
Abstract
Purpose
Lysine-specific demethylase 1 (LSD1) was highly expressed in several malignancies and had been implicated in pathological processes of cancer cells. However, the role of LSD1 in colorectal cancer (CRC) carcinogenesis, prognosis and treatment remains uncharacterized.
Methods
In this study, we examined LSD1 expression in paraffin-embedded CRC specimens from 295 patients, including 65 patients with paired samples of colorectal carcinoma, adjacent adenoma and normal colorectal tissues. Using an LSD1 inhibitor, CBB1003, as a probe, we studied the association between LSD1 and leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5), a CRC stem cell marker involved in carcinogenesis. The anti-tumor effects of CBB1003 on CRC cells were also examined.
Results
LSD1 expression was significantly elevated in colorectal tumor tissues compared with adjacent adenoma and normal colorectal tissues (P < 0.001), and LSD1 levels were significantly correlated with an advanced AJCC T stage (P = 0.012) and distant metastasis (P = 0.004). CBB1003 inhibited CRC cell proliferation and colony formation. In cultured CRC cells, inhibiting LSD1 activity by CBB1003 caused a decrease in LGR5 levels while overexpression of LGR5 reduced CBB1003-induced cell death. We also observed the inactivation of β-catenin/TCF signaling after CBB1003 treatment, consistent with the positive correlations among LSD1, LGR5, β-catenin and c-Myc expression in human colon tumor and adenoma tissues.
Conclusion
Our result suggested that LSD1 overexpression promotes CRC development and that the LSD1 inhibitor inhibits CRC cell growth by down-regulating LGR5 levels and inactivates the Wnt/β-catenin pathway. Thus, LSD1 and its inhibitor might provide a new target or a useful strategy for therapy of CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is one of the most common types of malignancy and the most frequent etiologies of cancer mortality worldwide (Jemal et al. 2011; Siegel et al. 2012). It has become a major health problem, and thus, extensive CRC-focused research has been conducted to improve the clinical outcome of CRC. Genetic alterations are important contributors to the multistep carcinogenesis process in sporadic and hereditary CRC (Saif and Chu 2010). However, in the past decade, the roles of epigenetic alterations, which are the regulation of gene expression that does not involve a change in the DNA sequence of the cell, have also begun to receive more attention in the development of CRC. Histone modification is one type of epigenetic alterations (Van Engeland et al. 2011) that includes histone acetylation, deacetylation, methylation and demethylation. Aberrant histone modifications have been shown to contribute to cancer initiation and progression by altering the pattern of gene transcription such as inactivating tumor suppressor genes (Konishi and Issa 2007) and by inducing genome instability due to direct effects in the higher order of chromatin, chromosome condensation and mitotic disjunction (Bannister and Kouzarides 2011).
Lysine-specific demethylase 1 (LSD1), the first histone demethylase that was discovered as a flavin adenine dinucleotide (FAD)-dependent amine oxidase, catalyzes the demethylation of histone H3-K4 and K9 (Shi 2007; Shi et al. 2004; Forneris et al. 2008). It is composed of several domains, including a SWIRM domain, a conserved motif shared by many chromatin regulatory complexes, an amine oxidase domain and a Tower domain found in BRCA2 (Shi and Whetstine 2007; Mimasu et al. 2008; Yang et al. 2007). The expression of LSD1 has been associated with tumor recurrence during therapy in various cancers (Lim et al. 2010; Hayami et al. 2011; Chen et al. 2007). Tissue cDNA microarray analysis also revealed up-regulation of LSD1 in lung and colorectal carcinomas (Hayami et al. 2011). The dysregulation of LSD1 activity in human has already been confirmed in several cancers (Lim et al. 2010; Wu et al. 2012; Schenk et al. 2012; Lv et al. 2012; Kauffman et al. 2011; Schutle et al. 2009; Huang et al. 2007). However, the expression and significance of LSD1 in CRC has not been fully elucidated. Moreover, CBB1003, a known LSD1 inhibitor, was reported to selectively inhibit the growth of pluripotent embryonic stem cells (ESCs) and pluripotent germ tumor cells (Wang et al. 2011). However, whether CBB1003 has any effect on the CRC cells remains unknown.
The leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5), also known as GPR49, belongs to the G-protein-coupled receptor family of proteins and is a target of Wnt signaling (O’Brien et al. 2007; Ricci-Vitiani et al. 2007; Visvader and Lindeman 2012). Barker et al. discovered that LGR5 is a marker of adult stem cells in the intestine (Visvader and Lindeman 2012). Subsequent studies observed that intestinal tumors arise from LGR5-positive cells, suggesting LGR5 marks intestinal cancer stem cells (Barker et al. 2009). Kemper et al. determined that LGR5 is a functional human CRC stem cells (CSCs) marker (Kemper et al. 2012). Our previous study showed that overexpression of LGR5 is associated with CRC progression and that knocking down LGR5 suppressed CRC cell proliferation and colony formation ability (Hsu et al. 2013).
Both LSD1 and LGR5 are associated with tumorigenesis, and both proteins have been implicated in the maintenance of cancer stem cells. However, the relationship between these two proteins has not been elucidated. In this study, we found that LSD1 expression was significantly up-regulated in CRC tissues compared with colorectal adenoma and normal colorectal tissue. In addition, high expression levels of LSD1 were significantly associated with high expression levels of LGR5 in CRC tissues. Furthermore, we observed that a known LSD1 inhibitor (CBB1003) inhibited growth of CRC cells by down-regulating LGR5 and subsequent inactivating the Wnt/β-catenin pathway. Using LSD1 inhibitors as a probe, we established that LSD1 involves in CRC tumorigenesis via regulation of LGR5 expression.
Materials and methods
Study population
Paraffin-embedded colorectal tumor specimens were obtained from a consecutive series of 295 patients who underwent radical tumor resection for colorectal carcinomas between 2008 and 2009 at the Department of Surgery, Chang Gung Memorial Hospital Tao-Yuan, Taiwan. These specimens were used in our previous LGR5 study [28]. All patients were followed every week to every 3 months until March 1, 2012, or until death. Mean follow-up time was 52 months, with a standard deviation of 18.4 months. The age at diagnosis of the patients ranged from 28 to 93 years (mean 63.5 ± 14.0). The associated subsites were the colon (182 patients) and rectum (113 patients). Tumor stages (based on the AJCC 2002 guidelines) included stage I (60 patients), stage II (91 patients), stage III (67 patients) and stage IV (77 patients). In addition, paired paraffin-embedded samples of colorectal carcinoma, adjacent adenomas and normal colorectal tissues were obtained from 65 of 295 patients. The adenoma described in the present study belonged to adenoma with low-grade dysplasia (most common adenoma, Category 3) according to Vienna classification of gastrointestinal epithelial neoplasia (Rubio et al. 2006). Written informed consent from all patients regarding tissue sampling was obtained. The study protocol was approved by the Institutional Review Board at Chang Gung Memorial Hospital (101-1990B).
Immunohistochemical staining
The immunohistochemical staining of LSD1, c-Myc, β-catenin and LGR5 was conducted using 5-μm sections from paraffin-embedded tissue blocks. The sections were de-paraffinized by xylene, rehydrated in graded concentrations of ethanol and boiled in a microwave oven for 5 min. Slides of consecutive sections were incubated with diluted LSD1 (1:400 dilution, Cell Signaling, Cat. No. #2184), c-Myc (1:1,000 dilution, Cell Signaling, Cat. No. #9402), β-catenin (1:1,000 dilution, Cell Signaling, Cat. No. #2698) and LGR5 antibody (1:100 dilution, Origene, Cat. No. TA503316) at room temperature for 2 h. After incubation, the slides were washed three times with PBS, incubated with horseradish peroxidase (HRP)-conjugated antibody (Invitrogen, Carlsbad, CA, USA) at room temperature for 10 min and developed by the addition of 3,3′-diaminobenzidine tetrahydrochloride (DAB) reagent (Dako, Glostrup, Denmark), with the chromogen and hematoxylin as the counter stain. Images of stained slides were obtained using a ScaneScope CT automated slide-scanning system (Aperio Technologies, Vista, CA, USA).
Quantification of LSD1 and Lgr5 immunohistochemical staining
All pathologic specimens were reviewed by dedicated pathologists at Chang Gung Memorial Hospital. LGR5 expression and staining intensity were evaluated in our previous study [28]. In the present study, evaluation of LSD1 expression and staining intensity was performed by Dr. Y. Liang, a pathologist in the Pathology Core of the Molecular Medicine Research Center (MMRC) of Chang Gung University (CGU), without prior knowledge of the clinical origin of the specimen. LSD1 expression was scored based on both the staining intensity and the percentage of stained cells. Absent, weak, moderate and strong LSD1 staining intensities were scored as 0, 1, 2, and 3, respectively. The percentage of cells that stained at a specific level was manually evaluated. The final LSD1 staining score was calculated as the sum of the percentage of stained cells multiplied by the intensity scores, resulting in an LSD1 immunohistochemistry score that ranged from 0 (no LSD1-positive cell in the entire slide) to 300 (all cells showed strong LSD1 staining) (Chang et al. 2010).
Cell culture and transfection
The CRC cell lines LoVo, HCT116 and HT29 were purchased from the American Type Culture Collection (ATCC, VA, USA) and the Bioresource Collection and Research Center (BCRC, Taiwan) and maintained in RPMI-1640 (Gibco, USA) supplemented with 10 % fetal bovine serum. The medium was changed every 3 days. For performing the LSD1 knockdown studies, LoVo and HT29 cells were transfected with 5 nM synthetic siRNA specifically targeting human LSD1 (si-LSD1) (5′-GGUCUUGGAGGGAAUCCUAtt-3′, Ambion, siRNA ID: 617) or 5 nM negative control siRNA (si-Ctrl) (Ambion, Cat. No. #4390483) using RNAiMax (Invitrogen, USA) according to the manufacturer’s instructions. Forty-eight hours after transfection, the cells were trypsinized and reseeded at an appropriate density for proliferation and colony formation assays. For LGR5 gain-of-function studies, HCT116 cells were transfected with 10 µg of plasmid expressing full-length human LGR5 (p-LGR5) (Cat. No. SC117852, OriGene) or LacZ control vector (LacZ) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. Twenty-four hours after transfection, cells were trypsinized for drug sensitivity assays.
RNA extraction and quantitative reverse transcription polymerase chain reaction (q-PCR)
RNA extraction was performed using TRIZOL® reagent (Ambion, USA) following the manufacturer’s instructions. Forty-eight hours after treatment, the total RNA of colon cancer cells was harvested for the quantitative measurement of mRNA; reverse transcription and q-PCR quantification were performed using previously established techniques (Hsu et al. 2013). About 1 µl of diluted RT product was used as the template in a 10-µl q-PCR reaction mixture using the following conditions: 95 °C for 10 min, followed by 45 cycles of 95 °C for 15 s and 60 °C for 1 min, and a dissociation stage. All of the q-PCR reactions were performed using an ABI Prism 7500 Fast Real-Time PCR system (Foster City, CA, USA).
Drug sensitivity assay
CBB1003 was obtained from Sigma and was dissolved according to the manufacturer’s instructions. Forty-eight hours after transfecting with expression vector, HCT116 cells were seeded at 1 × 104 cell/well in 96-well plates and treated with serial dilutions of LSD1 inhibitors in triplicate. Cell viability was measured using DAPI stain and counted using InCell 1000 analyzer. The percentage of cell survival in treated cells was normalized with untreated controls. IC50 was calculated using Prism 5.0 (GraphPad, USA).
Cell proliferation and colony formation assays
For proliferation analysis, HCT116 cells treated with CBB1003 were seeded in 96-well plates at 10,000 cells/well and cultured for 5 days. Cell numbers per well were counted daily for 5 days using the IN Cell Analyzer 1000 (GE Healthcare) after DAPI staining. For the colony formation assay, CBB1003-treated HCT116 cells were seeded in triplicate at 100 cells per well in 6-well plates. After 14 days, the plates were fixed, and colonies were stained using crystal violet. Colony numbers were quantified using ImageJ software.
Cell cycle analysis
To determine the effect of CBB1003 on the cell cycle distribution, HCT116 cells were seeded in 6-cm dishes and treated with 250 µM CBB1003 for 48 h. These cells were then stained with propidium iodide (PI) buffer. A total of 10,000 events were analyzed by flow cytometry using an excitation wavelength of 488 nm and an emission wavelength of 610 nm.
Promoter activity assay
To measure β-catenin/TCF-4 transcriptional activity, a pair of luciferase reporter plasmids (TOPflash and FOPflash; Upstate Biotechnology) were used. The pRL-TK luciferase reporter gene plasmid (Promega) was co-transfected for normalization purpose. Transient transfection was performed using a Lipo2000 (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. The cells were treated with CBB1003 for 24 h and, after 24 h, were subsequently co-transfected with the TOPflash luciferase reporter (or mutant control FOP flash vector) and pRL-TK. After an additional 24 h of incubation, the cells were harvested for luciferase activity measurement using the dual-luciferase reporter assay system (Promega). Three replicate experiments were performed.
Immunoblotting assay
Protein extracts were harvested using centrifugation, boiled with a 5× Laemmli buffer for 10 min before being loaded onto an SDS-PAGE and then transferred onto the equilibrated PVDF membranes. After blockade with 5 % skimmed milk in TBS–Tween-20 for 1 h, the membrane was incubated with anti-LSD1 (1:2,000 dilution), anti-LGR5 (1:500 dilution), anti-H3K4me2 (1:3,000 dilution, Cell Signaling, Cat. No. #9725), anti-H3K9me2 (1:3,000 dilution, Cell Signaling, Cat. No. #9753), anti-Histone H3 (1:10,000 dilution, Cell Signaling, Cat. No. #9715) or anti-GAPDH (1:10,000 dilution, BioWorld, Cat. No. AP0063) at 4 °C overnight. After washing with TBS–Tween-20, blots were probed for 2 h with a horseradish peroxidase-labeled goat anti-rabbit or anti-mouse IgG secondary antibody. Protein bands were visualized on X-ray film using an ECL and Western blotting detection system.
Statistical analysis
PASW version 18.0 for Windows (SPSS Inc., Chicago, IL., USA) was used for all calculations. Chi-square test was applied to examine the independence of LSD1 expression and clinical and pathologic parameters. The Kaplan–Meier method was used to estimate survival curves. Differences between survival curves were assessed using the log-rank test. The survival endpoint for cancer-specific survival (CSS) was the time from surgery until cancer-related death. Death caused by other factors was counted as a censored event. Dose–response curves were fitted by nonlinear regression, and IC50 values were calculated using Prism software (version 5, GraphPad). The statistical significance was evaluated using two-tailed t test. All P values were two-sided, and results were considered significant when P < 0.05.
Results
LSD1 expression in normal colons, adenomas and colorectal tumor tissues
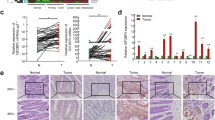
LSD1 expression has not been analyzed in neoplastic colorectal adenoma lesions before. Therefore, we compared LSD1 expression in colorectal tumors with the adjacent colorectal adenomas and normal colorectal areas. Using immunohistochemistry, we analyzed the expression of LSD1 protein in 65 paired paraffin-embedded samples of colorectal carcinoma, adjacent adenoma and normal colorectal tissues. Immunohistochemical staining of the colorectal specimens showed that LSD1 protein was mainly localized in the nuclei of cells. In healthy epithelia located in adjacent normal colorectal areas, LSD1-positive cells were rarely detected and constituted <1 % of the entire epithelia population. In the adenoma sections, nuclear LSD1 expression was stronger than that in normal colon tissue. LSD1 protein was abundantly expressed in colorectal carcinoma (Fig. 1a). Analysis of LSD1 scores in 65 paired normal tissues and colorectal tumors revealed a statistically significant difference in LSD1 expression (P < 0.001, Fig. 1b), with mean LSD1 scores of 76 ± 17 and 168 ± 29 for normal and CRC tissues, respectively. In addition, analysis of LSD1 scores in 65 paired normal and adenoma colorectal tissues also revealed a statistically significant difference in LSD1 expression (P < 0.001, Fig. 1b), with mean LSD1 scores of 76 ± 17 and 113 ± 31 for normal tissues and colorectal adenomas, respectively. Moreover, analyzing LSD1 scores of 65 paired colorectal adenomas and carcinomas revealed a statistically significant difference in LSD1 expression (P < 0.001, Fig. 1b), with mean LSD1 scores of 113 ± 31 and 168 ± 29 for colorectal adenomas and carcinomas, respectively. These data suggest that LSD1 level is increased in colon adenoma tissues and the level is further elevated in CRC tissues.

Immunohistochemical analysis of lysine-specific demethylase 1 (LSD1) in human colorectal tissues. a Representative LSD1 staining in de-paraffinized sections of normal colorectal, colorectal adenoma and carcinoma tissues. b Expression levels of LSD1 in paired normal colorectal tissues, colorectal adenoma and carcinoma tissues (n = 65). (C) Expression levels of LSD1 in colorectal cancer (CRC) according to AJCC stage. (*** P < 0.001, *P < 0.05)
Relationship between LSD1 expression and clinicopathological characteristics in CRC patients
The LSD1 scores of CRC tumors were positively correlated with advanced AJCC stage, with mean scores of 139 ± 29 and 148 ± 29 for stages I–III and stage IV samples, respectively (Fig. 1c, P = 0.018). No statistically significant difference in LSD1 scores was observed among stages I–III colorectal tumors (Fig. 1c).
To investigate whether increased LSD1 expression is correlated with various clinicopathological characteristics of CRC, the samples were grouped as LSD1 low (LSD1 score <160, n = 189) and LSD1 high (LSD1 score ≥160, n = 106) based on a cutoff score corresponding to 53 % cells with strong LSD1 staining or 80 % of cells with moderate LSD1 staining. Among the clinical parameters, LSD1 expression level was found significantly associated with T stage (P = 0.012) and M stage (P = 0.004) (Table 1). Thus, higher level of LSD1 expression is associated with advanced tumor stage.
During follow-up, Kaplan–Meier and multivariate analysis revealed no significant associations between CSS and LSD1 score (Supplementary Fig. 1 and Table 1). However, the estimated 3- and 5-year CSS rates in LSD1-low group were significantly higher than those in LSD1-high group (3-year CSS rate: LSD1-low vs LSD1-high = 80.4 vs 65.1 %, P = 0.004; 5-year CSS rate: LSD1-low vs LSD1-high = 67.7 vs 42.5 %, P < 0.001) (Supplementary Fig. 1).
The LSD1 inhibitor, CBB1003, is required for the survival of cultured CRC cells
LSD1 is a demethylase, and it catalyzes the specific demethylation of H3K4me1/2 or H3K9me1/2. Here, we used Western blotting to detect changes in the global methylation levels of H3K4me1/2 and H3K9me1/2 in the CRC cells under CBB1003 treatment. The results showed that loss of LSD1 in HCT116 cells did not affect the global levels of H3K4me2 or H3K9me2 (Supplementary Fig. 2). Because LSD1 expression level was positively associated with CRC formation, we investigated whether LSD1 activity is required for the survival of cultured CRC cells, HCT116. Blocking LSD1 activity by CBB1003 induced a dose-dependent growth inhibition in HCT116 cells (Fig. 2a). The CBB1003 IC50 was 250.4 µM for HCT116 cells. These results suggest that LSD1 enzymatic activity is required for the growth of CRC cells. We also used CBB1003 to investigate the role of LSD1 activity on the growth and cell cycle progression of cultured HCT116 cells. Cell proliferation was determined using both growth curve analysis and colony formation assay. As shown in Fig. 2b, c, CBB1003 significantly suppressed the growth and colony formation capability compared with the vehicle control (DMSO) in HCT116 cells. Flow cytometry analysis revealed that CBB1003 treatment (250 µM, 3 days) caused a change in cell cycle distribution, with the G1 phase population increased from 44.5 to 56.0 % while the S phase population decreased from 47.2 to 36.8 % and G2/M phase population decreased from 8.3 to 7.2 % (Fig. 2d).

The effects of CBB1003 (LSD1 inhibitor) on the growth and survival of cultured HCT116 cells. HCT116 cells were seeded at 1 × 104 cell/well in 96-well plates and treated with serial dilutions of CBB1003 for 3 days in triplicate. Cell viability was measured using DAPI stain and counted using an InCell 1000 analyzer. The percentage of cell survival in treated cells was normalized with untreated controls. a The CBB1003 IC50 was 250.4 µM in HCT116 cells. b Effects of CBB1003 on the growth of cultured CRC cells. Cultured CRC cells were seeded on 96-well plates and treated with CBB1003 (250 µM). Cell numbers were determined daily from day 1 to day 5. c HCT116 cells were treated with CBB1003 (250 µM) or DMSO and seeded at 100 cells per well in 6-well plates. After 14 days, the plates were fixed, and colonies were stained with crystal violet. Colony numbers were quantified using ImageJ software. Each experimental condition was performed in duplicate wells, and three independent experiments were performed. d Cell cycle distribution of HCT116 cells after propidium staining was analyzed using flow cytometry. HCT116 cells were treated with CBB1003 (250 µM) for 3 days. The image represents the staining profile for 10,000 cells per experiments
CBB1003 suppresses LGR5 expression and Wnt signaling
Our previous study showed that knocking down Lgr5 suppressed CRC cell proliferation and colony formation ability, while overexpression of Lgr5 in CRC cells increased cell proliferation (Hsu et al. 2013). Therefore, we tested whether the LSD1 inhibitor inhibits cell growth by reducing LGR5 expression. Treating HCT116 cells with CBB1003 caused a significant decrease in the transcript level and protein level of LGR5 (Fig. 3a, c). Similar decrease in LGR5 protein and transcript level was observed when HT29 and LoVo cells were treated with specific siRNA to deplete endogenous LSD1 (Fig. 3b, d). As HCT116 cells exhibit active β-catenin/TCF-4 transcriptional activity, we further investigated whether LSD1 regulates the Lgr5 expression level and affects the Wnt/β-catenin signaling. HCT116 cells were transiently transfected using a TCF reporter plasmid, TOPflash, which consists of three TCF-binding sites upstream of the a minimal tk promoter and the luciferase open reading frame, or a control plasmid, FOPflash, which is identical to TOPflash except that it contains mutant inactive TCF-binding sites. CBB1003 potently inhibited the luciferase activity in cells expressing the TOPflash reporter gene (Fig. 3e). In contrast, inhibiting LSD1 activity by CBB1003 showed no effect on FOPflash activity, indicating that inhibiting LSD1 activity specifically blocked the β-catenin/TCF signaling. Consequently, this result suggests that the LSD1 inhibitor reduced β-catenin/TCF signaling and decreased LGR5 expression in CRC cells.

Effects of LSD1 level and activity on leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5) expression and Wnt signaling in CRC cells. HCT116 cells were treated with 250 µM CBB1003. HT29 and LoVo cells were treated with 10 nM si-Ctrl or si-LSD1 for 48 h. At 48 h post-treatment, the cells were harvested and mRNA (a and b) and protein (c and d) expression was determined using qPCR and Western blot. e After CBB1003 treatment, the HCT116 cells were co-transfected with reporter genes harboring Tcf-4 binding sites (TOP flash) combined with pRL-TK. Luciferase activity was determined 24 h post-transfection and normalized against values for the corresponding pRL-TK activity
LGR5 prevents CBB1003-induced apoptosis in CRC cells
LGR5 expression has been implicated in CRC cell survival. Therefore, we tested whether ectopic expression of LGR5 may rescue CBB1003-induced cell growth inhibition. Treating HCT116 cells with the LGR5 plasmid to increase LGR5 expression significantly reduced CBB1003-induced cell death (Fig. 4). The observation that LGR5 can rescue growth inhibition caused by LSD1 inhibitor suggests that LSD1 and LGR5 may function through the same pathway to regulate the growth of CRC cells.

LGR5 overexpression prevents CBB1003-induced growth inhibition in CRC cells. HCT116 cells were transfected with LacZ and LGR5 plasmids for 24 h and reseeded at 2000 cells/well. Cells were treated with DMSO or CBB1003 (250 µM). Cell numbers were determined daily using DAPI stain and an InCell 1000 analyzer
Positive correlation between LSD1, LGR5, β-catenin and c-Myc expression in human colorectal tumor tissues
To confirm the hypothesis that LSD1 positively regulates the LGR5 level and Wnt signaling pathway in vivo, we examined the expression patterns of LSD1, LGR5, β-catenin and c-Myc in serial sections of human colon carcinoma tissues by immunohistochemical staining. The expression levels of LSD1, LGR5, β-catenin and c-Myc were much lower in adjacent normal tissues than in tumor tissues (Fig. 5). A similar result was observed in a comparison of colorectal adenoma tissues and carcinoma tissues (Fig. 5). In addition, a significantly positive correlation between LSD1 and LGR5 expression was observed in CRC tissues (P = 0.001, Table 1). All of these data were consistent with the data obtained from cultured CRC cells, providing strong support for the relationship between LSD1 and LGR5 in CRC tissues.

Positive correlation between LSD1, c-Myc, β-catenin and LGR5 expression in human colorectal tumor tissues. Expression levels of LSD1, LGR5, β-catenin and c-Myc in consecutive sections from normal colon, adenoma and CRC tumor tissues
Discussion
The adenoma–carcinoma sequence accounts for 60 % of CRC tumorigenesis (Perea et al. 2011). In this study, we used immunohistochemistry to demonstrate that LSD1 protein was elevated in colorectal carcinoma tissues than in adjacent normal colorectal tissues and colorectal adenoma tissues. Moreover, we observed a gradual increase of LSD1 expression during tumor formation from normal colorectal tissue to adenoma, and to tumor. This increase of LSD1 in adenoma had not been reported previously, suggesting that LSD1 might play a crucial role in the early process of colorectal tumorigenesis. The Western blot analysis of Huang et al. (2013) supported the observation that LSD1 is overexpressed in colorectal carcinomas compared with normal colorectal tissues.
Our study showed that elevated LSD1 expression levels were significantly correlated with advanced CRC, including advanced AJCC T stage and distant metastasis. This was compatible with the results of Jie et al. (2013). Moreover, 3- and 5-year CSS rates in LSD1-low group were significantly higher than those in LSD1-high group. However, high LSD1 levels were not found associated with shorter CSS in CRC patients when using Kaplan–Meier method. The lack of statistical significance in CSS is likely due to the short follow-up period (4–5 years). The survival curves in our study were similar to that of the first 5 years of the 7- to 8-years follow-up period of Jie et al., who reported that high LSD1 expression was correlated with poor overall survival in CRC patients. This suggests the difference in CSS between the LSD1-low and LSD1-high groups might become more significant after extending the follow-up period.
LSD1 is a demethylase, and it catalyzes the specific demethylation of H3K4me1/2 or H3K9me1/2. Therefore, blocking LSD1 activity by LSD1 inhibitors is expected to increase the methylation levels of H3K4me1/2 or H3K9me1/2. However, we did not observe any significant change in global methylation levels upon LSD1 inhibitor treatment, suggesting that some unknown mechanism may compensate for the loss of LSD1 demethylase catalytic activity. The result is similar to the study of Huang et al. (2013). Although Hunag et al. (2013) found that LSD1 contributes to colorectal tumorigenesis via activation of the Wnt/β-catenin pathway by down-regulating Dickkopf-1 (DKK1) and Ding et al. (Ding et al. 2013) reported that LSD1 promotes metastasis of colon cancer cells by down-regulating the expression of CDH-1, the role of LSD1 in CRC stem cell has not been elucidated. CBB1003, a known LSD1 inhibitor, was reported to selectively inhibit the proliferation of pluripotent ESCs and germ tumor cells that express pluripotent stem cell proteins (Wang et al. 2011). LSD1 inactivation was also found to selectively impair the growth of lung, breast and ovarian carcinoma cells that express Sox2, a pluripotent stem cell marker (Zhang et al. 2013). LGR5, a functional stem cell marker of CRC, can regulate the proliferation and colony formation of CRC cells (Kemper et al. 2012; Hsu et al. 2013). We therefore used CBB1003 as a probe to study the associations between LSD1 and LGR5 in CRC carcinogenesis. In addition, we evaluated the anti-tumor efficiency of CBB1003 in CRC cells. CBB1003 was found to inhibit cell growth and colony formation of CRC cells and to induce growth arrest of CRC cells at G1 phase; this has not been reported previously. We also found that inhibiting LSD1 activity in HCT116 cells by CBB1003 caused a significant decrease in the transcript and protein levels of LGR5. Immunohistochemical staining in CRC tissues confirmed this significant and positive correlation. Moreover, overexpressing LGR5 in CRC cells significantly reduced CBB1003-induced cell death. These results suggest that CBB1003 inhibits cell proliferation by depleting LGR5 expression in CRC cells.
Carmon et al. (2012) reported that LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/β-catenin signaling. In our study, we demonstrated that CBB1003 specifically inhibited Wnt/β-catenin/TCF signaling in CRC cells. In immunohistochemical study, the expression levels of LSD1, LGR5, c-Myc and β-catenin were much lower in adjacent normal and adenoma colorectal tissues than in colorectal tumor tissues, suggesting that LSD1 regulated β-catenin/TCF signaling and affected LGR5 expression in CRC cells.
LSD1 and its inhibitor have been reported to target cancer stem cell markers in several cancers (Wang et al. 2011; Zhang et al. 2013; Amente et al. 2013). Our study revealed that LSD1 is involved in CRC tumorigenesis by up-regulating the expression of LGR5, a functional CRC stem cell marker, which had been confirmed to promote cancer stemness in CRC cells in our previous study (Liu et al. 2013). Wang et al. reported that LSD1 inhibition can suppress Oct4 expression in F9 cells (Wang et al. 2011). Furthermore, Shen et al. found that microRNA-142-3p (miR-142-3p) inhibits the expression of LGR5 in colon cancer cells and OCT4 suppressed miR-142-3p (Shen et al. 2013). These results suggest that LSD1 may indirectly regulate LGR5 through Oct4 and miR-142-3p. While the mechanism through which LSD1 regulates LGR5 expression or β-catenin/TCF signaling requires further clarification, our results suggest that LSD1 may be involved in the maintenance of cancer stem cells in CRC. To the best of our knowledge, this is the first report that elucidates the impact of LSD1 on LGR5 and the anti-tumor efficiency of CBB1003 in CRC cells. Furthermore, these data suggest that LSD1 inhibition is a potential strategy for treating CRC.
References
Amente S, Lania L, Majello B (1829) The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim Biophys Acta. 10:981–986
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21:381–395
Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H et al (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457:608–611
Carmon KS, Lin Q, Gong X, Thomas A, Liu Q (2012) LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/β-catenin signaling. Mol Cell Biol. 32(11):2054–2064
Chang KP, Kao HK, Yen TC, Chang YL, Liang Y, Liu SC et al (2010) Overexpression of macrophage inflammatory protein-3alpha in oral cavity squamous cell carcinoma is associated with nodal metastasis. Oral Oncol 47:108–113
Chen PP, Li WJ, Wang Y, Zhao S, Li DY et al (2007) Expression of Cyr61, CTGF, and WISP-1 correlates with clinical features of lung cancer. PLoS ONE 2:e534
Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, Zhang Y, Yan ZS (2013) LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer. 109(4):994–1003
Forneris F, Binda C, Battaglioli E, Mattevi A (2008) LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem Sci 33:181–189
Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M et al (2011) Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer 128:574–586
Hsu HC, Liu YS, Tseng KC, Hsu CL, Liang Y et al (2013) Overexpression of Lgr5 correlates with resistance to 5-FU-based chemotherapy in colorectal cancer. Int J Colorectal Dis. 28(11):1535–1546
Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB et al (2007) Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci USA 104:8023–8028
Huang Z, Li S, Song W, Li X, Li Q, Zhang Z et al (2013) Lysine-specific demethylase 1 (LSD1/KDM1A) contributes to colorectal tumorigenesis via activation of the Wnt/β-catenin pathway by down-regulating Dickkopf-1 (DKK1). PLoS One 8:7
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Jie D, Zhongmin Z, Guoqing L, Sheng L, Yi Z, Jing W, Liang Z (2013) Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer. Dig Dis Sci. 58(6):1581–1589
Kauffman EC, Robinson BD, Downes MJ, Powell LG, Lee MM et al (2011) Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol Carcinog 50:931–944
Kemper K, Prasetyanti PR, De Lau W, Rodermond H, Clevers H et al (2012) Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 30:2378–2386
Konishi K, Issa JP (2007) Targeting aberrant chromatin structure in colorectal carcinomas. Cancer J 13:49–55
Lim S, Janzer A, Becker A (2010) Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31:512–520
Liu YS, Hsu HC, Tseng KC, Chen HC, Chen SJ (2013) Lgr5 promotes cancer stemness and confers chemoresistance through ABCB1 in colorectal cancer. Biomed Pharmacother. 67(8):791–799
Lv T, Yuan D, Miao X, Lv Y, Zhan P et al (2012) Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 7:e35065
Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S (2008) Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 A. Biochem Biophys Res Commun 366:15–22
O’Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110
Perea J, Lomas M, Hidalgo M (2011) Molecular basis of colorectal cancer: towards an individualized management. Rev Esp Enferm Dig 103(1):29–35
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M et al (2007) Identification and expansion of human colon-cancer-initiating cells. Nature 445:111–115
Rubio CA, Nesi G, Messerini L, Zampi GC, Mandai K, Itabashi M, Takubo K (2006) The Vienna classification applied to colorectal adenomas. J Gastroenterol Hepatol 21(11):1697–1703
Saif MW, Chu E (2010) Biology of colorectal cancer. Cancer J 16:196–201
Schenk T, Chen WC, Gollner S, Howell L, Jin L et al (2012) Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 18:605–611
Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J et al (2009) Lysine specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res 69:2065–2071
Shen WW, Zeng Z, Zhu WX, Fu GH (2013) MiR-142-3p functions as a tumor suppressor by targeting CD133, ABCG2, and Lgr5 in colon cancer cells. J Mol Med (Berl). 91(8):989–1000
Shi Y (2007) Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet 8:829–833
Shi Y, Whetstine JR (2007) Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 25:1–14
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA et al (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119:941–953
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics. CA A Cancer J Clin 62:10–29
Van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG (2011) Colorectal cancer epigenetics: complex simplicity. J Clin Oncol 29:1382–1391
Visvader JE, Lindeman GJ (2012) Cancer stem cells: current status and evolving complexities. Cell Stem Cell 10:717–728
Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R et al (2011) Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res 71:7238–7249
Wu CY, Hsieh CY, Huang KE, Chang C, Kang HY (2012) Cryptotanshinone down-regulates androgen receptor signaling by modulating lysine-specific demethylase 1 function. Int J Cancer 131:1423–1434
Yang M, Culhane JC, Szewczuk LM, Gocke CB, Brautigam CA, Tomchick DR, Machius M, Cole PA, Yu H (2007) Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat Struct Mol Biol 14:535–539
Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D et al (2013) Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 5(2):445–457
Acknowledgments
This work was supported by grants from Chang Gung Memorial Hospital, Taiwan (CMRPG3B1361) to H.C. Hsu. We thank the Pathology Core of the MMRC at CGU for assistance with immunohistochemical staining and quantification.
Conflict of interest
No potential conflicts of interest are disclosed.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hsu, HC., Liu, YS., Tseng, KC. et al. CBB1003, a lysine-specific demethylase 1 inhibitor, suppresses colorectal cancer cells growth through down-regulation of leucine-rich repeat-containing G-protein-coupled receptor 5 expression. J Cancer Res Clin Oncol 141, 11–21 (2015). https://doi.org/10.1007/s00432-014-1782-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-014-1782-4