
Abstract
Background
Ephrin-A2, a member of the Eph receptor subgroup, is used in diagnosing and determining the prognosis of prostate cancer. However, the role of ephrin-A2 in prostate cancer is remains elusive.
Methods
We established stable clones overexpressing or silencing ephrin-A2 from prostate cancer cells. Then, CCK-8 was used in analyzing the proliferation ability of cells. CD31 staining was used in evaluating angiogenesis. Migration and invasion assay were conducted in vivo and in vitro. The expression of EMT-related markers was evaluated in prostate cancer cells through Western blotting.
Results
We revealed that the ectopic expression of ephrin-A2 in prostate cancer cells facilitated cell migration and invasion in vitro and promoted tumor metastasis and angiogenesis in vivo and that the silencing of ephrin-A2 completely reversed this effect. Although ephrin-A2 did not affect tumor cell proliferation in vitro, ephrin-A2 significantly promoted primary tumor growth in vivo. Furthermore, to determine the biological function of ephrin-A2, we assayed the expression of EMT-related markers in stable-established cell lines. Results showed that the overexpression of ephrin-A2 in prostate cancer cells down-regulated the expression of epithelial markers (ZO-1, E-cadherin, and claudin-1) and up-regulated the expression of mesenchymal markers (N-cadherin, β-catenin, vimentin, Slug, and Snail), but the knocking out of ephrin-A2 opposed the effects on the expression of EMT markers.
Conclusions
These findings indicate that ephrin-A2 promotes prostate cancer metastasis by enhancing angiogenesis and promoting EMT and may be a potentially therapeutic target in metastatic prostate cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Prostate cancer (PCa), one of the most common malignancies, poses threat to the health of men worldwide (Smith et al. 2014). High metastasis and high recurrence rate after castration treatment are the dominant clinical features of PCa. Metastasis has become a constraint to the long-term survival of patients with PCa and is the key to conquering it. Therefore, a major clinical challenge is preventing PCa progression and metastasis.
As the largest subgroup family of receptor tyrosine kinase, the Eph receptor family contains 14 members in mammals (Manning et al. 2002). Ephrin-A2, belonging to Eph ligands, are widely expressed in normal tissues and has a vital function in tumors (Hafner et al. (2004); Fox and Kandpal 2004; Feng et al. 2010; Shi et al. 2015) For example, ephrin-A2 is involved in breast carcinoma, liver cancer, and PCa (Fox and Kandpal 2004; Feng et al. 2010). In addition, ephrin-A2 promotes the migration of MDA-MB-231 breast cancer cells and reduces E-cadherin expression (Shi et al. 2015). We previously reported that ephrin-A2 expression is dramatically high in the tissues and exosomes derived from patients with PCa (Li et al. 2016, 2018). Although ephrin-A2 has an ectopic expression in PCa patients, its function remains poorly investigated.
The abnormal activation of signaling pathways plays a significant role in tumor genesis, progression, and metastasis, and blocking key molecules is an approach for antitumor therapy. The Eph family can trigger EMT-related signaling pathways to regulate cancer (Hou et al. 2012; Yuan et al. 2009; Macrae et al. 2005). EMT and its related markers, including N-cadherin, vimentin, fibronectin, matrix metalloproteinases (MMPs), E-cadherin, β-catenin, snail, and slug, are associated with cancer development. When undergoing EMT, epithelial cells show polarity loss, changes in morphology and cytoskeletal organization, and dissolution of direct contacts. Hence, cell deterioration can be prevented by inhibiting the up-regulation of the expression of EMT-related markers, which might be novel anticancer therapeutic targets (Lahat et al. 2010).
In this study, we reported that ephrin-A2-stimulated tumor growth in vivo and promoted PCa angiogenesis and metastasis. We highlighted a novel method of using ephrin-A2 in altering the expression of EMT-related markers to promote metastasis. Ephrin-A2 may be a potential target for cancer therapy because it regulates EMT-related markers. This study provides novel insights into the role ephrin-A2 in PCa tumorigenesis.
Methods
Cell culture
LNCaP, PC-3, and DU145 were purchased from American Type Culture Collection (ATCC). All the cell lines were cultured in DMEM/F-12 media (Gibco, USA), each containing 10% fetal bovine serum (FBS, Gibco, USA), 100 U/mL penicillin, and 100 U/mL streptomycin, at 5% CO2, 95% atmosphere, and 37 °C.
Construction of plasmids and transfection
The full-length cDNA of human ephrin-A2 was cloned into lentiviruses vectors (pLent-U6-GFP-Puro) at certain site. Vectors within ephrin-A2 fragments or control were transfected into 293T cells at 70% confluency for the generation of the lentiviruses. Then, the lentiviruses were transfected into PC-3 cells at appropriate confluency with polybrene for 24 h. Subsequently, infected cells were selected with puromycin at 2 µg/mL for 1 week. Finally, the selected cells were harvested, and ephrin-A2 expression was confirmed through Western blotting.
RNA interference of ephrin-A2
Ephrin-A2 shRNA (shRNA-1, shRNA-2, and shRNA -3) or control shRNA were used for RNA interference. Briefly, lentivirus shRNA or control were transfected into the 293 T cells at 70% confluency to generate lentiviruses. Then, lentiviruses were transfected into LNCaP and DU145 cells at appropriate confluency with polybrene for 24 h. Subsequently, the cells were selected with proper puromycin (LNCaP for 1 µg/mL, DU145 for 4 µg/mL) for 1 week. Finally, the selected cells were harvested, and ephrin-A2 expression was confirmed through Western blotting.
Cell proliferation
A total of 3000 cells were plated and incubated for 24, 48, 72, and 96 h. CCK-8 reagent (Keygen Biotech, Nanjing, China) was added to the media and cultured for 3 h. The OD450 value was measured with an automatic micro-plate reader every 24 h for 4 days for the generation of a growth curve.
Evaluation of angiogenesis
CD31 staining was used in evaluating microvascular density (MVD) (Foote et al. 2005). Briefly, the slides were immunohistochemical staining against CD31 (Servicebio Technology, Wuhan, China) were identified (× 100). Five areas of MVD were selected for counting manually (× 200). Brown-stained endothelial cells and their clusters that were clearly separated from adjacent microvessels, tumor cells, and other connective tissues were considered separate countable vessels. The mean score of the five hot spots was set as the level of MVD in each animal. The criterion for a positive immune reaction for VEGF is a dark brown cytoplasmic precipitate. VEFG expression was assessed for intensity: negative, weak = less than 10%, moderate = 11 to 50%, and strong = more than 50%.
Wound healing assay
DU145, LNCaP and PC-3 cell lines were performed for wound healing assay. In brief, a scratch was made with a micropipette tip at 90% confluency. The wounded area was captured immediately after scratching and after 24 h with a microscope. Wound healing abilities were quantified by measuring the average gap area between 0 and 24 h with ImageJ.
Migration assay
Modified migration assays were performed as previously described (Su et al. 2010). Briefly, cells suspended in a 100 μL serum-free medium were added to the up-wells, and the lower chambers contained 10% FBS as a chemoattractant. The plates were incubated for 24 h. Then, the chamber membrane was fixed and stained with 0.1% crystal violet. Finally, the upper non-migrating cells were gently wiped off with cotton swabs. Six fields were counted randomly (× 200), and the average of three independent experiments was obtained.
Invasion assay
Modified invasion assays were performed as previously described (Su et al. 2010). Briefly, diluted Matrigel (Corning Costar, USA) with serum-free medium (1:5) was added to the upper chamber, and incubation was performed for 4 h at 37 °C. Cells suspended in a 100 μL serum-free medium were added into the upper wells, and the lower chambers contained 10% FBS. The plates were incubated for 24 h. Then, the well membrane was fixed and stained with 0.1% crystal violet. Finally, the upper non-migrating cells were gently wiped off with cotton swabs. Six fields were counted randomly (× 200), and the average of three independent experiments was used.
Xenograft models
Stable-established cell lines were cultured in a DMEM/F-12 medium containing 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin at 5%CO2, 95% atmosphere, and 37 °C. Single cell suspensions with a concentration of 5 × 107 cells were inoculated into nude mice at one side of the abdomen. Six BALB/C (nu/nu) nude mice aged 4–6 weeks were randomly divided into two groups of three. The inoculation site was observed, and whether the site was infected or not and whether the tumor had subsided naturally were checked daily. The tumor long diameter (a) and short diameter (b) were measured every week, then the tumor volume was calculated according to the formula: volume = length × width2 / 2 for the drawing of a growth curve. After 7 weeks, nude mice were sacrificed, and tumor tissues, forelimb axillary lymph nodes, and lung tissues were obtained, and tumor masses were weighed. The tumor tissues, left axillary lymph nodes, and left lung tissue were subjected to immunohistochemistry and HE staining. Paraffin sections were subjected to staining, and blood vessel density was assayed with Image Plus.
PCR-based detection of human Alu sequences
Human disseminated tumor cells (DTCs) were detected in lymph nodes and lungs with Alu-qPCR as previously described (Liu et al. 2011) under the following conditions: 95 °C for 10 min, 40 cycles at 95 °C for 5 s, 65 °C for 5 s, and 72 °C for 20 s. Numerical data were determined against a standard curve established using murine blood DNA containing log-fold dilutions of DNA from 1 × 106 prostate carcinoma cells (PC-3 or DU145) grown in culture. Negative controls were established for each tissue by testing DNA from species with similar sex and age. The independent experiments were repeated at least three times.
Immunohistochemistry
Immunohistochemical staining was performed according to a standard method. Briefly, primary tumor tissues were fixed in a 10% formalin buffer, embedded in paraffin, and sectioned at 4 μm. Then, the slides were deparaffinized, rehydrated, and microwaved for 30 min (pH 6.0) for the retrieval of antigen epitopes in citrate buffer. The slides were subsequently treated with 3% H2O2 for 10 min for the elimination of endogenous peroxidase. The slides were incubated overnight with a primary antibody (E-cadherin, N-cadherin, CD31). The section was washed with PBS and then incubated with secondary antibody for 30 min at room temperature. 3,3′-Diaminobenzidine (DAB) was used for color detection, and nuclei were counterstained with hematoxylin.
Western blotting assay
Total protein was extracted from cells, and protein concentrations were determined using a BCA protein reagent kit (Beyotime, Nantong, China). Lysates were separated and transferred to a polyvinylidene fluoride membrane (Millipore, Bedford, USA) and incubated with ephrin-A2 (Affinity Biosciences, Cincinnati, USA) and EMT primary antibodies (Cell Signaling Technology, Danvers, USA) at 4 °C for overnight. The membranes were washed three times, incubated with secondary antibodies for 2 h, and visualized using an enhanced chemiluminescent substrate (Millipore, Bedford, USA). Finally, the membranes were visualized again using another enhanced chemiluminescent substrate (Bio-Rad Laboratories Inc, California, USA).
Statistical analysis
SPSS16.0 (SPSS, Chicago, USA) was used in statistical analyses. Multiple groups were evaluated with one-way analysis of variance (ANOVA), Statistical between groups were determined through t test. A P value of less than 0.05 was considered statistically significant.
Results
Ephrin-A2 expression in stable-established PCa cell lines
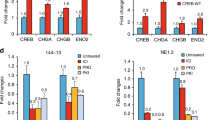
To explore ephrin-A2-exerted function in PCa, we first developed stable clones overexpressing or knocking down ephrin-A2 in PCa cell lines. The expression of ephrin-A2 in established cell lines was confirmed through Western blotting. Ephrin-A2 showed forced expression in overexpressed PC-3 cells. (Fig. 1a). For the purpose of the efficiency of inhibition, we selected three RNA interferences to block ephrin-A2 expression. Compared with shRNA-1 and shRNA-2 vectors, ephrin-A2 showed significantly inhibited expression in the DU145 and LNCaP cells infected with shRNA-3 lentiviral particles (Fig. 1b and c). Therefore, we used cells infected with shRNA-3 lentiviral particles for the next step.

Ephrin-A2 was assessed in established stable cell lines through Western blotting analysis: a Ephrin-A2 level was detected in PC-3 cell lines (Left panel). The relative expression of ephrin-A2 was normalized to that in PC-3 cells (Right panel). b Ephrin-A2 level was detected in DU145 cell lines (Left panel). Ephrin-A2 relative level expression was normalized to that in DU145/NC cells (Right panel) c Ephrin-A2 level was detected in LNCaP cell lines (Left panel). Ephrin-A2 relative level expression was normalized to that in LNCaP/NC cells (Right panel). *P < 0.05 compared with the control group
Ephrin-A2 contributes to tumor growth
To investigate whether ephrin-A2 functions in tumor growth, we established stable overexpressing ephrin-A2 in PC-3 cells and silencing ephrin-A2 in DU145 and LNCaP, respectively. No significant difference in proliferation between the control and experimental group was observed in vitro (Fig. 2a). To further verify the feature of this effect, a subcutaneous xenograft model in mice was established. Ephrin-A2 overexpression in the PC-3 cells generated xenografts that were larger than those in the control, consistent with the silencing of ephrin-A2 cells (Fig. 2b). This result indicates that ephrin-A2 contributes to tumor growth in vivo, rather than to proliferation in vitro.

Ephrin-A2 contributed to tumor growth in stable cell lines in vivo: a Ephrin-A2 showed no difference in cell proliferation according to the CCK‑8 assay in vitro. b Ephrin-A2 contributed to tumor growth in vivo. Tumor harvested from overexpressing ephrin-A2 PC-3 groups and ephrin-A2 knock-down DU145 group
Ephrin-A2 promotes tumor angiogenesis
Ephrin-A2 may affect tumor environments. By determining whether ephrin-A2 plays a role in tumor angiogenesis, we found that the level of CD31, an indicator of angiogenesis, showed a higher increase in the PC-3 group with overexpressed ephrin-A2 than that in the control (Fig. 3). The silencing of ephrin-A2 in the DU145 group decreased microvascular density. What is more, we analyzed the expression of VEGF in tumor tissues of tumor-bearing mice (DU145 group) using the immunohistochemistry, and found that VEGF expression was correlated significantly with expression of CD31 (Supporting Fig. 1). The data indicates that ephrin-A2 may promote tumor angiogenesis to increase tumor growth.

Ephrin-A2 promotes tumor angiogenesis. CD31 was evaluated in the xenograft models through immunohistochemical staining. Tissues harvested from overexpressing ephrin-A2 PC-3 groups and ephrin-A2 knock-down DU145 group. Scale bar, 100 µm
Ephrin-A2 led to tumor metastasis
To evaluate the metastasis potential of ephrin-A2, we examined the effect of ephrin-A2 on migration in PC-3 cells. The overexpression of ephrin-A2 in the PC-3 cells facilitated migration (Supporting Fig. 2 and Fig. 4a, P < 0.001). Invasion assay showed that the overexpression of ephrin-A2 in PC-3 cells promoted the migration of cells (Fig. 4b, P < 0.001). These data show that ephrin-A2 can promote tumor invasion in PC-3 cells. To further verify whether ephrin-A2 led to invasion in PCa, we performed migration and invasion assays in depletion of ephrin-A2 in LNCaP and DU145 cells. We found that ephrin-A2 enhanced the capability of migration and invasion in LNCaP cells (Fig. 4a and b ,P < 0.001, Supporting Fig. 3) and that the loss of ephrin-A2 significantly inhibited DU145 cell invasion (Fig. 4b). Nevertheless, no significantly changes in cell migration ability was observed in the DU145 cells (Supporting Fig. 4 and Fig. 4a, P > 0.05). These data suggest that ephrin-A2 promotes PC-3, LNCaP, and DU145 migration and invasion but has no effect on the migration capabilities of DU145.

Ephrin-A2 led to tumor metastatic. a The effect of ephrin-A2 on migration in three PCa cell lines. Ephrin-A2 enhanced PC-3 and LNCaP cell migration, but no effect on DU145. b The effect of ephrin-A2 on invasion in three PCa cell lines. Ephrin-A2 enhanced prostate cell invasion. c, d Ephrin-A2 promote metastasis in vivo in PC-3 and DU145 group. Tissues were harvested and the existence of metastatic tumor cells in local draining lymph nodes (Left panel) and lungs (Right panel) were assessed by qPCR-based detection of human Alu sequences and HE staining. Scale bar, 100 µm. Scale bar, 100 µm
Furthermore, we assessed whether ephrin-A2 can promote metastasis in vivo in a subcutaneous xenograft model using PC-3 and DU145 cells in nude mice. Tissues harvested from overexpressing ephrin-A2 PC-3 groups and ephrin-A2 knock-down DU145 group. HE-staining showed that ephrin-A2 significantly increased the incidence of metastases to local draining lymph nodes (LN) and lungs compared with that in the control group (Fig. 4c and Fig. 4d, P < 0.01). Collectively, these observations indicate that ephrin-A2 contributes to tumor metastasis.
Ephrin-A2 regulates EMT related markers
Ephrin-A2, as a member of the Eph receptor tyrosine kinase ligand family, regulates a series of downstream molecules. To determine whether the promotion of metastasis by ephrin-A2 is associated with the EMT family, we tested the expression of EMT-related markers (ZO-1, N-cadherin, E-cadherin, β-catenin, vimentin, slug, snail, and claudin-1) in stable PCa cells through Western blotting. The results show the down-regulated expression of epithelial markers (ZO-1, E-cadherin, and claudin-1) and up-regulated expression of mesenchymal markers (N-cadherin, β-catenin, vimentin, slug, and snail), but the knocking out of ephrin-A2 opposed the effect on the expression of EMT markers (Fig. 5). These data indicate that ephrin-A2-medicated tumor cell metastasis may be associated with the activation of EMT progress.

Ephrin-A2 altered the expression of EMT-related markers. a Western blotting analysis of EMT-related markers protein expression in PC-3 cell lines (Left panel). The relative expression of ephrin-A2 was normalized to that in PC-3 cells (Right panel). b Western blotting analysis of EMT-related markers protein expression in DU145 cell lines (Left panel). The relative expression of ephrin-A2 was normalized to that in DU145 cells (Right panel)
To confirm the hypothesis, subcutaneous xenograft mouse model was used. The mice were sacrificed, and related tissues were harvested for E-cadherin and N-cadherin expression analyses. In the overexpressing ephrin-A2 PC-3 groups, E-cadherin was down-regulated compared with that in the control group; whereas, N-cadherin was up-regulated (Fig. 6a and b). Similarly, the depletion of ephrin-A2 expression was consistent with a previous result (Fig. 6a and b). The results further show that the activation of EMT signaling pathway play a vital role in ephrin-A2-medicated PCa metastasis.

Ephrin-A2 affect the expression of E-cadherin and N-cadherin in xenograft models. a E-cadherin and N-cadherin were analyzed in xenograft models by immunohistochemical staining. Tissues harvested from overexpressing ephrin-A2 PC-3 groups. Scale bar, 50 µm. b E-cadherin were analyzed in xenograft models by immunohistochemical staining. Tissues harvested from knock down ephrin-A2 DU145 groups. Scale bar, 50 µm
Discussion
In this study, we first developed stable-established cell lines with overexpressing and knock-down, and our results demonstrated that ephrin-A2 promotes prostate cancer metastasis and angiogenesis. Furthermore, we provided novel insights into this mechanism, by which ephrin-A2 alters EMT-related markers to promote cancer metastasis. Moreover, ephrin-A2 may be a potential target for cancer metastasis therapy because it regulates EMT-related markers.
In our previous reports, we found that ephrin-A2 expression is significantly higher in the tissues and exosomes derived from patients with PCa (Li et al. 2016, 2018). However, in the present study, ephrin-A2 overexpression did not affect cell proliferation in PC-3 cell lines in vitro, and the depletion of ephrin-A2 in DU145 and LNCaP confirmed this finding. Ephrin-A2 contributed to tumor growth in vivo, and this result is consistent with the proliferation of liver cancer cells (Feng et al. 2010). These results show that ephrin-A2 may affect cells in tumor environments and promotes tumor growth, rather than in the tumors. Traditionally, tumor growth depends on multiple factors, such as blood vessel development, growth factors, and nutrition supply. Many studies reported that the Eph family is involved in tumor angiogenesis (Pasquale 2010; Nassar et al. 2013; Kandouz 2012). To further elucidate the biological functions of ephrin-A2, we detected CD31, an indicator of angiogenesis. Our results show that ephrin-A2 contribute to tumor growth in vivo, rather than in vitro, suggesting that tumor proliferation rate is not responsible for tumor growth. Simultaneously, the depletion of ephrin-A2 reduced angiogenesis. Hence, we speculated that ephrin-A2 can induce angiogenesis and promote tumor metastasis. Overall, these observations revealed that ephrin-A2 may promote PCa progression and metastasis by enhancing angiogenesis, at least in part.
Typically, an important feature of malignant tumor cells is invasion. In breast tumor cells, the ephrin-A2 gene promotes cell migration. Feng et al. (Feng et al. 2010) confirmed that ephrin-A2 can promote liver cancer cell invasion in vivo. Consistent with previous studies, our study also found that ephrin-A2 has a positive action on cell invasion in PCa in vitro and in vivo. Our results show that the activity of ephrin-A2 can enhance cell motility and invasion. Further, ephrin-A2 increased the incidence of metastases in lymph nodes (LN) and lungs in vivo, indicating that ephrin-A2 leads to tumor metastasis.
EMT, an important mechanism of invasion and metastasis, has been observed in many malignant tumors in humans, but the pathogenesis of this disease remains unclear (Colas et al. 2012). Zheng et al. (2016) found that after knocking out the CCAT2 gene, cell proliferation, invasion, and migration were significantly enhanced, suggesting that EMT is closely related to PCa invasion and metastasis. In our study, EMT-related markers were evaluated in stable-established cell lines through Western blotting. Our studies identified that ephrin-A2 can inhibit E-cadherin, ZO-1, and claudin-1 expression and enhance β-catenin, vimentin, snail, slug, and N-cadherin. E-cadherin is involved in the progression of human malignancies progression, and the low expression of E-cadherin is associated with lymphatic vessel invasion (Yuan et al. 2009; Saito et al. 2004). When EMT occurs, epithelial cells lose polarity and show enhanced ability to migrate and promote cytoskeletal rearrangement. The overexpression of Eph receptor promotes migration and invasion and inhibits E-cadherin-independent ligand-dependent mechanisms through the downstream singling pathways (Fang et al. 2008; Miura et al. 2009). This study reveals that ephrin-A2 down-regulates E-cadherin and loss cell adhesion to promote invasion and metastasis, suggesting that E-cadherin has potential as a tumor suppressor.
In many tumors, N-cadherin is overexpressed and is related to metastasis (Abufaraj et al. 2017; Nakajima et al. 2004). E-cadherin and N-cadherin belongs to the cadherin family. E-cadherin loss is always accompanied by the up-regulation of N-cadherin, which indicates poor prognosis (Kuphal and Bosserhoff 2006). N-cadherin promotes cancer metastasis to regulate adhesion and angiogenesis (Hazan et al. 2000; Gerhardt and Betsholtz 2003). Owing to the low expression of N-cadherin in DU145, we assayed N-cadherin in the overexpression ephrin-A2 group only. In this study, ephrin-A2 can increase N-cadherin expression but down-regulate E-cadherin. These data indicate that ephrin-A2 promotes PCa metastasis and angiogenesis by regulating EMT-related markers.
N-cadherin and vimentin not only promote tumor invasion and are associated with poor prognosis but also are therapeutic targets in cancer (Macrae et al. 2005; Mariotti et al. 2007; Satelli and Li 2011). Our study indicates that ephrin-A2 intensifies the expression of N-cadherin and vimentin, thereby accelerating tumor invasion and resulting in poor prognosis. Thus, ephrin-A2 is a promising target for PCa cancer therapy.
Another key factors, snail and slug, are linked to the promoter of E-cadherin and directly involved in inhibiting the expression of E-cadherin (Dissanayake et al. 2007; Cano et al. 2000). Ephrin-A2 probably interferes with Snail and Slug to down-regulate E-Cadherin.
In conclusion, we demonstrated that ephrin-A2 promotes PCa metastasis and angiogenesis by altering EMT-related markers. Furthermore, ephrin-A2 may be a potential target for cancer metastasis therapy because it regulates EMT-related markers.
Data availability statement
The raw data used and analyzed in the current study are available from the corresponding author upon a reasonable request.
References
Abufaraj M, Shariat SF, Haitel A et al (2017) Prognostic role of N-cadherin expression in patients with non-muscle-invasive bladder cancer. Urol Oncol 35(5):264–271
Cano A, Pérez-Moreno MA, Rodrigo I et al (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2(2):76–83
Colas E, Pedrola N, Devis L et al (2012) The EMT signaling pathways in endometrial carcinoma. Clin Transl Oncol 14(10):715–720
Dissanayake SK, Wade M, Johnson CE et al (2007) The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem 282(23):17259–17271
Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, Chen J (2008) Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J Cell Sci 121(Pt 3):358–368
Feng YX, Zhao JS, Li JJ et al (2010) Liver cancer: EphrinA2 promotes tumorigenicity through Rac1/Akt/NF-kappaB signaling pathway. Hepatology 51(2):535–544
Foote RL, Weidner N, Harris J et al (2005) Evaluation of tumor angiogenesis measured with microvessel density (MVD) as a prognostic indicator in nasopharyngeal carcinoma: results of RTOG 9505. Int J Radiat Oncol Biol Phys 61(3):745–753
Fox BP, Kandpal RP (2004) Invasiveness of breast carcinoma cells and transcript profile: Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application. Biochem Biophys Res Commun 318(4):882–892
Gerhardt H, Betsholtz C (2003) Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res 314(1):15–23
Hafner C, Schmitz G, Meyer S et al (2004) Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin Chem 50(3):490–499
Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA (2000) Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol 148(4):779–790
Hou F, Yuan W, Huang J et al (2012) Overexpression of EphA2 correlates with epithelial-mesenchymal transition-related proteins in gastric cancer and their prognostic importance for postoperative patients. Med Oncol 29(4):2691–2700
Kandouz M (2012) The Eph/Ephrin family in cancer metastasis: communication at the service of invasion. Cancer Metastasis Rev 31(1–2):353–373
Kuphal S, Bosserhoff AK (2006) Influence of the cytoplasmic domain of E-cadherin on endogenous N-cadherin expression in malignant melanoma. Oncogene 25(2):248–259
Lahat G, Zhu QS, Huang KL et al (2010) Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS ONE 5(4):e10105
Li S, Wu Z, Chen Y et al (2016) Diagnostic and prognostic value of tissue and circulating levels of Ephrin-A2 in prostate cancer. Tumour Biol 37(4):5365–5374
Li S, Zhao Y, Chen W et al (2018) Exosomal ephrinA2 derived from serum as a potential biomarker for prostate cancer. J Cancer 9(15):2659–2665
Liu W, Guan M, Hu T, Gu X, Lu Y (2011) Re-expression of AKAP12 inhibits progression and metastasis potential of colorectal carcinoma in vivo and in vitro. PLoS ONE 6:e24015
Macrae M, Neve RM, Rodriguez-Viciana P et al (2005) A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell 8(2):111–118
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298(5600):1912–1934
Mariotti A, Perotti A, Sessa C, Rüegg C (2007) N-cadherin as a therapeutic target in cancer. Expert Opin Investig Drugs 16(4):451–465
Miura K, Nam JM, Kojima C, Mochizuki N, Sabe H (2009) EphA2 engages Git1 to suppress Arf6 activity modulating epithelial cell-cell contacts. Mol Biol Cell 20(7):1949–1959
Nakajima S, Doi R, Toyoda E et al (2004) N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res 10(12):4125–4133
Nassar ZD, Moon H, Duong T et al (2013) PTRF/Cavin-1 decreases prostate cancer angiogenesis and lymphangiogenesis. Oncotarget 4(10):1844–1855
Pasquale EB (2010) Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 10(3):165–180
Saito T, Masuda N, Miyazaki T et al (2004) Expression of EphA2 and E-cadherin in colorectal cancer: correlation with cancer metastasis. Oncol Rep 11(3):605–611
Satelli A, Li S (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 68(18):3033–3046
Shi Z, Xiaofu Y, Xiaojia W, et al (2015) A primary study on ephrin-A2 promoted migration of MDA-MB-231 breast cancer cells. J Med Res **
Smith RA, Manassaram-Baptiste D, Brooks D et al (2014) Cancer screening in the United States, 2014: a review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin 64(1):30–51
Su B, Bu Y, Engelberg D, Gelman IH (2010) SSeCKS/Gravin/AKAP12 inhibits cancer cell invasiveness and chemotaxis by suppressing a protein kinase C- Raf/MEK/ERK pathway. J Biol Chem 285:4578–4586
Yuan W, Chen Z, Wu S et al (2009) Expression of EphA2 and E-cadherin in gastric cancer: correlated with tumor progression and lymphogenous metastasis. Pathol Oncol Res 15(3):473–478
Zheng J, Zhao S, He X et al (2016) The up-regulation of long non-coding RNA CCAT2 indicates a poor prognosis for prostate cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Biochem Biophys Res Commun 480(4):508–514
Funding
This study was supported by the grant from the Postdoctoral Science Foundation of China (No.2020M670076ZX), the National Natural Science Foundation of China (No. 81702061), the Natural Science Foundation of Jiangsu Province (No. BK20160233) and the Jiangsu Provincial Medical Youth Talent (No. QNRC2016781).
Author information
Authors and Affiliations
Contributions
SBL, YZ and CCC contributed to the idea, drafting and editing of the manuscript. LUS and JWW completed statistical analysis. MMZ and HLZ contributed to literature search and feeding animals and data analysis. PM and SBL revised the manuscript with critical reviews and comments and all the authors approved the final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical statement
The animals in this study were obtained in accordance with the ethical standards of the ethics committee of the affiliated Hospital of Xuzhou Medical University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
432_2021_3618_MOESM1_ESM.tif
Supplementary file1 Supporting Fig. 1 Ephrin-A2 promotes tumor angiogenesis. VEGF was evaluated in tumor tissues of tumor-bearing mice (DU145 group) through immunohistochemical staining. The criterion for a positive immune reaction for VEGF is a dark brown cytoplasmic precipitate. Scale bar, 100 µm. (TIF 1420 KB)
432_2021_3618_MOESM2_ESM.tif
Supplementary file2 Supporting Fig. 2 Effects of EphrinA2 on migration of PC-3 cells in vitro. Wound areas of overexpression ephrin-A2 PC-3 cells. The cells were cultured in regular medium and wound healing abilities were quantified by measuring the average gap area between 0 and 24 h with ImageJ. (TIF 179 KB)
432_2021_3618_MOESM3_ESM.tif
Supplementary file3 Supporting Fig. 3 Effects of EphrinA2 on migration of LNCaP cells in vitro. Wound areas of knockdown ephrin-A2 LNCaP cells. The cells were cultured in regular medium and wound healing abilities were quantified by measuring the average gap area between 0 and 24 h with ImageJ. (TIF 187 KB)
432_2021_3618_MOESM4_ESM.tif
Supplementary file4 Supporting Fig. 4 Effects of EphrinA2 on migration of DU145 cells in vitro. Wound areas of knockdown ephrin-A2 DU145 cells. The cells were cultured in regular medium and wound healing abilities were quantified by measuring the average gap area between 0 and 24 h with ImageJ. (TIF 159 KB)
Rights and permissions
About this article

Cite this article
Zhao, Y., Cai, C., Zhang, M. et al. Ephrin-A2 promotes prostate cancer metastasis by enhancing angiogenesis and promoting EMT. J Cancer Res Clin Oncol 147, 2013–2023 (2021). https://doi.org/10.1007/s00432-021-03618-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03618-2