
Abstract
Purpose
The parasites of genera such as Babesia and Theileria are called piroplasmids due to the pear-shaped morphology of the multiplying parasite stages in the blood of the vertebrate host. Because of the enormous number of parasite species and the challenges of multiplex PCR, initial screening of samples using piroplasmid-specific PCR may be a more cost-effective and efficient technique to identify parasite species, especially during epidemiological studies. Accordingly, 18S rRNA PCR was standardized and optimized on common piroplasmids of different animals like cattle, buffaloes, sheep, goats, dogs, horses, and leopards.
Methods
Bloods samples from 1250 animals were collected from different animals in Junagadh district of Gujarat, India. 18S rRNA PCR was standardized and optimized as a primary method for molecular screening of piroplasms in domestic and wild animals. The method was checked for its analytical sensitivity and specificity. Parasite species-specific PCR and sequencing was used to validate the test. Moreover, in-silico restriction enzyme (RE) analysis was also done to assess its applicability in PCR–RFLP.
Results
Piroplasm infections were recorded in 63.3% of animals in Junagadh. The 18S rRNA PCR detected the piroplasmid DNA in as low as 39 picograms (pg) of whole blood genomic DNA isolated from microscopically Theileria positive blood samples and no reactivity was recorded from common but unrelated haemoparasites viz., Trypanosoma evansi, Hepatozoon spp., Anaplasma spp., and Ehrlichia canis was observed. The 18S rRNA PCR assay findings were confirmed by species-specific PCR and sequencing. Analysis of different sequences generated using 18S rRNA PCR revealed that the amplicon size of Babesia spp. is nearly 400 bp (393–408 bp) whereas Theileria spp. were more than 400 bp (418–424 bp). The percentage of sequence divergence among Babesia and Theileria spp. was 7.3–12.2% and 0.7–12.2%, respectively. In-silico restriction enzyme (RE) analysis reveals the presence of at least one site for a commercially available RE in 18S rRNA fragments of every parasite, which can differentiate it from its congeners.
Conclusions
The presented universal oligonucleotide-based PCR assay provides a highly sensitive, specific, cost-effective, and rapid diagnostic tool for the initial screening of piroplasmids infecting domestic and wild animals and is potentially helpful for large-scale epidemiological studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In general, piroplasmosis is a disease condition in vertebrate hosts caused by the parasites belonging to the class Piroplasmea and order Piroplasmorida and is transmitted by ticks [1,2,3,4]. Piroplasms are pear-shaped intraerythrocytic stages. They primarily consist of hemoprotozoan parasites belonging to four related genera: Babesia, Theileria, Cytauxzoon, and Rangelia infecting both wild and domestic animals [1]. A variety of parasite species belonging to these genera are common and widespread blood parasites worldwide, with significant economic, medical, and veterinary consequences [1, 2, 5]. Different piroplasmids like B. bigemina, B. gibsoni, B. canis vogeli, T. equi, T. annulata, T. orientalis, T. lestoquardi, T. ovis and in some extent B. caballi, B. bovis, and T. luwenshuni have been reported among domesticated animals from India [6,7,8,9,10,11,12,13,14]. Although some are benign or low pathogenic, others can cause a wide range of symptoms and even death. A range of prevalence of the piroplasmids in different animals was recorded worldwide [reviewed by 4, 15–21].
Fever, anaemia, malaise, lethargy, and anorexia are common symptoms of piroplasmid infections in animals [1, 2, 5, 8, 22]. However, the pathogenesis and accordingly clinical signs and symptoms vary with parasite species and the animal-associated factors such as species, breed, age, immunological status, concurrent infections with other pathogens, and/or genetic factors. In most cases, clinical signs and symptoms caused by piroplasmid infections are confused with other protozoal, bacterial, or viral infections that can be identified through laboratory testing. On the diagnostic front, microscopic examination of blood smear has been widely used to detect the piroplasm stage of parasites in erythrocytes. However, they have serious limitations like very low sensitivity, the requirement of skilled person, inability to detect the subclinical and carrier animals, and inability to identify the species [16, 23]. Other modern diagnostic techniques like serodiagnostics utilize to detect parasite-specific antibodies have also suffered from some limitations like cross reactivity, inability to discriminate active from past-infection, and cannot be used as a test of cure [24]. However, the antigen-based serodiagnostics can overcome certain limitations but have challenging to identify the suitable antigens and compromised with sensitivity.
Alternatively, with the advancement of molecular biology, PCR-based diagnostics now give researchers and diagnosticians the ability to detect and identify numerous parasites down to the subspecies or strain level in clinical samples and their natural carrier [23]. The method can overcome the significant limitations of both microscopy and serology and has proven to be the most sensitive and specific method for detecting agents and new strains and being an essential tool for evaluating therapeutic efficacy [25, 26].
Various regions of DNA were targeted for molecular identification of Theileria and Babesia parasites, either at the species or genus level that includes 18S rRNA, internal transcribed spacer-1 (ITS-1), Apical membrane antigen-1 (AMA-1), equi merozoite antigen-1 (EMA1), equi merozoite antigen-2 (EMA2), 16S rRNA, major piroplasm surface protein (MPSP), Heat shock protein70 (HSP70), T. annulata merozoite surface antigen-1 (Tams-1), cytochrome b, rhoptry-associated protein-1c (rap-1c), thrombospondin-related anonymous protein (TRAP), spherical body protein-4 (SBP-4) [5, 27,28,29,30,31], etc. 18S rRNA is the most common target for PCR-based detection and identification in eukaryotic species. Piroplasmids have been identified at the genus and species levels using different regions of the 18S rRNA gene [27, 32,33,34,35].
At least 111 valid Babesia and 39 valid Theileria species had been described with morphological and biological characteristics. However, many more are still waiting to get species status after introducing molecular biology [5]. Because there are so many different species of Theileria and Babesia, identifying individual parasites at the species level using a single PCR is difficult, especially during epidemiological investigations. Recently, few multiplex PCR assays for the simultaneous and rapid detection of multiple pathogens have been achieved. However, this technique also has its inherent problems like challenges of designing effective primers to avoid the cross-primer dimer formation or false priming amplification, limitations in the size of target amplification (short-length amplification is more effective), and variable amplification efficiency to different targets/templates [36]. Even if these constraints are solved, PCR multiplexing can only be done on a limited number of organisms. So, instead of going straight to species identification, screening the samples for piroplasmids first will be more cost-effective and efficient.
Moreover, the same amplicons can be used for species identification through restriction enzyme (RE) and sequencing analysis. If not, species-specific PCR can be performed solely on piroplasmid-positive samples. Accordingly, the present investigation was designed to optimize and validate the short-length 18S rRNA PCR for rapid screening of animals suspected of piroplasmid infections.
Materials and Methods
Ethical Statement
Approval and necessary guidelines of Institute Animal Ethics Committee (IAEC), College of Veterinary Science and Animal Husbandry, Junagadh, Gujarat, India were obtained and followed during this study (F. No. 25/15/2018-CPCSEA dated 14/09/2018). All the field samples were collected with prior consent of the owners.
Sample Collection and Isolation of Whole Genomic DNA
The blood samples collected from different animals species presented at Veterinary Clinical Complex (VCC), Veterinary College, Junagadh were used in the present investigation. About 2–3 ml jugular vein blood was collected from each animal in a vial containing EDTA, and various animal parameters like age, sex, breed, and species were recorded. During 2015–2020, 1250 samples were collected from cattle, buffaloes, horses, dogs, sheep, goats, and wild felids. Randomly, 468 samples were chosen for microscopic examination (Supplementary Table 1). The samples found positive in microscopy were used as the positive control. The samples collected from apparently healthy cow calves maintained in the institute farm were used as the negative control.
The whole blood genomic DNA was isolated from 200 µl of blood sample using GeneJET Whole Blood Genomic DNA Purification Kit (Thermo Scientific, Lithuania) following the manufacturer's protocol. Finally, DNA was eluted in 200 µl of elution buffer and stored at − 20 °C till further use.
Microscopic Examination
Thin blood smear was prepared on the microscopy glass slide (Borosil, India), dried, and fixed by methanol (Merck, India) for 2–3 min. Subsequently, the smear was stained 20 times diluted Giemsa’s stain (Himedia, India) in distilled water for 30 min. The slide was washed over tap water, dried, and observed under 100 × oil emersion objectives of a compound light microscope (Labomed, USA).
Piroplasmid 18S rRNA PCR
The oligonucleotide primers used in the present investigation were previously used to identify subtropical case of human babesisois [32] and subsequently used to detect babesiosis in dogs [33]. The forward and reverse primer sequences: BaF-5ʹ AAT ACC CAA TCC TGA CAC AGG G 3ʹ and BaR- 5ʹ TTA AAT ACG AAT GCC CCC AAC 3ʹ, respectively, were analysed through Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome) which can amplify about 400 bp fragments of 18S rRNA of piroplasmids from various animals and custom synthesized (Eurofins Genomics India Pvt. Ltd., Bengaluru). Each PCR reaction of 25 µl was set in 200 µl tubes with 12.5 µl 2 × DreamTaq Green PCR master mix (Thermo Scientific, Lithuania), 1 µl each of forward and reverse primers (10 mM), 4 µl (15.8 to 28.5 ng/µl) genomic DNA, and 6.5 µl of nuclease-free water (NFW). The reaction mixture was loaded in a Thermal Cycler (Applied Bio Systems, USA) optimized for amplification of the targeted DNA sequence and the cycling protocol was programmed as: initial denaturation at 95 ºC for 3 min followed by 35 cycle of denaturation (96 ºC for 15 s), annealing (60 ºC for 20 s), and extension (72 ºC for 25 s). The final extension was kept at 72 ºC for 1 min at the end of the PCR cycle. Finally, to resolve the amplified products, 10 µl of the PCR product along with DNA ladder were electrophoresed in 1.5% agarose gel containing 0.5 µg/ml of Ethidium bromide (EtBr) in 1 × Tris–Acetate–EDTA (TAE) buffer at 120 V for 20 min. Subsequently, amplicons were visualized and documented in the Gel-documentation system (Syngene, UK).
Identification of Parasite Species Using Species-Specific PCR and Sequencing
Randomly, 5–6 piroplasmid-positive samples (confirmed by 18S rRNA PCR) from each animal species such as cattle, buffaloes, horses, dogs, and sheep were chosen for species-specific PCR. In-house standardised species-specific PCR was used to identify the common piroplasmids found in this region (Table 1). Three of the seven pairs of primers were previously published, while the remaining four were designed specifically for this study using protein coding genes like rhoptry-associated Protein 1c (rap-1c) gene for Babesia bigemina (Accession: AY146987), Major Piroplasm Surface Protein (MPSP) gene for Theileria orientalis/buffali (Accession: D11047), erythrocyte/equine merozoite antigen-1 (EMA1) for Theileria equi (Accession: MF447154) and Cytochrome b gene in Theileria lestoquardi (Accession: MN481239). The alignment of sequences for primers design are presented in Supplementary Figs. 1–4. Standard 25 µl PCR reaction was set up in 200 µl tubes using 12.5 µl of 2 × DreamTaq Green PCR master mix (Thermo Scientific, Lithuania), 1 µl each of forward and reverse primers (10 mM), 4 µl (15.8 to 28.5 ng/µl) genomic DNA, and 6.5 µl of nuclease-free water (NFW). Non-template control (NTC) was used as the negative control. The amplification was done in Gradient Thermal Cycler (Applied Biosystems, USA) with the cycling condition as initial denaturation at 95 ºC for 3 min followed by 35 cycle of denaturation (96 ºC for 15 s), annealing (X ºC for 30 s) and extension (72 ºC for 40 s). The final extension was kept at 72 ºC for 5 min at the end of the PCR cycle. Finally, to resolve the amplified products, 10 µl of the PCR product and DNA ladder were electrophoresed in 1.2% agarose gel containing 0.5 µg/ml of EtBr in 1 × TAE buffer at 120 V for 20 min. The amplicons were visualized and documented in the Gel-documentation system (Syngene, UK).
The amplified product was purified from 50 µl PCR reaction using GeneJET Gel Extraction Kit (Thermo Scientific, Lithuania), and 10 µl was submitted along with primers used for amplification of targeted gene to the commercial house (Eurofins Genomics India Pvt. Ltd., Bengaluru) for bi-directional Sanger sequencing. Upon receiving of sequences, the quality of sequences was checked in BioEdit programme, both forward and reverse sequences were aligned, and a correct consensus sequence was obtained. Subsequently, the sequence analysis and similarity searches were performed with the basic local alignment search tool available in GenBank data in National Centre for Biotechnology Information, USA, and species were confirmed. The sequences generated from different species of parasites were submitted to GenBank (NCBI, USA), and accession numbers were obtained.
Sensitivity and Specificity of Piroplasmid 18S rRNA PCR
The analytical sensitivity of 18S rRNA PCR was done on whole blood genomic DNA positive for T. annulata. The concentration of DNA was measured by Qubit® dsDNA BR assay kit in Qubit™ 4 Fluorometer (Thermo Scientific, Singapore) as per the manufacturer’s instructions. The sample with 10 ng/µl genomic DNA was serially double-fold diluted in NFW, and 2 µl was used as a template DNA from each dilution in an optimized PCR to amplify 18S rRNA DNA fragments.
The analytical specificity of the 18S rRNA PCR was determined by including the total genomic DNA in the reactions extracted from whole blood of different animals infected with other common haemoparasites viz., Trypanosoma evansi, Hepatozoon spp., Anaplasma spp., and Ehrlichia canis maintained in the laboratory.
Piroplasmid 18S rRNA Sequence Analysis
Randomly, 18S rRNA PCR positive samples from each species of animals, whether confirmed through species-specific PCR or not, were used to amplify the targeted DNA using BaF and BaR primers in a 50 µl reaction at optimized PCR conditions. The amplified product was purified using GeneJET Gel Extraction Kit (Thermo Scientific, Lithuania), and 10 µl was submitted along with primers used for amplification of targeted gene to the commercial house (Eurofins Genomics India Pvt. Ltd., Bengaluru) for bi-directional Sanger sequencing.
Upon receiving the sequences, the quality of sequences was checked in the BioEdit programme. Both forward and reverse sequences were aligned to achieve the right consensus sequence. Subsequently, sequences were analysed on the BLASTn programme (NCBI, USA) and confirmed their identity. The sequences generated from different species of parasites were submitted to GenBank (NCBI, USA), and accessions were obtained. Further, to see the similarity among the newly generated and other published sequence, they were aligned using the ClustalW programme (Lasergene, DNA STAR, USA). In phylogenetic analysis, the GenBank sequence Accessions L24381 (Toxoplasma gondii) and XR_003001211 (Plasmodium vivax) were used as outgroup species to root the tree for the 18S rRNA gene of piroplasm.
In-Silico Restriction Enzyme Analyses of 18S rRNA DNA Fragments of Piroplasms
The newly generated 18S rRNA piroplasm sequences of bovines, horses, dogs, sheep, and goats (MZ573175, MZ573172, MZ573176, MZ573174, MZ573177, MZ573173, MZ573171, MZ701980, MZ573178) as well as available sequences in GenBank (MN629354, MH257729) were used to identify the unique restriction enzymes (RE) sites (GenScript Restriction Enzyme Map Analysis Tools, https://www.genscript.com/tools/restriction-enzyme-map-analysis). The group of parasites affecting an animal species was further analyzed to identify the unique RE enzymes for easy identification and differentiation.
Results
Piroplasmid 18S rRNA PCR and Microscopy
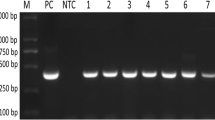
Primer_BLAST analysis revealed reactivity of published primers (BaF/BaR) to wider range of piroplasmids. However, primers showed matching with the sequences of few species of Cryptosporidium and Sarcocystis, and Besnoitia besnoiti under the apicomplexan group of mammals. Alignment of 18S rRNA sequences of piroplasmid with other apicomplexan parasites revealed that the sequences of reverse primer region is conserved but forward primer region of other apicomplexan parasites varied by only a few nucleotides. (Supplementary Fig. 5). The PCR assay successfully amplified the expected size of targeted DNA from blood samples found positive for various piroplasmids collected from different animals (Fig. 1) that was confirmed by sequencing and BLASTn analysis (NCBI, USA). All microscopically piroplasmid-positive samples also had positive 18S rRNA PCR results. The amplicon size of Babesia spp. was somewhat smaller than Theileria spp., especially B. bigemina, compared to T. annulata. Among the 1250 samples collected, 63.3% of animals were detected positive for piroplasmids infections through 18S rRNA PCR. In contrast, 23.3% of animals were found positive by microscopy in 468 samples examined (Supplementary Table 1). The time required for PCR reaction was merely 58 min.

Amplification of targeted nucleotide sequences through 18S rRNA PCR assay. 1 and 6: B. bigemina, 2: T. lestoquardi, 3: T. luwenshuni, 4: T. annulata, 5: T. orientalis, 7: B. canis vogeli, 8: B. gibsoni, 9: T. equi, 10: T. ovis, 11: Babesia sp. Leopard and 12: NTC. L-100 bp plus DNA ladder (Thermo Scientific, Lithuania) band size (from lower to upper): 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1200, 1500, 2000, 3000 bp
Species Identification Through Species-Specific PCR
The three steps confirmation of species have been done first, by the use of species-specific PCR primers; second, by sequencing and NCBI–nBLAST analysis and third, the primers were reacted on microscopically and piroplasmid 18S rRNA PCR positive samples. Species-specific PCR assays successfully detected the common parasites available in this region like B. bigemina, T. annulata, T. buffali/orientalis, T. equi, B. canis vogeli, B. gibsoni, and T. lestoquardi (Supplementary Fig. 6). To further confirm the species of parasites, sequences were generated, submitted to GenBank, and accession numbers were obtained (MZ665956, MZ665960, MZ720762, MZ665961, MZ646048, MZ665957, MZ665959).
Sensitivity and Specificity of Piroplasmid 18S rRNA PCR
The PCR was able to amplify the 18S rRNA gene fragment of T. annulata from as low as 39 picograms (pg) of whole blood genomic DNA microscopically positive for the same parasites. On the other hand, no amplification of such gene fragments was recorded from common but unrelated haemoparasites viz., Trypanosoma evansi, Hepatozoon spp., Anaplasma spp., and Ehrlichia canis positive samples (Fig. 2).

Assessment of analytical sensitivity (A) and specificity (B) of 18S rRNA PCR assay. A Amplification of targeted gene from double-fold serially diluted T. annulata positive sample DNA (1st to 11th dilution; Lane 1st—20 ng, 2nd—10 ng, 3rd—5 ng, 4th—2.5 ng, 5th—1.25 ng, 6th—625 pg, 7th—321.5 pg, 8th—156.25 pg, 9th—78.13 pg, 10th—39.06 pg, 11th—19.5 pg DNA, 12th—NTC). B 18S rRNA PCR on sample positive for 1. Trypanosoma evansi, 2. Hepatozoon canis, 3. Anaplasma marginale, 4. Ehrlichia canis, 5. T. annulata, and 6. haemoparasite negative control DNA. L-100 bp plus DNA ladder (Thermo Scientific, Lithuania) band size (from lower to upper): 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1200, 1500, 2000, 3000 bp
Sequence Analysis of 18S rRNA Gene Fragments of Piroplasmids
The sequence analysis revealed two different size ranges of 18S rRNA gene fragments. The Babesia spp. sequences were nearly 400 bp (393–408 bp), whereas; Theileria spp. were more than 400 bp (418–424 bp). BLASTn analysis revealed more than 99% identity with distinct parasite species of Theileria and Babesia. The resultant consensus sequences were submitted to GenBank (National Center for Biotechnology Information, USA), and accessions were acquired as B. bigemina (MZ573175), T. annulata (MZ573172), T. buffali/orientalis (MZ573176), T. equi (MZ573174), B. canis vogeli (MZ573177), B. gibsoni (MZ573173), T. luwenshuni (MZ701916). After blast analysis, the sequences obtained from 18S rRNA gene fragments and species-specific PCR amplicons indicated the same parasite species. The sequence divergence among the different species of Babesia and Theileria were recorded as 7.3–12.2% and 0.7–12.2%, respectively (Fig. 3A). Sequence alignment report revealed that the forward and reverse primer regions of the 18S rRNA gene of various piroplasmids are highly conserved. In phylogenetic analysis, sequences of all members of Theileriade and Babesidiae formed two distinct clads. T. annulata and T. lestoquardi form a close group. Similarly, T. orientalis and T. luwenshuni are very close to each other. T. ovis is very much closer to T. annulata compared to T. orientalis. Similarly, in the Babesidae group, B. gibsoni is closer to B. canis vogeli than B. bigemina but farther away from Babesia sp. Leopard. Toxoplasma gondii, an apicomplexan parasite with a unique lineage, forms a separate clad along with Theileriade group while Plasmodium vivax showed almost equal distance to both Theileriade and Babesidae groups and form a separate clad (Fig. 3B).

Analysis of nucleotides sequences of 18S rRNA gene fragments of various piroplasms of animals generated in the present study. A sequence identity/divergence analysis and B sequence phylogenetic analysis using MEGA X software (statistical Method: Maximum-likelihood; model: Tamura 3-parameter model + Gamma distribution (+ G); bootstrap value:500; Neighbor-Join and BioNJ algorithms)
In-Silico RE Analysis of 18S rRNA Gene Fragments of Piroplasmids
Based on in-silico analysis of 18S rRNA gene fragments of various piroplasmid, few unique RE sites were identified for an individual parasite of an animal species. None of the RE sites for T. luwenshuni of sheep and goat was recorded those were available commercially, which can differentiate it from T. ovis and/or T. lestoquardi. Otherwise, each parasite has at least one commercially available unique RE that distinguishes it from its congeners in the same animal species (Table 2).
Discussion
Babesia and Theileria spp. are apicomplexan parasites transmitted by ticks. Traditional morphological and contemporary molecular studies substantiated these genera as close relatives and, correspondingly, constitute the order Piroplasmida [39]. Piroplasmid infections are detrimental to the health of various wild and domestic animals. They are a major cause of economic losses in the animal husbandry industry in tropical and subtropical countries worldwide [40].
Animals suffering from acute babesiosis or theileriosis can have various symptoms such as fever, haemolytic anaemia, haemoglobinuria, oculo-nasal discharge, anaemia, malaise, lethargy, increased heart rate, increased respiratory rate, and even death in severe case. Although these symptoms are very typical, they are not pathognomonic, and animals with chronic infections can be asymptomatic carriers [22, 41]. Carrier animals with no clinical symptoms are thought to be a key reservoir of infection for ticks that can spread the infection to other animals.
The laboratory diagnosis of piroplasmosis was based on the microscopic examination of Giemsa-stained blood smears to detect piroplasmid inclusions in erythrocytes. However, species identification by microscopy is challenging because different parasites have similar morphologies, making identification even more difficult if mixed infections occur. Furthermore, identifying parasites in carrier animals with low parasite counts and in acute instances at the onset of the disease might be difficult. Additionally, it needs special diagnostic knowledge [16]. Some serological assays, such as the complement fixation test (CFT) and the indirect fluorescent antibody test (IFAT), can help to detect prior infections, however, there have been reports of cross-reactions between species [42, 43].
Furthermore, these tests can produce erroneous positive and negative results. Molecular techniques, such as polymerase chain reaction (PCR) and its variants, have shown to be the most sensitive and specific tools for diagnosis and study in recent decades and have been widely employed for detection and discrimination between Theileria and Babesia species [44,45,46,47,48]. Although uniplex PCR assays are designed to detect single species at a time, they can be time-consuming and expensive when applied to many samples co-infected with more than one pathogen species. On the contrary, multiplex PCR (mPCR) shows unparalleled advantages, including detecting multiple pathogens in a single reaction and saving time [49]. However, this technique also has inherent problems like designing effective primer, limitation in the target size amplification, and variable amplification efficiency [36]. Reverse line blotting method had also been used for the simultaneous detection and differentiation of Babesia and Theileria spp. infecting ruminants [45, 50, 51]. However, Reverse line blot (RLB) assay requires expertise, specialized equipment, and the protocol is very labor-intensive and due to its high cost, this technique is not feasible [52]. None of these approaches can be deemed superior to the others. There is a need to develop efficient diagnostic strategies to deal with this problem in resource-poor countries and developed countries facing piroplasmosis as an extensive burden. So, screening the samples for piroplasmids first will be more cost-effective and efficient than proceeding straight to species identification. The present study focused on standardisation and validation of a very convenient and universal PCR assay that can detect piroplasms infective to domestic and wild animals, which can be used for diagnosis, quarantine, and epidemiological survey. Accordingly, the current study was planned to optimize and validate the short-length 18S rRNA PCR for rapid screening of animals suspected of piroplasmid infections. The eukaryotic 18S rRNA gene has both conserved and variable regions. Due to its high specificity and sequence conservation, it has been used as a universal biomarker to screen closely related species and biodiversity studies [7, 53,54,55]. Universal oligonucleotide primers based on 18S rRNA has also been used for initial screening of piroplasmids infecting horses and Bactrian camels north eastern Mongolia [56].
Most of the earlier studies on 18S rRNA gene screening commonly prevalent Babesia/Theileria spp. were limited to their related host species [32]. The novelty of the current investigation is to standardize and optimize 18S rRNA PCR for the initial screening of common piroplasmids in different animals like cattle, buffaloes, sheep, goats, dogs, horses, and leopards.
Using 18S rRNA PCR, 63.3 percent of the 1250 tested samples were detected positive for piroplasmid infections. However, in 468 samples examined randomly, 23.3 percent of animals were positive by microscopy and more than 33% samples were found false negative in microscopy in comparison to 18S rRNA PCR. That indicate the higher sensitivity of molecular diagnostics like PCR in comparison to microscopy which is very common [57, 58]. The amplicon size of Babesia spp. was somewhat smaller than Theileria spp., especially B. bigemina, compared to T. annulata. Piroplasmid positive samples from each animal species such as cattle, buffaloes, horses, dogs, and sheep were chosen at random for species-specific PCR. Species-specific PCR assays successfully detected the common parasites available in this region like B. bigemina, T. annulata, T. buffali/orientalis, T. equi, B. canis vogeli, B. gibsoni, and T. lestoquardi further confirmed through sequence analysis.
Sequence analysis of 18S rRNA gene fragments revealed two different size ranges of amplicons. The Babesia spp. sequences were nearly 400 bp (393–408 bp), whereas Theileria spp. were more than 400 bp (418–424 bp). After blast analysis, the sequences obtained from 18S rRNA gene fragments and species-specific PCR amplicons revealed the same parasite species. Phylogenetic analysis and sequence distance report further confirm the current findings. Similar observations based on molecular phylogenetic tree of 18S rRNA gene sequences were reported by previous researchers among different Theileria and Babesia spp. infecting domestic animals [55]. These studies further reveal that neither Theileria nor Babesia are monophyletic taxonomic units and systematic re-examination is required to determine the generic diversity of the piroplasmids [54, 59,60,61].
Uniplex PCR targeting the 18S rRNA genetic markers has been shown to have good analytical sensitivity and specificity compared to other taxonomic markers, for identifying several piroplasms species [62]. The PCR assay standardised in the present study was able to amplify the 18S rRNA gene fragment of T. annulata from as low as 39 pico grams (pg) of whole blood genomic DNA microscopically positive for the same parasites. On the other hand, no amplification of such gene fragments was recorded from common but unrelated haemoparasites. However, apart from piroplasmids, primer-BLAST research revealed plausible primer reactivity to a few species of Cryptosporidium, Sarcocystis, and Besnoitia besnoiti in mammals. Yet, the likelihood of these parasites in circulation is remote. These results support the reliability of 18S rRNA-specific oligonucleotides used in the present study as universal primers for screening piroplasmids infecting livestock. The current findings are consistent with the previously published report [7, 56]. Sequence alignment report revealed that both the forward and reverse primers region of the 18S rRNA gene of various piroplasmids are highly conserved.
Moreover, BLASTn analysis, further demonstrated that the sequences obtained from 18S rRNA gene fragments and species-specific PCR amplicons indicated the same piroplasmid. In-silico RE analysis of 18S rRNA gene fragments revealed that each parasite has at least one commercially available unique RE that distinguishes it from its congeners in the same animal species. These findings support the hypothesis that 18S rRNA-based PCR assay is a highly specific, sensitive, cost-effective, and rapid diagnostic tool for initial screening of piroplasmids infecting domestic and wild animals and can be helpful for large-scale epidemiological studies.
Conclusion
This study standardized and optimized an 18S rRNA PCR assay to detect common piroplasmids of different animals like cattle, buffaloes, sheep, goats, dogs, horses, and leopards. The presented universal oligonucleotide-based PCR assay provides a highly sensitive, cost-effective, and rapid diagnostic tool for the initial screening of piroplasmids infecting domestic and wild animals and is potentially helpful for large-scale epidemiological studies.
References
Alvarado-Rybak M, Solano-Gallego L, Millán J (2016) A review of piroplasmid infections in wild carnivores worldwide: importance for domestic animal health and wildlife conservation. Parasites Vectors 9:538. https://doi.org/10.1186/s13071-016-1808-7
Homer MJ, Aguilar-delfin I, Telford S III, Krause PJ, Persing DH (2000) Babesiosis. Clin Microbiol Rev 13:451–469
Jefferies R, Ryan UM, Irwin PJ (2007) PCR–RFLP for the detection and differentiation of the canine piroplasm species and its use with filter paper-based technologies. Vet Parasitol 144:20–27
Inácio EL, Pérez-Macchi S, Alabi A, Bittencour P, Müller A (2019) Prevalence and molecular characterization of piroplasmids in domestic dogs from Paraguay. Ticks Tick-borne Dis 10(2):321–327
Yabsley MJ, Shock BC (2012) Natural history of Zoonotic Babesia: role of wildlife reservoirs. Int J Parasitol Parasites Wildl 2:18–31. https://doi.org/10.1016/j.ijppaw.2012.11.003
Singh A, Singh H, Singh NK, Singh ND, Rath SS (2014) Canine babesiosis in northwestern India: molecular detection and assessment of risk factors. BioMed Res Int 2014:741785. https://doi.org/10.1155/2014/741785
George N, Bhandari V, Reddy DP, Sharma P (2015) Molecular and phylogenetic analysis revealed new genotypes of Theileria annulata parasites from India. Parasit Vectors 8:468. https://doi.org/10.1186/s13071-015-1075-z
Maharana BR, Tewari AK, Saravanan BC, Sudhakar NR (2016) Important hemoprotozoan diseases of livestock: challenges in current diagnostics and therapeutics: an update. Vet World 9(5):487–495. https://doi.org/10.14202/vetworld.2016.487-495
Sumbria D, Singla LD, Sharma A (2016) Theileria equi and Babesia caballi infection in equines in Punjab: a study on serological and molecular prevalence. Trop Anim Health Pro 48:45–52
Diallo T, Singla LD, Sumbria D, Kaur P, Bal MS (2018) Conventional and molecular diagnosis of haemo-protozoan infectionsin cattle and equids from Republic of Guinea and India. Indian J Anim Res 52:1206–1211
Zheng W, Liu M, Moumouni PF, Liu X, Efstratiou A, Liu Z, Liu Y, Tao H, Guo H, Wang G, Gao Y, Li Z, Ringo AE, Jirapattharasate C, Chen H, Xuan X (2017) First molecular detection of tick-borne pathogens in dogs from Jiangxi, China. J Vet Med Sci 79(2):248–254. https://doi.org/10.1292/jvms.16-0484
Nagaraj HV, Lakshmanan B, Lonappan GV, Shameem H, Jose J, Sabu L (2019) Molecular identification of caprine carriers of theileriosis in South India. Vet Arh 89(3):367–378. https://doi.org/10.24099/vet.arhiv.0227
Ganguly A, Maharana BR, Ganguly I (2020) Pentaplex PCR assay for rapid differential detection of Babesia bigemina, Theileria annulata, Anaplasma marginale and Trypanosoma evansi in cattle. Biologicals 63:81–88. https://doi.org/10.1016/j.biologicals.2019.10.011
Devi G, Ajith Y, Mal G, Dimri U, Preena P, Jairath G, Kattoor JJ, Jacob SS, Singh B, Dhar JB (2021) Migratory Gaddi sheep and goats as potential carriers of Theileria infection: a molecular survey. Trop Anim Health Prod 53(2):302. https://doi.org/10.1007/s11250-021-02742-y
Bock R, Jackson L, de Vos A, Jorgensen W (2004) Babesiosis of cattle. Parasitol 129(Suppl):S247–S269. https://doi.org/10.1017/s0031182004005190
Solano-Gallego L, Sainz Á, Roura X, Estrada-Peña A, Miró G (2016) A review of canine babesiosis: the European perspective. Parasit Vectors 9(1):336. https://doi.org/10.1186/s13071-016-1596-0
Alvarado-Rybak M, Solano-Gallego L, Millán J (2016) A review of piroplasmid infections in wild carnivores worldwide: importance for domestic animal health and wildlife conservation. Parasit Vectors 9(1):538. https://doi.org/10.1186/s13071-016-1808-7
Vishwakarma P, Nandini M (2020) Overview of canine babesiosis. In: Bekoe SO, Saravanan M, Adosraku RK, Ramkumar PK (eds) Veterinary medicine and pharmaceuticals. IntechOpen, London. https://doi.org/10.5772/intechopen.82243
Onyiche TE, Suganuma K, Igarashi I, Yokoyama N, Xuan X, Thekisoe O (2019) A review on equine piroplasmosis: epidemiology, vector ecology, risk factors, host immunity, diagnosis and control. Int J Environ Res Public Health 16:1736. https://doi.org/10.3390/ijerph16101736
Krishnamoorthy P, Akshata LG, Jaco SS, Suresh KP, Roy P (2021) Theileriosis prevalence status in cattle and buffaloes in India established by systematic review and meta-analysis. Indian J Anim Sci 91(4):269–279
Chen Y, Chen YY, Liu G, Lyu C, Hu Y, An Q, Qiu HY, Zhao Q, Wang CR (2022) Prevalence of Theileria in cattle in China: a systematic review and meta-analysis. Microb Pathog 162:105369
Köster LS, Lobetti RG, Kelly P (2015) Canine babesiosis: a perspective on clinical complications, biomarkers, and treatment. Vet Med (Auckl) 6:119–128. https://doi.org/10.2147/VMRR.S60431
Momčilović S, Cantacessi C, Arsić-Arsenijević V, Otranto T-O (2019) Rapid diagnosis of parasitic diseases: current scenario and future needs. Clin Microbiol Infect 25(3):290–309
Aboge GO, Jia H, Terkawi MA, Goo Y, Kuriki K, Nishikawa Y, Igarashi I, Suzuki H, Xuan X (2007) A novel 57-kDa merozoite protein of Babesia gibsoni is a prospective antigen for diagnosis and serosurvey of canine babesiosis by enzyme-linked immunosorbent assay. Vet Parasitol 149:85–94. https://doi.org/10.1016/j.vetpar.2007.06.025
Nakaghi ACH, Machado RZ, Costa MT, André MR, Baldani CD (2008) Canine ehrlichiosis: clinical, hematological, serological and molecular aspects. Cienc Rura 38:766–770
Yang S, Rothman RE (2004) PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis 4(6):337–348. https://doi.org/10.1016/S1473-3099(04)01044-8
d’Oliveira C, van der Weide M, Habela MA, Jacquiet P, Jongejan F (1995) Detection of Theileria annulata in blood samples of carrier cattle by PCR. J Clin Microbiol 33(10):2665–2669
Ibrahim HM, Adjou Moumoun PF, Mohammed-Geba K, Sheir SK, Hashem IS, Cao S, Terkawi MA, Kamyingkird K, Nishikawa Y, Suzuki H, Xua X (2013) Molecular and serological prevalence of Babesia bigemina and Babesia bovis in cattle and water buffalos under small-scale dairy farming in Beheira and Faiyum Provinces, Egypt. Vet Parasitol 198(1–2):187–192
Sivakumar T, Tattiyapong M, Fukushi S, Hayashida K, Kothalawala H, Silva S, Vimalakumar SC, Kanagaratnam R, Meewewa AS, Suthaharan K, Puvirajan T, de Silva WK, Igarashi I, Yokoyama N (2014) Genetic characterization of Babesia and Theileria parasites in water buffaloes in Sri Lanka. Vet Parasitol 200(1–2):24–30
Ricciardi A, Ndao M (2015) Diagnosis of parasitic infections: what’s going on? J Biomol Screen 20(1):6–21
He L, Miao X, Hu J, Huang Y, He P, He J, Yu MN, Shi L, Zhao J (2017) First molecular detection of Babesia gibsoni in dogs from Wuhan, China. Front Microbiol 8:1577
Olmeda AS, Armstrong PM, Rosenthal BM, Valladares B, del Castillo A, de Armas F, Miguelez M, González A, Rodríguez Rodríguez JA, Spielman A, Telford SR 3rd (1997) A subtropical case of human babesiosis. Acta Trop 67(3):229–234. https://doi.org/10.1016/s0001-706x(97)00045-4
Cardoso L, Costa A, Tuna J, Vieira L, Eyal O, Yisaschar-Mekuzas Y, Baneth G (2008) Babesia canis canis and Babesia canis vogeli infections in dogs from northern Portugal. Vet Parasitol 156(3–4):199–204
Mohamed SB, Alagib A, AbdElkareim TB, Hassan MM, Johnson W, Hussein HE, Taus N, Ueti MW (2018) Molecular detection and characterization of Theileria spp. infecting cattle in Sennar state, Sudan. Parasitol Res 117(4):1271–1276. https://doi.org/10.1007/s00436-018-5775-0
Gargano V, Blanda V, Gambino D, La Russa F, Di Cataldo S, Gentile A, Schirò G, Torina A, Millán J, Vicari D (2021) Serological survey and molecular characterization of Theileria annulata in Sicilian cattle. Pathogens 10(2):101. https://doi.org/10.3390/pathogens10020101
Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE (2000) Multiplex PCR: optimization and application in diagnostic virology. Clin Microbiol Rev 13(4):559–570. https://doi.org/10.1128/CMR.13.4.559
Bilgic HB, Karagenç T, Shiels B, Tait A, Eren H, Weir W (2010) Evaluation of cytochrome b as a sensitive target for PCR based detection of T. annulata carrier animals. Vet Parasitol 174(3–4):341–347. https://doi.org/10.1016/j.vetpar.2010.08.025
Kundu K, Kumar S, Maurya PS, Mandal M, Ram H, Garg R, Pawde AM, Raina OK, Banerjee PS (2012) PCR based identification of Babesia canis vogeli in clinically affected dogs. J Vet Parasitol 26(2):167–169
Allsopp MT, Cavalier-Smith T, De Waal DT, Allsopp BA (1994) Phylogeny and evolution of the piroplasms. Parasitol 108(Pt2):147–152. https://doi.org/10.1017/s0031182000068232
Jongejan F, Uilenberg G (2004) The global importance of ticks. Parasitol 129(Suppl):S3–S14. https://doi.org/10.1017/s0031182004005967
Gomes J, Soares R, Santos M, Santos-Gomes G, Botelho A, Amaro A, Inácio J (2013) Detection of Theileria and Babesia infections amongst asymptomatic cattle in Portugal. Ticks Tick Borne Dis 4(1–2):148–151. https://doi.org/10.1016/j.ttbdis.2012.07.002
Bruning A (1996) Equine piroplasmosis an update on diagnosis. Br Vet J 152:139–151
Papadopoulos B, Brossard M, Perie NM (1996) Piroplasms of domestic animals in the Macedonia region of Greece. 3 Piroplasms of small ruminants. Vet Parasitol 63:67–74
Nagore D, García-Sanmartín J, García-Pérez AL, Juste RA, Hurtado A (2004) Detection and identification of equine Theileria and Babesia species by reverse line blotting: epidemiological survey and phylogenetic analysis veterinary assistant. Vet Parasitol 123:41–54
Schnittger L, Yin H, Qi B, Gubbels MJ, Beyer D, Nieman S, Jongejan F, Ahmed JS (2004) Simultaneous detection and differentiation of Theileria and Babesia parasites infecting small ruminants by reverse line blotting. Parasitol Res 92(3):189–196
Aktaş M, Altay K, Dumanli N (2005) Development of a polymerase chain reaction method for diagnosis of Babesia ovis infection in sheep and goats. Vet Parasitol 133(4):277–281. https://doi.org/10.1016/j.vetpar.2005.05.057
Alhassan A, Pumidonming W, Okamura M, Hirita H, Battsetseg B, Fujisaki F, Yokoyama N, Igarashi I (2005) Development of a single-round and multiplex PCR method for the simultaneous detection of Babesia caballi and Babesia equi in horse blood. Vet Parasitol 129:43–49
Altay K, Dumanli N, Aktas M (2007) Molecular identification, genetic diversity and distribution of Theileria and Babesia species infecting small ruminants. Vet Parasitol 147:161–165
Edwards MC, Gibbs RA (1994) Multiplex PCR: advantages, development, and applications. PCR Methods Appl 3:S65-75
Gubbels JM, de Vos AP, van der Weide M, Viseras J, Schouls LM, de Vries E, Jongejan F (1999) Simultaneous detection of bovine Theileria and Babesia species by reverse line blot hybridization. J Cli Microbiol 37(6):1782–1789
Georges K, Loria GR, Riili S, Greco A, Caracappa S, Jongejan F, Sparagano O (2001) Detection of haemoparasites in cattle by reverse line blot hybridisation with a note on the distribution of ticks in Sicily. Vet Parasitol 99(4):273–286
Bilgiç HB, Karagenç T, Simuunza M, Shiels B, Tait A, Eren H, Weir W (2013) Development of a multiplex PCR assay for simultaneous detection of Theileria annulata, Babesia bovisand Anaplasma marginale in cattle. Exp Parasitol 133(2):222–229. https://doi.org/10.1016/j.exppara.2012.11.005
Van de Peer Y, Baldauf SL, Doolittle WF, Meyer A (2000) An updated and comprehensive rRNA phylogeny of (crown) eukaryotes based on rate-calibrated evolutionary distances. J Mol Evol 51:565–576
Lack JB, Reichard MV, Van Den Bussche RA (2012) Phylogeny and evolution of the Piroplasmida as inferred from 18S rRNA sequences. Int J Parasitol 42(4):353–363. https://doi.org/10.1016/j.ijpara.2012.02.005
Schnittger L, Ganzinelli S, Bhoora R, Omondi D, Nijhof AM, Florin-Christensen M (2022) The Piroplasmida Babesia, Cytauxzoon, and Theileria in farm and companion animals: species compilation, molecular phylogeny, and evolutionary insights. Parasitol Res 121(5):1207–1245. https://doi.org/10.1007/s00436-022-07424-8
Sloboda M, Jirků M, Lukešová D, Qablan M, Batsukh Z, Fiala I, Hořín P, Modrý D, Lukeš J (2011) A survey for piroplasmids in horses and Bactrian camels in North-Eastern Mongolia. Vet Parasitol 179(1–3):246–249. https://doi.org/10.1016/j.vetpar.2011.01.064
Almeria S, Castellà J, Ferrer D, Ortuño A, Estrada-Peña A, Gutiérrez JF (2001) Bovine piroplasms in Minorca (Balearic Islands, Spain): a comparison of PCR-based and light microscopy detection. Vet Parasitol 99(3):249–259. https://doi.org/10.1016/s0304-4017(01)00464-2
Kaur R, Yadav A, Rafiqi SI et al (2021) Epidemiology, haematology and molecular characterization of haemoprotozoon and rickettsial organisms causing infections in cattle of Jammu region, North India. BMC Vet Res 17:219. https://doi.org/10.1186/s12917-021-02915-9
Criado-Fornelio A, Martinez-Marcos A, Buling-Sarana A, Barba-Carretero JC (2003) Molecular studies on Babesia, Theileria and Hepatozoon in southern Europe part II. Phylogenetic analysis and evolutionary history. Vet Parasitol 114:173–194
Morrison DA (2009) Evolution of the Apicomplexa: where are we now? Trends Parasitol 25:375–382
Yam J, Gestier S, Bryant B, Campbell-Ward M, Bogema D, Jenkins C (2017) The identification of Theileria bicornis in captive rhinoceros in Australia. Int J Parasitol Parasites Wildl 7(1):85–89. https://doi.org/10.1016/j.ijppaw.2017.12.003
Brown C (2008) Tropical theileriosis. In: Brown C, Torres A (eds) Foreign animal diseases, 7th edn. Publications Boca, Florida USA, pp 401–404
Acknowledgements
Authors are heartily thankful to the Director of Research, Kamdhenu University, Gandhinagar and Director of Research, Junagadh Agricultural University, Junagadh, Gujarat for proving necessary facilities and funds for research work. Authors are also thankful to Principal and Dean, College of Veterinary Science and Animal Husbandry, Junagadh, Gujarat for providing necessary administrative support during research. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kumar, B., Maharana, B.R., Thakre, B. et al. 18S rRNA Gene-Based Piroplasmid PCR: An Assay for Rapid and Precise Molecular Screening of Theileria and Babesia Species in Animals. Acta Parasit. 67, 1697–1707 (2022). https://doi.org/10.1007/s11686-022-00625-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11686-022-00625-2