
Abstract
Purpose
Pegfilgrastim is a long-acting granulocyte colony-stimulating factor indicated for prevention of febrile neutropenia in patients receiving myelosuppressive chemotherapy by promoting neutrophil recovery.
Methods
This phase 1, randomized, double-blind, three-way crossover trial in healthy volunteers evaluated the pharmacokinetics (PK), pharmacodynamics (PD), safety, and tolerability of the proposed biosimilar, comparing MYL-1401H, reference pegfilgrastim (Neulasta®, Amgen Inc, Thousand Oaks, CA, USA) sourced from the European Union, and reference pegfilgrastim sourced from the USA. Primary PK end points were peak plasma concentration of pegfilgrastim (Cmax) and area under the plasma concentration–time curve from the time of dosing to infinity (AUC0−inf). Primary PD end points were area under the curve above baseline for absolute neutrophil counts (ANC AUC0−t) and maximum change from baseline for ANC (ANC Cmax). Adverse events were also recorded.
Results
The primary PK and PD end points were similar across all groups. For the PK parameters, the 90% confidence intervals (CIs) of the ratios of geometric means ranged between 0.91 and 1.18, which were within the predefined bioequivalence interval of 0.8000 to 1.2500 for all comparisons. For the PD parameters, the 95% CIs of the ratios of geometric means ranged between 0.94 and 1.06 for all comparisons, which were within the predefined PD equivalence interval of 0.8500 to 1.1765. The safety profiles were similar, with the most common adverse events being back pain and headache.
Conclusions
MYL-1401H demonstrated similar PK, PD, and safety to reference pegfilgrastim in healthy volunteers and may be an equivalent option for the prevention of febrile neutropenia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The prevention of chemotherapy-induced neutropenia can reduce infections, decrease mortality, and prevent reductions and delays in chemotherapy doses (Kuderer 2011; Dale 2002). Clinically, filgrastim and its long-acting pegylated analog, pegfilgrastim, are used for the prevention of febrile neutropenia (FN) in patients receiving myelosuppressive chemotherapy (Neulasta 2016; Neupogen 2016). Filgrastim and pegfilgrastim are recombinant granulocyte colony-stimulating factors (G-CSFs) that act in the same manner as the endogenous G-CSF protein to stimulate the production of neutrophils (Neulasta 2016; Neupogen 2016). Recombinant G-CSFs have been used clinically for more than 25 years, and a significant amount of evidence demonstrates the long-term safety, efficacy, and value of these products.
As many biologics begin to lose patent protection, the development of biosimilars may serve to improve access to reliable treatments such as G-CSFs (Mellstedt et al. 2008; Blackstone and Joseph 2013). Filgrastim was among the first drugs to have biosimilar versions approved by the European Medicines Agency (EMA; in 2008) and the US Food and Drug Administration (FDA; in 2015) (Raedler 2016; Minghetti et al. 2012). Randomized controlled clinical studies demonstrated that biosimilar filgrastim has comparable efficacy and safety to originator filgrastim (Blackwell et al. 2015; del Giglio et al. 2008; Gascon et al. 2010; Waller et al. 2010b). Accordingly, guidelines from major medical organizations indicate that filgrastim biosimilars are an effective option for the primary prophylaxis of FN in patients receiving myelosuppressive chemotherapy (Smith et al. 2015; Aapro et al. 2011).
Filgrastim needs to be administered daily for several days after each chemotherapy cycle. Pegfilgrastim, however, is a longer-acting pegylated analog that only requires administration once per chemotherapy cycle, with more than 15 days between administrations (Neulasta 2016; Smith et al. 2015; Green et al. 2003; Holmes et al. 2002a, b). Although significant efforts are being made to develop pegfilgrastim biosimilars (Harbeck et al. 2016; Park et al. 2013), neither the EMA nor the FDA have currently approved biosimilar versions of pegfilgrastim. Here, we present preclinical data demonstrating the analytical similarity of the proposed biosimilar, MYL-1401H, and reference pegfilgrastim as well as data from a phase 1 trial evaluating the pharmacokinetic (PK) and pharmacodynamic (PD) equivalence of MYL-1401H and reference pegfilgrastim.
Materials and methods
Preclinical characterization
To compare the biological activity of MYL-1401H with that of originator pegfilgrastim (Neulasta®, Amgen Inc, Thousand Oaks, CA, USA) sourced from the European Union (EU-reference pegfilgrastim) and from the USA (US-reference pegfilgrastim), a cell proliferation assay using a murine myelogenous leukemia cell line [M-NFS-60 (ATCC® CRL-1838™) purchased from the American Type Culture Collection (Manassas, VA, USA)] was used. The in vitro assay compared the relative ability of MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim to induce proliferation of the cell line. The bioactivity data were reported as relative potency to the internal reference standard, calibrated against an international reference standard. Additionally, the binding affinities of MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim with the G-CSF receptor were evaluated by surface plasmon resonance (SPR).
Study design
This phase 1, randomized, double-blind, three-way crossover trial in healthy volunteers evaluated the PK, PD, safety, and tolerability of MYL-1401H compared with those of EU-reference and US-reference pegfilgrastim. The primary objective of this study was to compare the PK and PD of MYL-1401H with reference pegfilgrastim. It was estimated that with 180 evaluable subjects, the study would have a combined power of over 90% to establish PK and PD equivalence for each of the three pairwise comparisons. The sample size calculation was based on intrasubject variability from a pilot study [area under the curve above baseline for absolute neutrophil count (ANC AUC0−t), 14%; pegylated G-CSF (PEG-G-CSF) area under the plasma concentration–time curve from the time of dosing to infinity (AUC0−inf), 36%; PEG-G-CSF peak plasma concentration (Cmax), 50%] and the assumption that ANC AUC0−t, PEG-G-CSF AUC0−inf, and PEG-G-CSF Cmax met predefined criteria for equivalence.
The study was conducted at a single site (PRA Health Sciences, Groningen, The Netherlands). Male and female subjects were screened for eligibility in the trial within 3 weeks before the first administration of the study drug. Subjects were eligible if they were 18–65 years of age and deemed healthy as determined by clinical screening. Subjects were excluded if they had a history of medical conditions that would potentially increase risks or could affect the evaluation of study results, including evidence of clinically relevant pathology (e.g., sickle cell disorders, hematologic malignancies) and history of relevant drug or food allergies. Subjects were also excluded if they had any previous exposure to any G-CSF product.
This was the first in-human study conducted with MYL-1401H, so no human safety data were available before the start of the study. MYL-1401H was not expected to pose a significant safety risk based on the known pharmacologic mechanism of action and results from nonclinical studies comparing MYL-1401H with reference pegfilgrastim in rats. Additionally, based on the content and purity of the drug, physicochemical similarity, and functional comparability demonstrated during characterization studies, the safety profiles of MYL-1401H and reference pegfilgrastim were expected to be similar. A 2-mg dose was used in this study, which is less than the clinical dose of reference pegfilgrastim (6 mg), but is on the steep part of the dose–response curve for the PD parameters and more sensitive than a 6-mg dose for detecting differences in PD between MYL-1401H and reference pegfilgrastim. This dose was initially evaluated in one sentinel subcohort of six subjects, with two subjects receiving 2 mg of MYL-1401H, two subjects receiving 2 mg of EU-reference pegfilgrastim, and two subjects receiving 2 mg of US-reference pegfilgrastim. No safety concerns were identified in the sentinel group; thus, the remaining subjects were enrolled and dosed.
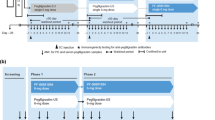
After randomization to one of six treatment sequences, subjects were administered MYL-1401H or one of the two reference pegfilgrastim products in period 1. In the two crossover periods, subjects were given alternative therapies (Fig. 1). Subjects received a single 2-mg subcutaneous injection of each drug followed by a washout period of 4 weeks. Subjects were in the clinic for all three treatment periods, beginning 2 days before drug administration and remaining in the clinical research center for approximately 96 h after each drug administration.

Study design. Patients were administered a single 2-mg SC dose of MYL-1401H, EU-reference pegfilgrastim, or US-reference pegfilgrastim during the first treatment period. Each of the other drugs was administered in the following crossover periods after a washout of 4 weeks. SC subcutaneous, XO crossover
PK/PD analyses
Blood samples (2.5 mL) were collected for PK/PD analyses immediately before dose administration (0 h) and at 2, 4, 6, 8, 10, 12, 16, 20, 24, 48, 72, 96, 120, 144, 168, 192, 264, 336, and 504 h after administration. The primary PK end points were pegfilgrastim Cmax and AUC0−inf. Other key PK end points included AUC0−t, the time of maximum serum concentration (tmax), terminal elimination rate constant (kel), apparent terminal elimination half-life (t1/2), and apparent volume of distribution (Vd/F). Analyses were performed on the PK parameters using the general linear model analyses of variance (GLM ANOVA). The bioequivalence criterion was that the 90% confidence intervals (CIs) of least squares mean ratios of Cmax and AUC0−inf were bounded within 0.8000 to 1.2500 for the natural log-transformed data.
The primary PD end points were ANC AUC0−t and maximum change from baseline for ANC (ANC Cmax). Other key PD end points included area under the curve above baseline for CD34+ cell counts (CD34+ AUC0−t), the time of maximum change from baseline for ANC and CD34+ cell counts (ANC tmax and CD34+ tmax, respectively), and maximum change from baseline for CD34+ cell counts (CD34+ Cmax). Pharmacodynamic parameters were calculated using nonparametric techniques. Statistical analyses were performed using GLM ANOVA. The PD equivalence criterion was that the 95% CI of least squares mean ratios of ANC AUC0−t and ANC Cmax were bounded within 0.8500 to 1.1765 for the natural log-transformed data.
Safety and tolerability analyses
Safety and tolerability assessments included adverse events (AEs), clinical laboratory parameters, vital signs, 12-lead electrocardiograms (ECGs), local tolerability, and physical examination. The safety set included all subjects who received at least one dose of the study medication. All AEs between signing of informed consent and completion of the follow-up visit were recorded. Any clinically significant observations that were made on clinical laboratory parameters, 12-lead ECGs, vital signs, local tolerability, or physical examinations were recorded as AEs. Adverse events were graded for severity using the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.
For immunogenicity measurements, two blood samples (5 mL each) were collected at baseline, on days 7–9 of each treatment period, and at follow-up. Antidrug antibody (ADA) analysis was conducted using a validated, sensitive, and specific analytical method on the MesoScale Discovery® Platform (Rockville, MD, USA). Positive samples were validated using a confirmatory assay that tested antibodies against MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim. Samples confirmed as positive for ADA were further tested for the presence of neutralizing antibodies (NAb) using a validated cell-based assay (Cirion BioPharma Research Inc., Laval, QC, Canada). Because immunogenicity data from the second and third treatment periods are confounded with earlier treatments, only immunogenicity data from the first treatment period are presented.
Ethical approval
This study was approved by the independent ethics committee of the Evaluation of Ethics in Biomedical Research Foundation (Assen, The Netherlands) and conducted in accordance with the ethical principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonisation E6 Guideline for Good Clinical Practice and the European Union Clinical Trial Directive. All patients provided written informed consent before any study-related procedures were started.
Results
Preclinical characterization
MYL-1401H showed high similarity to EU-reference pegfilgrastim and US-reference pegfilgrastim lots in the potency assay (Fig. 2). Similarly, the relative potency of EU-reference pegfilgrastim was equivalent to US-reference pegfilgrastim. Additionally, the equilibrium dissociation constants (KD) of MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim to the G-CSF receptor, as evaluated by SPR, were highly similar. Binding constants for all MYL-1401H lots fell within the quality range (QR) of EU-reference pegfilgrastim and US-reference pegfilgrastim lots. Overall, these data suggest a high degree of similarity between MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim.

a Scatter plot of the relative potency for various lots of MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim. b Scatter plot for the equilibrium dissociation constant (KD) of G-CSF receptor binding with MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim. The lines represent the ranges from the observed data for the EU-reference pegfilgrastim (dark gray) lots and US-reference pegfilgrastim (light gray) lots. G-CSF granulocyte colony-stimulating factor, KD equilibrium dissociation constant
Patient disposition and baseline characteristics
A total of 372 subjects were screened, 216 of whom were enrolled in the study (Online Resource 1). All 216 subjects who were enrolled received at least one dose of pegfilgrastim and were included in the safety set. Of these, 208 subjects were administered at least two of the three doses of pegfilgrastim and were included in the PK and PD analysis sets. Over the course of the entire study, 20 subjects discontinued because of protocol violation (n = 8), withdrawal of consent (n = 8), AEs (n = 3), or missing too many visits because of illness that was considered unrelated to the study drug (n = 1).
More male (n = 170) than female (n = 46) subjects participated in the study (Online Resource 2). The median age was 33 years, and the median body mass index was 24.4 kg/m2. Baseline characteristics were similar in the safety and PK/PD analysis sets and across all six treatment sequences (Online Resource 3).
Pharmacokinetics
The shape of the serum concentration–time profile for pegfilgrastim was similar across all three treatments (Fig. 3a). The mean [coefficient of variation (%CV)] Cmax of pegfilgrastim was 36.7 pg/mL (72.1), 34.2 pg/mL (72.1), and 37.3 pg/mL (67.6) in the MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim groups, respectively (Table 1). The mean (%CV) AUC0−inf was 869 h·ng/mL (69.1), 833 h·ng/mL (70.1), and 876 h·ng/mL (66.3) in the MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim groups, respectively. When comparing the primary PK end points across all three treatment groups, GLM ANOVA results showed that the 90% CIs of the ratios of geometric means for these PK parameters ranged between 0.91 and 1.18, and were all contained within the predefined bioequivalence interval of 0.8000 to 1.2500 for each of the comparisons.

a Mean serum pegfilgrastim concentration versus time profile (PK analysis set). b ANC versus time profile (PD analysis set). ANC absolute neutrophil count, PD pharmacodynamic, PEG-G-CSF pegylated granulocyte colony-stimulating factor, PK pharmacokinetic
Secondary PK end points (i.e., AUC0−t, tmax, kel, t1/2, and Vd/F) were also similar across all treatment groups. The least squares mean estimates and the corresponding 90% CIs of the geometric mean ratios were close to 1 for the secondary PK parameters AUC0−t, kel, t1/2, and Vd/F of pegfilgrastim, with 90% CIs ranging between 0.85 and 1.13 for each treatment group comparison. Across all three treatment groups, the intrasubject %CV was 44.7, 29.3, 29.3, and 57.2% for AUC0−t, kel, t1/2, and Vd/F, respectively.
Pharmacodynamics
The primary PD parameters (i.e., ANC AUC0−t and ANC Cmax) and overall ANC profiles were similar across all three treatment groups (Table 2; Fig. 3b). The geometric mean (%CV) ANC AUC0−t was 2784.4 h·109/L (29.0), 2792.6 h·109/L (30.7), and 2744.7 h·109/L (30.8) in the MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim groups, respectively. The geometric mean (%CV) ANC Cmax was 22.5 × 109/L (25.7), 22.7 × 109/L (25.9), and 22.5 × 109/L (26.4) in the MYL-1401H, EU-reference pegfilgrastim, and US-reference pegfilgrastim groups, respectively. When comparing the primary PD end points across all three treatment groups, GLM ANOVA results showed that the 95% CIs of the ratios of geometric means for these PD parameters ranged between 0.94 and 1.06, and were all contained within the predefined PD equivalence interval of 0.8500 to 1.1765 for each of the comparisons. Additionally, there were no meaningful differences in median ANC tmax, a secondary PD parameter, across all three treatment groups.
Safety
During the course of the study, 97% (210/216) of subjects in the safety set reported at least one treatment-emergent AE (TEAE). From a total of 1733 TEAEs, 1339 (77%) were of grade 1 intensity, 393 (23%) were of grade 2 intensity, and 1 (< 1%) was of grade 3 intensity. The total number of TEAEs and percentage of subjects reporting TEAEs were comparable across all three treatment groups (Table 3). Overall, the most frequently reported TEAEs were back pain (81%), headache (63%), pain in the extremity (36%), and nasopharyngitis (22%). Musculoskeletal complaints (e.g., back pain, headache, pain in the extremity) were likely manifestations of bone pain resulting from expansion of bone marrow. Three TEAEs led to subject withdrawal: appendicitis, which occurred after dosing with US-reference pegfilgrastim, and abnormal liver function test and arthropod bite, both of which occurred after dosing with EU-reference pegfilgrastim.
Subcutaneous injections were well tolerated and local reactions were minimal in all treatment groups. Mild injection site reactions were observed in seven subjects after administration of MYL-1401H, in four subjects after administration of EU-reference pegfilgrastim, and in three subjects after administration of US-reference pegfilgrastim. One subject receiving MYL-1401H experienced a moderate injection site reaction.
The number of subjects with ADA was comparable among treatment groups. At baseline, 7% (16/216) of all subjects were confirmed positive for ADA. The proportion of subjects with treatment-emergent ADA (excluding those who were ADA positive at baseline) after the first treatment period was comparable among the MYL-1401H (14/63; 22%), EU-reference pegfilgrastim (16/68; 24%), and US-reference pegfilgrastim groups (21/69; 30%; Online Resource 4). Of the subjects who were NAb negative at baseline, two in the MYL-1401H group and one each in the EU-reference pegfilgrastim and US-reference pegfilgrastim groups were treatment-induced NAb positive during period 1. Of the two NAb-positive subjects in the MYL-1401H group, one was positive for PEG only and the other was positive for both PEG and G-CSF; both had very low titers of ADA (8–10 ng/mL). The NAb-positive subject in the EU-reference pegfilgrastim group was positive for both PEG and G-CSF. The NAb-positive subject in the US-reference pegfilgrastim group was positive for PEG only. The presence of NAb was not associated with any clinically relevant changes in ANC. Overall, there was no evidence of loss of efficacy or serious treatment-induced, immune-related AEs in the ADA- or NAb-positive subjects.
Discussion
Here, preclinical and clinical data demonstrate similarity of MYL-1401H with EU-reference pegfilgrastim and US-reference pegfilgrastim. Overall, the results of this study are similar to those from other PK/PD studies of G-CSF products, including the proposed biosimilars (Harbeck et al. 2016; Waller et al. 2010a; Buchner et al. 2014; Crobu et al. 2014). The study met its primary PK and PD end points, demonstrating bioequivalence of MYL-1401H to EU-reference pegfilgrastim and US-reference pegfilgrastim in healthy volunteers. All products were generally well tolerated, with the majority of TEAEs being bone pain, the most common side effect of G-CSF products (Smith et al. 2015). Serious but rare side effects of G-CSF therapy (Neulasta 2016) such as splenomegaly, acute respiratory distress syndrome, capillary leak syndrome, and severe allergic reactions were not observed in this study. No significant safety concerns and no relevant differences in safety or tolerability were observed among the treatments. It should be noted that this study was conducted in healthy subjects with doses lower than those used clinically. Still, the general profile of AEs appears similar to what has been reported for other G-CSFs in studies of patients receiving myelosuppressive anticancer drugs (Holmes et al. 2002b; Park et al. 2013). It is possible that differences in the safety profiles of MYL-1401H and reference pegfilgrastim products could emerge with increased exposure and under clinical conditions.
Pegfilgrastim is currently indicated for the reduction in duration of neutropenia and the incidence of FN in patients treated with cytotoxic chemotherapy for malignancy (Neulasta 2016). For the prevention of FN, pegfilgrastim acts by binding to receptors on the surface of hematopoietic cells, thereby stimulating the production of neutrophils. MYL-1401H consistently stimulated the production of neutrophils in a manner similar to approved pegfilgrastim products. Thus, the proposed pegfilgrastim biosimilar, MYL-1401H, may be an effective treatment option for the prevention of chemotherapy-induced neutropenia.
References
Aapro MS, Bohlius J, Cameron DA et al (2011) 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer 47:8–32
Blackstone EA, Joseph PF (2013) The economics of biosimilars. Am Health Drug Benefits 6:469–478
Blackwell K, Semiglazov V, Krasnozhon D et al (2015) Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol 26:1948–1953
Buchner A, Lammerich A, Abdolzade-Bavil A, Müller U, Bias P (2014) Lipegfilgrastim: pharmacodynamics and pharmacokinetics for body-weight-adjusted and 6 mg fixed doses in two randomized studies in healthy volunteers. Curr Med Res Opin 30:2523–2533
Crobu D, Spinetti G, Schrepfer R et al (2014) Preclinical and clinical phase I studies of a new recombinant filgrastim (BK0023) in comparison with Neupogen®. BMC Pharmacol Toxicol 15:7
Dale DC (2002) Colony-stimulating factors for the management of neutropenia in cancer patients. Drugs 62(suppl 1):1–15
del Giglio A, Eniu A, Ganea-Motan D, Topuzov E, Lubenau H (2008) XM02 is superior to placebo and equivalent to Neupogen™ in reducing the duration of severe neutropenia and the incidence of febrile neutropenia in cycle I in breast cancer patients receiving docetaxel/doxorubicin chemotherapy. BMC Cancer 8:332
Gascon P, Fuhr U, Sörgel F et al (2010) Development of a new G-CSF product based on biosimilarity assessment. Ann Oncol 21:1419–1429
Green MD, Koelbl H, Baselga J et al (2003) A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 14:29–35
Harbeck N, Lipatov O, Frolova M et al (2016) Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12:1359–1367
Holmes FA, Jones SE, O’Shaughnessy J et al (2002a) Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 13:903–909
Holmes FA, O’Shaughnessy JA, Vukelja S et al (2002b) Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with high-risk stage II or stage III/IV breast cancer. J Clin Oncol 20:727–731
Kuderer NM (2011) Meta-analysis of randomized controlled trials of granulocyte colony-stimulating factor prophylaxis in adult cancer patients receiving chemotherapy. Cancer Treat Res 157:127–143
Mellstedt H, Niederwieser D, Ludwig H (2008) The challenge of biosimilars. Ann Oncol 19:411–419
Minghetti P, Rocco P, Cilurzo F, Del Vecchio L, Locatelli F (2012) The regulatory framework of biosimilars in the European Union. Drug Discov Today 17:63–70
Neulasta [package insert] (2016) Amgen Inc, Thousand Oaks
NEUPOGEN [package insert] (2016) Amgen Inc, Thousand Oaks
Park KH, Sohn JH, Lee S et al (2013) A randomized, multi-center, open-label, phase II study of once-per-cycle DA-3031, a biosimilar pegylated G-CSF, compared with daily filgrastim in patients receiving TAC chemotherapy for early-stage breast cancer. Invest New Drugs 31:1300–1306
Raedler LA (2016) Zarxio (filgrastim-sndz): first biosimilar approved in the United States. Am Health Drug Benefits 9(spec feature):150–154
Smith TJ, Bohlke K, Lyman GH et al (2015) Recommendations for the use of WBC growth factors: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol 33:3199–3212
Waller CF, Bronchud M, Mair S, Challand R (2010a) Comparison of the pharmacodynamic profiles of a biosimilar filgrastim and Amgen filgrastim: results from a randomized, phase I trial. Ann Hematol 89:971–978
Waller CF, Semiglazov VF, Tjulandin S, Bentsion D, Chan S, Challand R (2010b) A phase III randomized equivalence study of biosimilar filgrastim versus Amgen filgrastim in patients receiving myelosuppressive chemotherapy for breast cancer. Onkologie 33:504–511
Acknowledgements
This study was supported by Mylan Inc, Canonsburg, PA, and Biocon Ltd, Bangalore, India. Editorial assistance was provided under the direction of the authors by Scott Houck, PhD, and Jennifer Rossi, MA, ELS, MedThink SciCom, and funded by Mylan Inc. The investigators thank the volunteers in this study for their contribution.
Funding
This study was funded by Mylan Inc, Canonsburg, PA, and Biocon Ltd, Bangalore, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Cornelius F. Waller is a consultant/advisory board member for Mylan Inc. Renger G. Tiessen is a paid employee of PRA Health Sciences, the contract research organization that conducted the study. Tracey E. Lawrence, Andrew Shaw, Mark Shiyao Liu, Mark Baczkowski, Catherine E. Micales, Abhijit Barve, Gopinath M. Ranganna, and Eduardo J. Pennella are paid employees of Mylan Inc and may hold stock with the company. Rajiv Sharma was a paid employee of Mylan Inc at the time of analysis and may hold stock with the company. Mudgal A. Kothekar is a paid employee of Biocon Research Ltd and may hold stock with the company.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Waller, C.F., Tiessen, R.G., Lawrence, T.E. et al. A pharmacokinetics and pharmacodynamics equivalence trial of the proposed pegfilgrastim biosimilar, MYL-1401H, versus reference pegfilgrastim. J Cancer Res Clin Oncol 144, 1087–1095 (2018). https://doi.org/10.1007/s00432-018-2643-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-018-2643-3