis testing (data not shown), the partitioned Bayesian tree was most likely, although the other three alternative hypotheses cannot be statistically rejected. As shown in Fig. 1, there is no subclade diversification within Clade A. However, Clade B contains two major subclades (B1 and B2) supported by high posterior probabilities (PP = 0.98 and 0.91, respectively). In the subclade B2, three distinct subclades can be identified, i.e., B2_1, B2_2, and B2_3. In addition, all the resultant trees from different methods support that there is marked incongruence between the intra-relationships of the two species, although they have similar distribution patterns.

A major rule consensus tree obtained from partitioned Bayesian analyses in MrBayes 3.1 based on ITS1–5.8S–ITS2 sequences. Values above the branch represent posterior probabilities. Values below the branch are proportions of 1,000 bootstrap pseudoreplicates in which the node was recovered for NJ/MP/ML, respectively. For MP, tree length = 315, CI = 0.9587, RI = 0.9822. For ML, −ln L = 3423.31629, and the substitution model HKY + G was used according to the Modeltest result. DH1, DH2, DH3, DH8, DH9, DH14, and DH22 were the same haplotype. DV1, DV2, DV6, DV8, DV20, DV21, DV22, DV23, and DV24 were the same haplotype. DV4, DV14, DV25, DV28, DV29, DV31, DV32, DV33, and DV38 were the same haplotype
Genetic diversity and haplotype network
A total of 37 haplotypes were identified from 60 individuals (Tables 3 and 4), including 21 haplotypes of D. vaneyi and 16 haplotypes of D. hefeiensis. We found 21 haplotypes in 38 individuals of D. vaneyi defined by 44 polymorphic positions, 3 haplotypes were found in multiple individuals, and 18 haplotypes were represented by single individuals. The most frequently sampled haplotype, designated as “3b” in Table 3, was found in 9 of 38 individuals and was present in 5 of the 7 host species. In contrast, we found 16 haplotypes in 22 individuals of D. hefeiensis defined by 43 polymorphic positions; only 1 haplotype was found in multiple individuals, and 15 haplotypes were represented by single individuals. The most frequently sampled haplotype, designated as “1b” in Table 4, was found in 7 of 22 individuals and was present in 4 of the 5 host species. There was no shared haplotype between the two species. They exhibited similarly relatively high haplotypic diversity (h = 0.896 for D. vanyei and 0.909 for D. hefeiensis), whereas different nucleotide diversity (π = 0.00744 for D. vanyei and 0.00404 for D. hefeiensis). This pattern is also apparent in the unrooted parsimony networks of haplotypes (Fig. 2) that show that only approximately one third of the D. hefeiensis sampled shared the single most common haplotype, whereas all other individuals had unique haplotypes. The haplotype network of D. vaneyi reveals four haplotype clusters that are separated by long interlinking branches, which correspond exactly to B1, B2_1, B2_2, and B2_3 in the phylogenetic tree. However, the structure of the haplotype network in D. hefeiensis differs from that of D. vaneyi in having only one most common haplotype and having no evident differentiation.


Unrooted parsimony networks of ITS1–5.8S–ITS2 sequence haplotypes of two bucephalids from several host species. In each network, ovals indicate sampled haplotypes, small circles indicate unsampled or extinct haplotypes, and lines between haplotypes represent single nucleotide substitutions. Numbers inside ovals indicate the number of individuals carrying the haplotype, and different filled patterns represent the corresponding host species from which the haplotype was sampled. B1, B2_1, B2_2, and B2_3 refer to the corresponding to subclade in Fig. 1
In the two species, haplotype parsimony networks (Fig. 2) revealed a lack of host species’ structure to genetic variation, with parasites from different host species sharing haplotypes and no clustering of haplotypes from any host species. Similarly, it indicated a lack of geographic and microhabitat’s structure to genetic variation, with parasites from different geographical origin or habitat site sharing haplotypes and no clustering of haplotypes (data not shown).
Discussion
This study is the first attempt to carry out molecular identification and phylogenetic analysis of two closely related species from different species of fish hosts collected mainly in Hubei and Hunan Provinces but also in Jiangxi Province, China. Phylogenetic analyses revealed two robustly supported clades, one corresponding to D. vaneyi and the other corresponding to D. hefeiensis. The average divergence between the two clades (4.66%) is much higher than that among specimens of D. vaneyi or D. hefeiensis (0.92 and 0.62%, respectively). Although there is no yardstick for recognizing species boundaries by using DNA sequence differences, previous studies on the digeneans (Luton et al. 1992; Bell et al. 2001) have shown the magnitude of interspecific nucleotide differences in the ITS region was much lower than the high level of the nucleotide variation between D. vaneyi and D. hefeiensis specimens observed in the present study. Given the high sequence divergence and the phylogenetic tree, it is reasonable to consider that the D. hefeiensis represents a valid species, discrete from D. vaneyi.
In contrast to other digeneans (e.g., Tkach et al. 2000; Bell et al. 2001), the ITS2 region was much more variable than the ITS1 region. Despite the high level of intra-genetic conservation, however, sufficient variation has been found within ITS1 and ITS2 to differentiate between the two pairs of related species. In support of Anderson and Barker (1998) and Galazzo et al. (2002), we suggest that the ITS2 may also be sufficiently variable to permit discrimination at the species level, although this may not be the case for all digeneans. Thus, both the separated and combined ITS1 and ITS2 can distinguish the two pairs of related species in this study, although the ITS1 is more conservative than ITS2.
The intraspecies structure of genetic variation was found to vary between the two species. Whereas D. hefeiensis shows no subdivision as evidenced by the phylogenetic tree and parsimony relationships of haplotypes (Fig. 2), D. vaneyi has a higher level of subdivision. We postulate that the two bucephalid digeneans exhibit significantly different intra-relationships, as revealed by the clades and the phylogenetic tree itself as well as the haplotype networks. Moreover, the haplotype networks imply that the population structure between the two species exhibits little geographic and host-specific structure among them, with parasites from different host species or geographical origin sharing haplotypes and no clustering of haplotypes from any host species. This further indicates that the two species may have had a more complex evolutionary history than expected. On the one hand, most parasites are intimately dependent on one or a few hosts. Because of this host fidelity, parasites are expected to track speciating hosts by speciating themselves. This process, known as co-speciation, will lead to co-cladogenesis, the topological matching of symbiont phylogenies. Parasite and host phylogenies are rarely identical; however, forces, such as duplication (parasite speciation in the absence of host speciation), sorting events (host speciation without commensurate parasite speciation), and host-switching (parasites begin to use a new host; Johnson et al. 2003; Page 2003), can generate discordance between the phylogenies of hosts and their symbionts. On the other hand, co-distributed parasite species may display either congruent phylogeographic patterns, indicating similar responses to a series of shared host speciation events, or discordant patterns, indicating independent responses to shared evolutionary events due to different ecologies and life histories (e.g., Taberlet et al. 1998; Michaux et al. 2005; Rocha et al. 2005), or “pseudo-incongruence” in which co-distributed species respond independently to different evolutionary events occurring at different times (Donoghue and Moore 2003). Incongruence among population-level phylogenies may also be due to variation in the microevolutionary processes, such as host-switching or effective population size, that are responsible for generating the patterns of population-level divergence (Mason-Gamer and Kellogg 1996). Thus, many more samples from other fishes throughout their geographic range would be needed to investigate the phylogeographic patterns in the two species and to test whether the genetic structure of populations is consistent with current observations.
Identification of species via DNA sequences is the basis for DNA taxonomy and DNA barcoding. Currently, there is a strong focus on using a mitochondrial marker for this purpose, in particular a fragment from the cytochrome oxidase I gene (Hebert et al. 2003). While there is ample evidence that this marker is indeed suitable across a broad taxonomic range to delineate species, it has also become clear that a complementation by a nuclear marker system could be advantageous. Our results also echo those of Pons et al. (2006) in that DNA taxonomy for a particular group of organisms may be based on one or more regions of mitochondrial DNA or nuclear DNA and can be derived from phylogenetic and clustering methods using any gene region.
In conclusion, the molecular characteristics of ITS1–5.8S–ITS2 region is useful in the identification of the two closely related bucephalid digeneans and in the understanding of relationships between species of Dollfustrema, even Bucephalidae. However, further research is needed to fully understand the phylogenetic relationships of the members of this genus. Other genes and a much wider range of host species that harbor D. vaneyi and D. hefeiensis, as well as other species of Dollfustrema, need to be included. Further comparative phylogeographic studies are also required to understand the complex phylogeographic patterns and coevolution between the parasites and host species.
References
Anderson GR, Barker SC (1998) Inference of phylogeny and taxonomy within the Didymozoidae (Digenea) from the second internal transcribed spacer (ITS2) of ribosomal DNA. Syst Parasitol 41:87–94
Bell AS, Somerville C, Valtonen ET (2001) A molecular phylogeny of the genus Ichthyocotylurus (Digenea, Strigeidae). Int J Parasitol 31:833–842
Brandley MC, Schmitz A, Reeder TW (2005) Partitioned Bayesian analyses, partition choice, and the phylogenetic relationships of scincid lizards. Syst Biol 54:373–390
Buckley TR (2002) Model misspecification and probabilistic tests of topology: evidence from empirical data sets. Syst Biol 51:509–523
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Donoghue MJ, Moore BR (2003) Toward an integrative historical biogeography. Integr Comp Biol 43:261–270
Felsensten JP (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Galazzo DE, Dayanandan S, Marcogliese DJ, McLaughlin JD (2002) Molecular systematics of some North American species of Diplostomum (Digenea) based on rDNA sequence data and comparisons with European congeners. Can J Zool 80:2207–2217
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proc R Soc Lond B 270:313–321
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogeny. Bioinformatics 17:754–755
Jermiin LS, Ho SYW, Ababneh F, Robinson J, Larkum AWD (2004) The biasing effect of compositional heterogeneity on phylogenetic estimates may be underestimated. Syst Biol 53:638–643
Johnson KP, Adams RJ, Page RDM, Clayton DH (2003) When do parasites fail to speciate in response to host speciation? Syst Biol 52:37–47
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Briefings in Bioinformatics 5:150–163
Luo H-Y, Nie P, Zhang Y-A, Wang G-T, Yao W-J (2002) Molecular variation of Bothriocephalus acheilognathi Yamaguti, 1934 (Cestoda: Pseudophyllidea) in different fish host species based on ITS rDNA sequences. Syst Parasitol 52:159–166
Luton K, Walker D, Blair D (1992) Comparisons of ribosomal internal transcribed spacers from two congeneric species of flukes (Platyhelminthes: Trematoda: Digenea). Mol Biochem Parasitol 56:323–328
Mason-Gamer RJ, Kellogg EA (1996) Testing for phylogenetic conflict among molecular data sets in the tribe Triticeae (Gramineae). Syst Biol 45:524–545
McManus DP, Bowles J (1996) Molecular genetic approaches to parasite identification: their value in diagnostic parasitology and systematics. Int J Parasitol 26:687–704
Michaux JR, Libois R, Filippucci MG (2005) So close and so different: comparative phylogeography of two small mammal species, the yellow-necked fieldmouse (Apodemus xavicollis) and the woodmouse (Apodemus sylvaticus) in the Western Palearctic region. Heredity 94:52–63
Niewiadomska K, Laskowski Z (2002) Systematic relationships among six species of Diplostomum Nordmann, 1832 (Digenea) based on morphological and molecular data. Acta Parasitol 47:1230–1237
Nylander JA, Ronquist F, Huelsenbeck JP, Nieves-Aldrey JL (2004) Bayesian phylogenetic analysis of combined data. Syst Biol 53:47–67
Page RDM (2003) Tangled trees: phylogeny, cospeciation and coevolution. University of Chicago Press, Chicago
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP (2006) Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55:595–609
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Rocha LA, Robertson DR, Roman J, Bowen BW (2005) Ecological speciation in tropical reef fishes. Proc R Soc Lond B 272:573–579
Ronquist F, Huelsenbeck JP (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DNAsp, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, New York
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Shimodaria H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–598
Stunzenas V, Cryan JR, Molloy DP (2004) Comparison of rDNA sequences from colchicine treated and untreated sporocysts of Phyllodistomum folium and Bucephalus polymorphus (Digenea). Parasitol Int 53:223–228
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (* and other methods), version 4. Sinauer, Sunderland, MA
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The Clustal X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tkach VV, Pawlowski J, Sharpilo VP (2000) Molecular and morphological differentiation between species of the Plagiorchis vespertilionis group (Digenea, Plagiorchiidae) occurring in European bats, with a re-description of P. vespertilionis (Muller, 1780). Syst Parasitol 47:9–22
Wang G-T, Wang W-J (1998a) Taxonomy of the family Bucephalidae (Digenea). Acta Hydrobiol Sin 22 (suppl):89–99 (in Chinese)
Wang G-T, Wang W-J (1998b) Taxonomy and keys to the bucephalid species in China, with descriptions of three new species. Acta Hydrobiol Sin 22(suppl):100–110 (in Chinese)
Wang G-T, Wang W-J (2000) The life cycle of Dollfustrema vaneyi. Acta Hydrobiol Sin 24:644–647 (In Chinese)
Zhang J-Y, Qiu, Z-Z, Ding X-J et al (1999) Parasites and parasitic diseases of fishes. Science Press, Beijing, China (in Chinese)
Acknowledgments
The first author thanks Dr. Q. Gao for assistance in collecting specimens. We are grateful to Dr. X. Guo (Chengdu Institute of Biology, Chinese Academy of Sciences) for his kind help on the Bayesian hypothesis testing and helpful comments on multiple drafts of this manuscript. We also gracefully acknowledge the two anonymous reviewers and the editor whose critical review and insightful suggestions have helped to improve the clarity and focus of the manuscript. This study was supported by grants (project no. 30130150) from the National Natural Science Foundation of China and a grant [project no. (2005) 192] from the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, D., Wang, G., Yao, W. et al. Utility of ITS1–5.8S–ITS2 sequences for species discrimination and phylogenetic inference of two closely related bucephalid digeneans (Digenea: Bucephalidae): Dollfustrema vaneyi and Dollfustrema hefeiensis . Parasitol Res 101, 791–800 (2007). https://doi.org/10.1007/s00436-007-0549-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-007-0549-0