
Summary
Backgrounds A pegylated form of recombinant granulocyte-colony stimulating factor (G-CSF) was developed for prophylactic use in breast cancer. The aim of this study was to evaluate the efficacy and safety of once-per-cycle DA-3031 in patients receiving chemotherapy for breast cancer. Methods A total of 61 patients receiving docetaxel, doxorubicin, and cyclophosphamide (TAC) chemotherapy were randomized in cycle 1 to receive daily injections of filgrastim (100 μg/m2) or a single subcutaneous injection of pegylated filgrastim DA-3031 at a dose of either 3.6 mg or 6 mg. Results The mean duration of grade 4 neutropenia in cycle 1 was comparable among the treatment groups (2.48, 2.20, and 2.05 days for filgrastim, DA-3031 3.6 mg and 6 mg, respectively; P = 0.275). No statistically significant differences were observed in the incidence of febrile neutropenia between the treatment groups (9.5 %, 15.0 %, and 5.0 % for filgrastim, DA-3031 3.6 mg and 6 mg, respectively; P = 0.681) in cycle 1. The incidences of adverse events attributable to G-CSF were similar among the treatment groups. Conclusions Fixed doses of 3.6 mg or 6 mg DA-3031 have an efficacy comparable to that of daily injections of filgrastim in ameliorating grade 4 neutropenia in patients receiving TAC chemotherapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Myelosuppression is often the principal dose-limiting toxicity of cytotoxic chemotherapy and the reason for dose reduction in cancer patients. In early breast cancer, there is evidence supporting a close correlation of sustained relative dose intensity (RDI) of adjuvant chemotherapy and clinical outcomes of patients [1, 2]. Thus, substantial reductions in RDI may compromise the efficacy of adjuvant chemotherapy. Since the duration of grade 4 neutropenia is known to be a risk factor for life-threatening infectious complications, recombinant granulocyte-colony stimulating factor (G-CSF) has been developed for prophylactic use to reduce the severity and duration of chemotherapy-induced neutropenia.
Recently, TAC (docetaxel/doxorubicin/cyclophosphamide) and dose-dense (every 2-week) chemotherapy have been accepted as standard care because of their superior clinical benefit in early stage breast cancer patients [3, 4]. However, successful clinical implementation of these effective adjuvant treatments should be supported by prophylactic G-CSF treatment [3, 5–7]. After the initial development of G-CSF by recombinant technology, further modification of filgrastim by chemical addition of polyethylene glycol (PEG) resulted in increased plasma half-life, thus sustaining the pharmacologic effect in cancer patients [8]. With a single injection of pegfilgrastim per chemotherapy cycle, patients receive the same benefit as daily injections of filgrastim after chemotherapy [9, 10]. The development of this convenient biologic agent has led to better compliance of patients and fewer burdens for both patients and health professionals. However, the high cost of this biologic limits its routine use in clinical practice.
DA-3031, a pegylated human recombinant G-CSF which is biosimilar to the reference pegfilgrastim, Neulasta, has been developed for subcutaneous administration in the treatment of chemotherapy-induced neutropenia. The manufacturing process was developed by and preclinical and phase I studies have been conducted by Dong-A Pharmaceuticals.
The aim of this study was to evaluate the efficacy and safety of once-a-cycle DA-3031 at two dose levels in comparison with daily filgrastim in patients receiving TAC chemotherapy for early stage breast cancer.
Patients and methods
Patients
This study was approved by the institutional review boards of each participating center and the Korean Food and Drug administration. All patients gave written informed consent before any study-related procedure was performed. Patients were eligible for study enrollment if they met the following inclusion criteria: ≥18 years of age; diagnosis of high-risk (as defined by the investigator) stage II or III breast cancer; Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1; absolute neutrophil count (ANC) ≥ 1.5 × 109/L; platelet count ≥100 × 109/L; adequate renal, hepatic (i.e., bilirubin < 1.5 × the upper limit of normal and AST, ALT, or both < 1.5 × the upper limit of normal concomitant with alkaline phosphatase < 2.5 × the upper limit of normal), and cardiac function; and chemotherapy-naive.
Patients were excluded if they: had been enrolled into or had not yet completed other investigational drug trials within 30 days before randomization into this study; were still receiving other investigational agents; had previous exposure to filgrastim or pegfilgrastim within 6 weeks of randomization into this study; were pregnant or breast-feeding; had received systemic antibiotics within 72 h of chemotherapy; had prior bone marrow or stem-cell transplantation; or had undergone prior radiation therapy within 4 weeks of randomization into this study.
Study drug
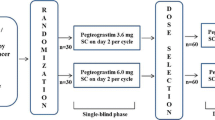
Patients randomized to filgrastim received daily subcutaneous injections of filgrastim 100 μg/m2/d beginning approximately 24 h after chemotherapy and continuing until documented ANC 5 × 109/L after nadir or up to 10 days. Patients randomized to pegfilgrastim received a single subcutaneous injection of DA-3031 at fixed doses of either 3.6 mg or 6 mg per chemotherapy cycle on day 2 of each cycle approximately 24 h after completion of chemotherapy. Both filgrastim (Leucostim, Dong-A pharmaceuticals, Seoul, Korea) [11] and pegfilgrastim (DA-3031, Dong-A pharmaceuticals, Seoul, Korea) were produced by recombinant DNA technology. DA-3031 was generated by adding PEG to filgrastim in order to prolong half-life and to enhance in vivo stability.
Study design
This study was a randomized, multi-center, open-label, dose-finding phase II study to compare the safety and efficacy between once-a-cycle pegfilgrastim and daily filgrastim. Patients at each center were randomized separately and were assigned in a 1:1:1 ratio to filgrastim100 μg/m2/d or pegfilgrastim 3.6 mg or 6 mg treatment groups by using a stratified permuted-block randomization schedule with body weight (≥60 kg versus <60 kg) as the stratification variable.
Chemotherapy
Patients received chemotherapy on day 1 of each cycle, which consisted of doxorubicin at 50 mg/m2, cyclophosphamide at 500 mg/m2, and docetaxel at 75 mg/m2, infused in that order. Chemotherapy was repeated every 3 weeks for up to six cycles.
End points
The primary efficacy end point was the duration of grade 4 neutropenia (defined as ANC < 0.5 × 109/L) in chemotherapy cycle 1. Secondary end points were the depth of ANC nadir, the time to ANC recovery to ≥ 2 × 109/L, the rate of febrile neutropenia, and the incidence of patients requiring intravenous antibiotics in the first cycle of chemotherapy.
Statistical analysis
The planned sample size for this study was approximately 60 patients, which was calculated using the confidence interval to ensure that the width of the 95 % confidence interval for the difference between the pegfilgrastim dose group and the filgrastim group for the mean duration of grade 4 neutropenia was 2 days and that the standard deviation was 1.5 days.
Efficacy analyses included all randomized patients who took at least one dose of the study drug and who had at least one post-baseline measurement. Safety analyses included all randomized patients who received at least one dose of study drug and any safety data were collected.
For efficacy analyses, descriptive statistics were performed for each efficacy endpoint. The means and standard deviations (SD) in each group were calculated for continuous variables, and 95 % two-sided confidence intervals (CIs) were calculated for the difference between each pegfilgrastim dose group and the filgrastim group for each efficacy endpoint. The treatment-group comparisons were performed using analysis of variance (ANOVA) or the Kruskal-Wallis test. The frequencies and percentages were calculated for categorical variables and the treatment-group comparisons were performed using the chi-square test or Fisher’s exact test.
For safety analyses, adverse events were classified according to the NCI Common Terminology Criteria for Adverse Events (CTCAE version 4.01). The number and percentage of patients who experienced adverse events were summarized, and the treatment-group comparisons were performed using the chi-square test or Fisher’s exact test.
All significance tests were two-tailed with a nominal significance level of 0.05.
Results
Patients
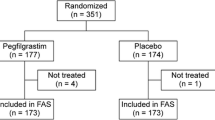
A total of 65 patients were screened from 9 centers, with 4 patients not passing the screening process. Sixty-one patients were randomized and received the study drug in the first cycle of chemotherapy. The groups were well-balanced for demographics, body size, and disease stage (Table 1). The patients in this study were all Korean women, and the mean age was 44.92 ± 7.26 years. Twenty-one patients were randomized to receive daily injections of filgrastim (100 μg/m2/d), 20 patients were randomized to receive DA-3031 3.6 mg, and the other patients received DA-3031 6 mg. Three patients erroneously received another study drug instead of the allocated one: filgrastim was given to a DA-3031 6 mg group patient, and DA-3031 6 mg was given to patients in the filgrastim and DA-3031 3.6 mg groups, respectively. Efficacy analysis for these patients was based on the initial randomized assignment and not on the treatment received. Safety analysis was based on the treatment received, not as randomized. All of the patients completed the study, and filgrastim was provided until the last cycle of chemotherapy for each group of patients.
Efficacy
The mean duration of G4 neutropenia in cycle 1 was comparable among the treatment groups (2.48 ± 1.03 days versus 2.20 ± 1.47 days versus 2.05 ± 1.05 days for filgrastim versus DA-3031 3.6 mg versus DA-3031 6 mg, respectively; P = 0.275; Table 2). Subgroup analysis by stratification factor and body weight showed a trend for shorter duration of G4 neutropenia with DA-3031 6 mg in patients who weighed 60 kg or higher. However, the trend was not statistically significant (P = 0.409).
Single injections of pegfilgrastim DA-3031 at the two different dose levels showed similar ANC profiles when compared with daily injections of filgrastim treatment (Fig. 1). The ANC nadir was observed on approximately day 7 of cycle 1 of chemotherapy. The mean nadir ANCs of the DA-3031 6 mg group and the 3.6 mg group were 139.2 ± 114.9/mm3 and 136.7 ± 183.2/mm3, respectively. These values tended to be higher than that of the filgrastim group; however, the difference was not statistically significant (96.0/mm3; P = 0.361; Table 3). The mean time to ANC recovery to ≥ 2 × 109/L was similar among the treatment groups (9.8 ± 0.8 days versus 10.1 ± 1.8 days versus 9.9 ± 1.6 days for filgrastim versus DA-3031 3.6 mg versus DA-3031 6 mg, respectively; P = 0.681; Table 3).

Mean ANC profiles in cycle 1
The overall incidence of febrile neutropenia was 9.8 % (6/61). In the DA-3031 3.6 and 6 mg dose groups, three (15.0 %) and one (5.0 %) patients, respectively, and two patients in the filgrastim group (9.5 %) experienced febrile neutropenia in cycle 1. No statistically significant differences were observed between the incidence of febrile neutropenia in any DA-3031 dose group and the filgrastim group (P = 0.681).
Safety
All of the participating patients reported at least one adverse event; however, most of the adverse events were attributed to complications from the TAC chemotherapy. Table 4 shows the adverse events that were considered to be possibly related to the study drugs by the investigators (22/61, 36.1 %), and all of these adverse events were grade 1 or 2. Musculoskeletal pain in the back, muscles, and extremities was the most frequently-reported adverse event in this study (Table 4). In the DA-3031 3.6 and 6 mg dose groups, four (21.1 %) and eight (38.1 %) patients, respectively, and six patients (28.6 %) in the filgrastim group experienced mild pain in cycle 1. Although the musculoskeletal pain in the DA-3031 group showed a dose effect, the difference among the study groups was not statistically different. Other adverse events attributable to the study drugs were headache (2/21 in the filgrastim group, 1/19 in the DA-3031 3.6 mg group), palpitation (1/21 in the filgrastim group), allergic reaction (1/21 in filgrastim group, 1/19 in DA-3031 3.6 mg group), and pelvic pain (1/19 in pegfilgrastim 3.6 mg group). One serious adverse event was reported by the patient who received 3.6 mg of DA-3031, however, the event was not considered to be related to the study drug.
Discussion
Pegfilgrastim (Neulasta, Amgen) is known to provide substantial protective effects against febrile neutropenia in patients receiving TAC, and prophylactic pegfilgrastim significantly reduced the incidence of febrile neutropenia from >20 % to 7 % [5]. The guidelines of the American Society of Clinical Oncology and the European Organization for Research and Treatment of Cancer recommend the use of colony-stimulating factor (CSF) in the first cycle of chemotherapy for treatments in which the risk of febrile neutropenia is greater than 20 % [12, 13]. Thus, the rational use of pegfilgrastim can protect patients from severe neutropenia and potentially life-threatening infection in an adjuvant TAC chemotherapy setting. The wide clinical use and pending expiry of the patent have stimulated the development of filgrastim biosimilars in order to reduce costs and improve accessibility to this agent. Currently, several agents have been approved and are being used in clinical practice [14].
In the present study, a single fixed dose of DA-3031, a pegfilgrastim biosimilar, demonstrated a similar efficacy and safety profile to that of the daily injections of the reference drug, filgrastim, in patients receiving TAC. The duration of grade 4 neutropenia in cycle 1 was 2.48, 2.20, and 2.05 days in the filgrastim, DA-3031 3.6 mg, and DA-3031 6 mg groups, respectively. In addition, the incidence of febrile neutropenia in cycle 1 was 9.5 %, 15.0 %, and 5.0 % in the filgrastim, DA-3031 3.6 mg, and DA-3031 6 mg groups, respectively. There was a decreasing trend in the duration of grade 4 neutropenia and the incidence of febrile neutropenia, which were not statistically significant.
The most common adverse event that was possibly related to the study drugs was musculoskeletal pain in the back, muscles, and extremities, which is a known effect of G-CSF. The severity of the pain was low grade, and the trend for higher incidence in the DA-3031 6 mg group was not statistically significant. No other unexpected adverse reactions related to the study medication were reported. Overall, administration of DA-3031 at two dose levels showed a safety profile comparable to that of the reference drug, filgrastim.
Pegfilgrastim is recommended for use as a single, fixed dose of 6 mg, as that dose was proven to provide the same benefit as daily filgrastim, regardless of body weight, in a randomized trial [9]. In this study, we used fixed doses of DA-3031 3.6 mg and 6 mg to determine the adequate dose in patients receiving myelosuppressive chemotherapy. The two different dosages were determined based on the results of a phase I study which showed a similar pharmacodynamics as determined by maximum ANC and the area under the curve of ANC for 264 h after study drug administration (unpublished data). Both doses of DA-3031 seemed to have a similar efficacy as the reference drug, filgrastim, for which the dosage was determined by body surface area as based on the approval labeling. Moreover, a predefined subgroup analysis according to body weight (≥60 kg versus < 60 kg) showed a comparable efficacy and safety profile in the two patient groups. Although the differences were not statistically significant, there was a decreasing trend of duration of G4 neutropenia and incidence of febrile neutropenia with 6 mg of DA-3031. In addition, the incidence of febrile neutropenia in the DA-3031 6 mg group seemed to be comparable to that of the original drug, Neulasta, in patients receiving TAC (5.0 % for DA 3031 6 mg versus 7 % for Neulasta) [5]. Thus, it is reasonable to compare 6 mg of DA-3031 to compare with reference drug in future clinical studies.
In this study, we could not determine if there was antibody formation against the study drug, DA-3031, since this study involved only the first cycle of treatment, and thus patients received daily filgrastim for subsequent treatment cycles as recommended by the Korean FDA. An additional phase III randomized trial comparing DA-3031 6 mg with daily injections of filgrastim in the same patient population as in this study is ongoing (NCT01674855). A confirmative study demonstrating clinical benefit and long-term safety, including antibody formation, may support the use of biosimilar pegfilgrastim, DA-3031, in clinical practice.
In conclusion, fixed doses of 3.6 mg or 6 mg DA-3031 have similar efficacy to daily injections of filgrastim in ameliorating grade 4 neutropenia and febrile neutropenia in patients receiving TAC chemotherapy for early stage breast cancer. DA-3031 showed a similar safety profile in the doses applied in this study.
References
Wood WC, Budman DR, Korzun AH, Cooper MR, Younger J, Hart RD, Moore A, Ellerton JA, Norton L, Ferree CR et al (1994) Dose and dose intensity of adjuvant chemotherapy for stage II, node-positive breast carcinoma. N Engl J Med 330(18):1253–1259. doi:https://doi.org/10.1056/NEJM199405053301801
Budman DR, Berry DA, Cirrincione CT, Henderson IC, Wood WC, Weiss RB, Ferree CR, Muss HB, Green MR, Norton L, Frei E 3rd (1998) Dose and dose intensity as determinants of outcome in the adjuvant treatment of breast cancer. The Cancer and Leukemia Group B. J Natl Cancer Inst 90(16):1205–1211
Eiermann W, Pienkowski T, Crown J, Sadeghi S, Martin M, Chan A, Saleh M, Sehdev S, Provencher L, Semiglazov V, Press M, Sauter G, Lindsay MA, Riva A, Buyse M, Drevot P, Taupin H, Mackey JR (2011) Phase III study of doxorubicin/cyclophosphamide with concomitant versus sequential docetaxel as adjuvant treatment in patients with human epidermal growth factor receptor 2-normal, node-positive breast cancer: BCIRG-005 trial. J Clin Oncol 29(29):3877–3884. doi:https://doi.org/10.1200/JCO.2010.28.5437
Citron ML, Berry DA, Cirrincione C, Hudis C, Winer EP, Gradishar WJ, Davidson NE, Martino S, Livingston R, Ingle JN, Perez EA, Carpenter J, Hurd D, Holland JF, Smith BL, Sartor CI, Leung EH, Abrams J, Schilsky RL, Muss HB, Norton L (2003) Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol 21(8):1431–1439. doi:https://doi.org/10.1200/JCO.2003.09.081
von Minckwitz G, Kümmel S, du Bois A, Eiermann W, Eidtmann H, Gerber B, Hilfrich J, Huober J, Costa SD, Jackisch C, Grasshoff ST, Vescia S, Skacel T, Loibl S, Mehta KM, Kaufmann M, German Breast G (2008) Pegfilgrastim +/− ciprofloxacin for primary prophylaxis with TAC (docetaxel/doxorubicin/cyclophosphamide) chemotherapy for breast cancer. Results from the GEPARTRIO study. Ann Oncol 19(2):292–298. doi:https://doi.org/10.1093/annonc/mdm438
Gogas H, Dafni U, Karina M, Papadimitriou C, Batistatou A, Bobos M, Kalofonos HP, Eleftheraki AG, Timotheadou E, Bafaloukos D, Christodoulou C, Markopoulos C, Briasoulis E, Papakostas P, Samantas E, Kosmidis P, Stathopoulos GP, Karanikiotis C, Pectasides D, Dimopoulos MA, Fountzilas G (2012) Postoperative dose-dense sequential versus concomitant administration of epirubicin and paclitaxel in patients with node-positive breast cancer: 5-year results of the Hellenic Cooperative Oncology Group HE 10/00 phase III trial. Breast Cancer Res Treat 132(2):609–619. doi:https://doi.org/10.1007/s10549-011-1913-4
Findlay B, Tonkin K, Crump M, Norris B, Trudeau M, Blackstein M, Burnell M, Skillings J, Bowman D, Walde D, Levine M, Pritchard KI, Palmer MJ, Tu D, Shepherd L (2007) A dose escalation trial of adjuvant cyclophosphamide and epirubicin in combination with 5-fluorouracil using G-CSF support for premenopausal women with breast cancer involving four or more positive nodes. Ann Oncol 18(10):1646–1651. doi:https://doi.org/10.1093/annonc/mdm277
Zamboni WC (2003) Pharmacokinetics of pegfilgrastim. Pharmacotherapy 23(8 Pt 2):9S–14S
Green MD, Koelbl H, Baselga J, Galid A, Guillem V, Gascon P, Siena S, Lalisang RI, Samonigg H, Clemens MR, Zani V, Liang BC, Renwick J, Piccart MJ (2003) A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 14(1):29–35
Fox E, Widemann BC, Hawkins DS, Jayaprakash N, Dagher R, Aikin AA, Bernstein D, Long L, Mackall C, Helman L, Steinberg SM, Balis FM (2009) Randomized trial and pharmacokinetic study of pegfilgrastim versus filgrastim after dose-intensive chemotherapy in young adults and children with sarcomas. Clin Cancer Res 15(23):7361–7367. doi:https://doi.org/10.1158/1078-0432.CCR-09-0761
Lee DH, Suh C, Park K, Kim TW, Kim JG, Kim WS, Kang WK, Heo DS, Bang YJ, Kim NK (1999) The effectiveness and safety of DA-3030 (rhG-CSF) for chemotherapy-induced neutropenia: a randomized controlled trial. J Korean Cancer Assoc 31(5):995–1002
Aapro MS, Cameron DA, Pettengell R, Bohlius J, Crawford J, Ellis M, Kearney N, Lyman GH, Tjan-Heijnen VC, Walewski J, Weber DC, Zielinski C, European Organisation for R, Treatment of Cancer Granulocyte Colony-Stimulating Factor Guidelines Working P (2006) EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphomas and solid tumours. Eur J Cancer 42(15):2433–2453. doi:https://doi.org/10.1016/j.ejca.2006.05.002
Smith TJ, Khatcheressian J, Lyman GH, Ozer H, Armitage JO, Balducci L, Bennett CL, Cantor SB, Crawford J, Cross SJ, Demetri G, Desch CE, Pizzo PA, Schiffer CA, Schwartzberg L, Somerfield MR, Somlo G, Wade JC, Wade JL, Winn RJ, Wozniak AJ, Wolff AC (2006) 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 24(19):3187–3205. doi:https://doi.org/10.1200/JCO.2006.06.4451
Niederwieser D, Schmitz S (2011) Biosimilar agents in oncology/haematology: from approval to practice. Eur J Haematol 86(4):277–288. doi:https://doi.org/10.1111/j.1600-0609.2010.01566.x
Acknowledgments
This study was funded by Dong-A Pharm. Co., LTD.
Conflicts of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Park, K.H., Sohn, J.H., Lee, S. et al. A randomized, multi-center, open-label, phase II study of once-per-cycle DA-3031, a biosimilar pegylated G-CSF, compared with daily filgrastim in patients receiving TAC chemotherapy for early-stage breast cancer. Invest New Drugs 31, 1300–1306 (2013). https://doi.org/10.1007/s10637-013-9973-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-9973-4