
Abstract
Recent advances in understanding the aetiology of the disorders that make up the haemolytic uraemic syndrome (HUS) permit a revised classification of the syndrome. With appropriate laboratory support, an aetiologically-based subgroup diagnosis can be made in all but a few cases. HUS caused by enterohaemorrhagic Escherichia coli remains by far the most prevalent subgroup, and new insights into this zoonosis are discussed. The most rapidly expanding area of interest is the subgroup of inherited and acquired abnormalities of complement regulation. Details of the pathogenesis are incomplete but it is reasonable to conclude that local activation of the alternative pathway of complement in the glomerulus is a central event. There is no evidence-based treatment for this diagnostic subgroup. However, in circumstances where there is a mutated plasma factor such as complement factor H, strategies to replace the abnormal protein by plasmapheresis or more radically by liver transplantation are logical, and anecdotal successes are reported. In summary, the clinical presentation of HUS gives a strong indication as to the underlying cause. Patients without evidence of EHEC infection should be fully investigated to determine the aetiology. Where complement abnormalities are suspected there is a strong argument for empirical and early plasma exchange, although rapid advances in this field may provide more specific treatments in the near future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Classification
Haemolytic uraemic syndrome (HUS) is diagnosed on the simultaneous features of microangiopathic haemolytic anaemia, thrombocytopenia, and acute renal failure. It is worth noting that there is an overlap between the clinical presentations of HUS and thrombotic thrombocytopenic purpura (TTP), even though they have clearly different aetiologies. Patients with an acquired or inherited deficiency of the von Willebrand protease, ADAMTS13, are much more likely to present with insidious or fluctuating neurological signs of TTP, rather than the abrupt renal failure that typifies HUS. But this is not absolute.
In 2006, the European Paediatric Study Group for HUS [2] published a revised classification of HUS, TTP, and related disorders, based on a contemporary understanding of causation (summarised in Table 1). In brief, patients can be classified at two levels. In level 1, the cause is well established, even if details of the pathogenesis are incomplete. In level 2, disease or drug associations may be described but causation is not proven. In practice the diagnostic subgroups in level 1 segregate well. However, occasionally an individual patient, or a particular episode in an individual, may have more than one aetiology. The classification permits this. It fits with the idea that HUS can be caused by either, or both, an external environmental trigger and/or an intrinsic inherited risk factor. This is referred to as a “two hits” hypothesis. For example, a patient with a complement factor H mutation might present with an episode of HUS precipitated by infection, perhaps enterohaemorrhagic Escherichia coli infection, or pregnancy. With comprehensive investigation, almost all patients can be allocated to an aetiologically-based subgroup diagnosis. However, some of the investigations dealt with below are costly and not easily accessed. A guideline has therefore been proposed that uses clinical presentation in the first instance to indicate which advanced investigations are needed, and what immediate treatment should be considered while awaiting diagnostic results.
Investigation
The diagnosis of HUS caused by enterohaemorrhagic E. coli or invasive pneumococcal infections [55] is generally straightforward, based upon the clinical presentation supported by locally available microbiological tests [3, 40]. These two groups account for at least 90% of all childhood HUS [31]. The remainder, commonly referred to as “atypical” for want of a better description, need comprehensive investigation including mutation analysis of specific complement genes [27] and the activity of von Willebrand factor cleaving protease, ADAMTS13 [53]. A subgroup diagnosis can be expected in about 50% of these cases. A list of relevant investigations for the atypical subgroup is given in the guideline and is updated on the website of the European Paediatric Study Group for HUS at www.espn.cardiff.ac.uk/registries. The guideline also gives contact details of specialist laboratories that provide complement gene mutation analysis and investigation of ADAMTS13 activity.
Enterohaemorrhagic E. coli infection
Enterohaemorrhagic E.coli (EHEC) cause about 90% of all childhood HUS in developed countries [11, 31]. EHEC have various virulence factors implicated in causing haemorrhagic colitis and HUS in humans, but an essential property is the production of certain Shiga toxins (Stx). Not all Stx producing E.coli (STEC) are pathogenic in man, and therefore they are not all EHEC [38]. The forms of Stx implicated in human disease are mostly Stx-2 and its variant Stx-2c, and less frequently Stx-1 (Stx-1 is identical to Shiga toxin, the exotoxin of Shigella dysenteriae type 1) [17, 19, 26, 29, 30]. EHEC with more than one toxin plasmid, for example, Stx-1 plus Stx-2, are well recognized. Organisms producing Stx 2d had been thought not to cause human disease but recently a toxin variant has been described that is activated by enzymes in intestinal mucus to the form Stx-2d(activatable) which is implicated in HUS [3]. Stx 2e is the toxin associated with Edema disease in piglets, and Stx-2f -producing E. coli occur in avians with no evidence to date that they can intoxicate humans.
The majority of EHEC express intimin on their surface and can deliver the intimin receptor into host enterocytes by a type 3 secretion system [13]. This is an important virulence factor and is the property that defines enteropathogenic E. coli (EPEC). It permits the organisms to bind tightly to the host surface, and the enterocyte responds by cytoskeletal changes. This induces loss of villi and pedestal formation, the attachment and effacement lesion. The loss of absorptive villi is the explanation for the watery diarrhea induced by EPEC. Understanding the molecular basis for attachment and effacement and the host signalling mechanisms that subtend it has progressed rapidly [7]. While this is recognised as an important virulence factor for EHEC, probably by allowing exotoxin to be produced in the immediate vicinity of the enterocyte, it is not essential. Thus any strategy to immunize against intimin or other components of the type 3 secretion might reduce but not abolish the risk of EHEC.
Being a zoonosis, EHEC reside in other species, often without causing disease in them. Transmission to humans has been described via a wide range of foods, but only recently has the importance of direct contact with animals, particularly petting farm animals, been shown [56]. It is not known why some individuals develop colitis and HUS after EHEC exposure while others do not. Humans acquire antibodies against Stx [30], and experimental models show that Stx antibody can protect against toxic manifestations [51]. The incidence of HUS is greatest in children between 6 months and 5 years of age and this might be explained by their lack of immunity to toxin. A strategy to prevent human disease by active immunization with relevant Stx toxoids is logical, and novel approaches to the development of such agents are being pursued.
At present there is no proven active treatment for EHEC infection or HUS. The role of antibiotics used during the diarrhoeal phase of the disease remains controversial. A recent meta-analysis has not shown adverse effects of antibiotics [45]. However, the conclusions are weakened because different antibiotics were given, and there was variable correction for severity of disease in the studies. Some antibiotics cause a burst of toxin release from the affected bacteria in culture, while others are thought not to do so [25]. Given that EHEC are typically not enteroinvasive and the infection is self-limiting, there is no direct indication for their use, and with hypothetical reasons why they might be harmful, most paediatric nephrologists do not use them.
Supportive therapy, including careful fluid and electrolyte balance remains the mainstay of treatment of this form of HUS [1]. It is vital that children with confirmed or suspected STEC infection are monitored carefully for their intravascular volume status. The development of oligoanuric renal failure during HUS indicates a poorer long term outcome [18], and a recent study suggests that early intravascular volume expansion with isotonic saline preceding the oligoanuric phase may reduce the incidence of oligoanuric renal failure [1].
Therapeutic proposals to sequester Stx in the intestine have not shown clinical benefit [52]. However, passive immunisation with anti-Stx antibodies has been shown to prevent or arrest disease in laboratory animals and is an attractive idea for humans [33]. Humanised monoclonal antibodies against Stx-1 and Stx 2 (Shiga-mab, Thallion Pharmaceuticals Inc., Canada) are now being tested in phase 2 trials. The initial studies propose to administer the antibody in the diarrhoeal stage of the illness before HUS occurs to show whether HUS can be prevented. A prerequisite for such studies will be the ability to positively diagnose EHEC infection at an early stage. Selective stool culture techniques with subsequent identification of Shiga-like toxin or Stx bacteriophage are too slow for this strategy. Rapid, near-to-patient techniques are required and under development. If successful this would revolutionize the ability of clinicians to identify and intervene early in EHEC infections.
The mortality from Stx-HUS is now about 2% [31]. It is important to note that although the glomerular filtration rate returns to normal in most patients, there may be permanent nephron loss, which may have longer term sequelae [9, 32]. A recent systematic review and meta-analysis of the intermediate outcome of Stx-HUS described permanent and serious renal sequelae (hypertension, proteinuria, declining GFR) in 5–25% of patients [18].
Disorders of complement regulation
Although cases of HUS with complement abnormalities were described more than 20 years ago, the significance of this was not appreciated until the seminal report of Warwicker et al. [54]. Since then a number of different gene mutations have been described in patients with HUS, all within genes encoding proteins of the alternative complement pathway [5, 6, 12, 15, 16, 35, 36, 39, 43, 44, 50].
The alternative complement pathway
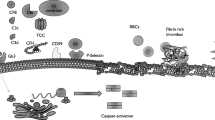
The alternative complement pathway is a network of plasma proteins that provides antibody-independent defence against micro-organisms. The goal of the pathway is to quickly remove invading pathogens by coating them with the opsonin, C3b, and by promoting terminal complement pathway activation on their surface. The activation and regulation of the alternative pathway is shown in Fig. 1.

The alternative complement pathway showing the centrality of C3b. The alternative pathway is continuously activated through the spontaneous hydrolysis of C3 (black dashed line) to a form of C3b that is able to combine with factors B and D to form a C3 convertase. This generates further C3b from C3 and a stable C3 convertase (C3bBb) is formed, amplifying the production of C3b. This C3b can participate in further C3 convertase formation and C5 convertase formation (the addition of one C3b molecule to the C3 convertase). Cleavage of C5 results in the C5a fragment (an anaphylotoxin) and the C5b fragment, which combines with C6, C7, C8, and multiple C9 molecules to form C5b-9, the membrane attack complex (MAC). The end results of alternative pathway activation are opsonisation with C3b, chemotaxis through anaphylotoxin C5a, and cell lysis through MAC. Host cells possess membrane-bound complement regulatory proteins (e.g. membrane cofactor protein, MCP, decay accelerating factor, DAF) and are rich in polyanions which promote the binding of plasma regulator factor H (FH). These regulators inactivate C3b (red dashed line) or dissociate the C3 convertase (dotted red line) so that amplification is limited
Since the alternative complement pathway is spontaneously activated by the hydrolysis of fluid phase complement C3, and positively amplified, its regulation is essential for host cell protection. This is provided by fluid-phase and membrane bound complement regulatory proteins. In the fluid phase, C3b is inactivated by the serine protease factor I (F1) which is dependent on cofactor activity from other regulators. Membrane cofactor protein (MCP) is a resident transmembrane protein expressed on almost every human cell except erythrocytes. It is a cofactor for factor I in the cleavage of C3b and C4b (formed during classical and lectin pathway activation) and also promotes the decay of C3 convertases (decay acceleration). Factor H is a fluid-phase regulator that has cofactor activity for the cleavage of C3b by factor I and also has decay acceleration activity. In addition to its role as a fluid phase regulator, FH is the main mechanism by which the alternative pathway discriminates between foreign and host surfaces. Unlike many pathogens, host surfaces are rich in polyanions such as sialic acid and glycosaminoglycans. FH has high affinity for these anionic sites, particularly when C3b is deposited during complement activation on the cell surface.
Mutations in FH, MCP, and FI are all implicated in the pathogenesis of HUS and the incidences of these mutations are summarised in Table 2. There are now reports emerging that mutations in C3 and factor B have also been detected in patients with HUS [15].
Complement gene mutations in HUS
FH mutations associated with HUS are mostly heterozygous missense mutations affecting the C-terminal domain of the protein which is important for host cell recognition [49]. In such cases, plasma FH and C3 concentrations are often normal. Mutant FH proteins exhibit reduced binding to C3b [23, 48], to heparin [22, 23], and to endothelial cells [23]. Most work normally as a cofactor for the cleavage of C3b in the fluid phase but show impaired complement regulation on cell surfaces including endothelial cells and erythrocytes [20, 47]. Other FH mutations affect the structure of the protein causing impaired secretion and low plasma concentration of factor H [37]. Plasma C3 concentration may be normal or low. Patients with FH mutations tend to have early onset of disease (70% onset before 12 years of age), follow a relapsing course, and 70% of cases progress to end-stage renal failure or death [6, 50]. Renal transplantation also has a poor outcome. Graft loss, mostly due to recurrent disease or graft thrombosis, occurs in about 80% of cases [4].
MCP mutations are usually heterozygous and appear to result in heterozygous MCP deficiency or impaired binding to C3b [6]. HUS associated with MCP mutation follows a relapsing course with gradual progression to end-stage renal failure. Recurrence after renal transplantation is rare and it is likely that the donor kidney is protected from recurrence because of donor MCP expression in the vasculature of the graft, although endothelial donor-host chimerism is a potential risk [14].
The FI mutations so far described are heterozygous and associated with either normal or reduced FI concentration and a variable reduction of complement C3. Certain mutant proteins demonstrate reduced inactivation of C3b and C4b [28]. The clinical course is severe with progression to irreversible renal failure. Renal grafts have been lost to recurrent HUS in the majority of those who have undergone renal transplantation [6].
Mutations have also been described in factor B and C3 and these appear to result in gain of function by rendering the C3 convertase more resistant to decay [12, 15]. In addition, some patients have antibodies to FH that impede its regulatory activity [10, 24].
In combination, these findings point to increased activation of the alternative complement pathway as a central phenomenon in this form of HUS. This is supported by animal models in which the terminal complement pathway is essential for developing glomerular thrombotic microangiopathy [34]. However, plasma concentrations of C3 in patients are often normal or only marginally reduced. It is therefore assumed that the escape of complement activation that underlies the disease is seldom systemic but takes place as a local event within the glomerular microcirculation.
Novel therapeutic strategies
Knowing the cause of atypical HUS allows a theoretical approach to therapy even though details of the pathogenesis are unconfirmed. In practice, the complex laboratory investigations needed to confirm aetiology take several weeks, and therefore initial treatment has to be empirical. The European Study Group for HUS argues for plasma exchange, replacing with fresh frozen plasma or a standardised whole plasma product such as Octaplas, as a catch-all, first line treatment. This is suggested on the basis that it would replace mutant complement proteins responsible for the disease and remove autoantibodies to factor H or ADAMTS13. These authors justify the urgency and invasive nature of this approach on the fact that glomerular thrombotic microangiopathy is a destructive process and a first episode of HUS may lead rapidly to end-stage failure. At present there are anecdotes but no evidence to back up this proposal. In patients with MCP mutations, plasma therapy is not expected to have a direct impact on the disease as MCP is a resident membrane protein not a circulating plasma factor.
It seems likely that specific treatments will be needed in different groups once the cause has been identified. In patients with genetic FH abnormalities, and based on the idea that the disease is caused by haplotype insufficiency, it seems logical to give normal factor H. Plasma derived factor H concentrate is being developed commercially with that intention. The same logic applies to HUS associated with FI mutation. Both factor H and factor I are produced in the liver, and successful liver transplantation would theoretically restore normal complement regulation. So far six cases of FH associated HUS have been treated by liver or liver plus kidney transplantation. Poor outcome occurred in the first three cases [8, 41, 42] but the three survivors have good organ function, normal complement activity, and no recurrence of HUS [21, 46].
Summary
The clinical presentation of HUS gives a strong indication as to the underlying cause. EHEC infection remains the most prevalent. The virulence factors of EHEC in humans are increasingly understood, as is the ecology of these organisms in the wider environment. Patterns of transmission are better recognised. New tools for microbiological diagnosis are in production, and there are prospects for early intervention by targeting Stx.
Patients with atypical HUS (familial or relapsing, or not associated with EHEC or Shigella dysenteriae or invasive Streptococcus pneumoniae infection) should be fully investigated to determine the cause. These patients are difficult to manage and should be transferred to a paediatric nephrology service urgently. There is a strong argument for empirical and early plasma exchange to replace defective proteins or remove causative antibodies. Knowledge of the cause of disease, particularly in the group with abnormal complement regulation, opens up the possibility of specific treatments in the near future.
Abbreviations
- HUS:
-
Haemolytic uraemic syndrome
- TTP:
-
Thrombotic thrombocytopenic purpura
- ADAMTS13:
-
A dysintegrin and metalloprotease with thrombospondin type motifs 13
- EHEC:
-
Enterohaemorrhagic Escherichia coli
- STEC:
-
Shiga toxin-producing Escherichia coli
- EPEC:
-
Enteropathogenic Escherichia coli
- Stx:
-
Shiga toxin
- GFR:
-
Glomerular filtration rate
- MCP:
-
Membrane cofactor protein (CD46)
- FH:
-
Complement factor H
- FI:
-
Complement factor I
References
Ake JA, Jelacic S, Ciol MA, Watkins SL, Murray KF, Christie DL, Klein EJ, Tarr PI (2005) Relative nephroprotection during Escherichia coli O157:H7 infections: association with intravenous volume expansion. Pediatrics 115:e673–e680
Besbas N, Karpman D, Landau D, Loirat C, Proesmans W, Remuzzi G, Rizzoni G, Taylor CM, Van de Kar N, Zimmerhackl LB (2006) A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int 70:423–431
Bielaszewska M (2007) Shiga toxin-mediated hemolytic uremic syndrome: time to change the diagnostic paradigm. PLoS ONE 2:e1024
Bresin E, Daina E, Noris M, Castelletti F, Stefanov R, Hill P, Goodship TH, Remuzzi G (2006) Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol 1:88–99
Caprioli J, Bettinaglio P, Zipfel PF, Amadei B, Daina E, Gamba S, Skerka C, Marziliano N, Remuzzi G, Noris M (2001) The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol 12:297–307
Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G (2006) Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108:1267–1279
Caron E, Crepin VF, Simpson N, Knutton S, Garmendia J, Frankel G (2006) Subversion of actin dynamics by EPEC and EHEC. Curr Opin Microbiol 9:40–45
Cheong HI, Lee BS, Kang HG, Hahn H, Suh KS, Ha IS, Choi Y (2004) Attempted treatment of factor H deficiency by liver transplantation. Pediatr Nephrol 19:454–458
Dieguez S, Ayuso S, Brindo M, Osinde E, Canepa C (2004) Renal functional reserve evolution in children with a previous episode of hemolytic uremic syndrome. Nephron Clin Pract 97:c118–c122
Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, Weiss L, Fridman WH, Fremeaux-Bacchi V (2005) Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol 16:555–563
Elliott EJ, Robins-Browne RM, O’Loughlin EV, Bennett-Wood V, Bourke J, Henning P, Hogg GG, Knight J, Powell H, Redmond D (2001) Nationwide study of haemolytic uraemic syndrome: clinical, microbiological, and epidemiological features. Arch Dis Child 85:125–131
Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, Lopez-Trascasa M, Sanchez-Corral P, Rodriguez de Cordoba S (2005) Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet 14:703–712
Frankel G, Phillips AD, Rosenshine I, Dougan G, Kaper JB, Knutton S (1998) Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol Microbiol 30:911–921
Fremeaux-Bacchi V, Arzouk N, Ferlicot S, Charpentier B, Snanoudj R, Durrbach A (2007) Recurrence of HUS due to CD46/MCP mutation after renal transplantation: a role for endothelial microchimerism. Am J Transplant 7:2047–2051
Fremeaux-Bacchi VGT, Regnier CH, Dragon-Durey MA, Janssen B, Atkinson J (2007) Mutations in complement C3 predispose to development of atypical haemolytic uraemic syndrome. In: XIth European meeting on complement in human disease. Elsevier, Cardiff, UK
Fremeaux-Bacchi V, Kemp EJ, Goodship JA, Dragon-Durey M-A, Strain L, Loirat C, Deng H-W, Goodship THJ (2005) The development of atypical HUS is influenced by susceptibility factors in factor H and membrane cofactor protein—evidence from two independent cohorts. J Med Genet 42(11):852–856
Friedrich AW, Bielaszewska M, Zhang WL, Pulz M, Kuczius T, Ammon A, Karch H (2002) Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J Infect Dis 185:74–84
Garg AX, Suri RS, Barrowman N, Rehman F, Matsell D, Rosas-Arellano MP, Salvadori M, Haynes RB, Clark WF (2003) Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. JAMA 290:1360–1370
Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB (2002) Clinical course and the role of shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis 186:493–500
Heinen S, Jozsi M, Hartmann A, Noris M, Remuzzi G, Skerka C, Zipfel PF (2007) Hemolytic uremic syndrome: a factor H mutation (E1172Stop) causes defective complement control at the surface of endothelial cells. J Am Soc Nephrol 18:506–514
Jalanko H, Peltonen S, Koskinen A, Puntila J, Isoniemi H, Holmberg C, Pinomaki A, Armstrong E, Koivusalo A, Tukiainen E, Makisalo H, Saland J, Remuzzi G, de Cordoba S, Lassila R, Meri S, Jokiranta TS (2008) Successful liver-kidney transplantation in two children with aHUS caused by a mutation in complement factor H. Am J Transplant 8:216–221
Jokiranta TS, Cheng ZZ, Seeberger H, Jozsi M, Heinen S, Noris M, Remuzzi G, Ormsby R, Gordon DL, Meri S, Hellwage J, Zipfel PF (2005) Binding of complement factor H to endothelial cells is mediated by the carboxy-terminal glycosaminoglycan binding site. Am J Pathol 167:1173–1181
Jozsi M, Heinen S, Hartmann A, Ostrowicz CW, Halbich S, Richter H, Kunert A, Licht C, Saunders RE, Perkins SJ, Zipfel PF, Skerka C (2006) Factor H and atypical hemolytic uremic syndrome: mutations in the C-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol 17:170–177
Jozsi M, Licht C, Strobel S, Zipfel SLH, Richter H, Heinen S, Zipfel PF, Skerka C (2008) Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 111:1512–1514
Karch H, Strockbine NA, O’Brien AD (1986) Growth of Escherichia coli in the presence of trimethoprim-sulfamethoxazole facilitates detection of Shiga-like toxin producing strains by colony blot assay. FEMS Microbiol Lett 35:141–145
Karmali MA (1989) Infection by verocytotoxin-producing Escherichia coli. Clin Microbiol Rev 2:15–38
Kavanagh D, Richards A, Fremeaux-Bacchi V, Noris M, Goodship T, Remuzzi G, Atkinson JP (2007) Screening for complement system abnormalities in patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 2:591–596
Kavanagh D, Richards A, Noris M, Hauhart R, Liszewski MK, Karpman D, Goodship JA, Fremeaux-Bacchi V, Remuzzi G, Goodship TH, Atkinson JP (2008) Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol 45:95–105
Kleanthous H, Smith HR, Scotland SM, Gross RJ, Rowe B, Taylor CM, Milford DV (1990) Haemolytic uraemic syndromes in the British Isles, 1985–8: association with verocytotoxin producing Escherichia coli. Part 2: microbiological aspects. Arch Dis Child 65:722–727
Ludwig K, Karmali MA, Sarkim V, Bobrowski C, Petric M, Karch H, Muller-Wiefel DE (2001) Antibody response to shiga toxins Stx2 and Stx1 in children with enteropathic hemolytic-uremic syndrome. J Clin Microbiol 39:2272–2279
Lynn RM, O’Brien SJ, Taylor CM, Adak GK, Chart H, Cheasty T, Coia JE, Gillespie IA, Locking ME, Reilly WJ, Smith HR, Waters A, Willshaw GA (2005) Childhood hemolytic uremic syndrome, United Kingdom and Ireland. Emerg Infect Dis 11:590–596
Moghal NE, Ferreira MA, Howie AJ, Milford DV, Raafat E, Taylor CM (1998) The late histologic findings in diarrhea-associated hemolytic uremic syndrome. J Pediatr 133:220–223
Mukherjee J, Chios K, Fishwild D, Hudson D, O’Donnell S, Rich SM, Donohue-Rolfe A, Tzipori S (2002) Human Stx2-specific monoclonal antibodies prevent systemic complications of Escherichia coli O157:H7 infection. Infect Immun 70:612–619
Nangaku M, Alpers CE, Pippin J, Shankland SJ, Kurokawa K, Adler S, Johnson RJ, Couser WG (1997) Renal microvascular injury induced by antibody to glomerular endothelial cells is mediated by C5b-9. Kidney Int 52:1570–1578
Neumann HP, Salzmann M, Bohnert-Iwan B, Mannuelian T, Skerka C, Lenk D, Bender BU, Cybulla M, Riegler P, Konigsrainer A, Neyer U, Bock A, Widmer U, Male DA, Franke G, Zipfel PF (2003) Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet 40:676–681
Noris M, Brioschi S, Caprioli J, Todeschini M, Bresin E, Porrati F, Gamba S, Remuzzi G (2003) Familial haemolytic uraemic syndrome and an MCP mutation. Lancet 362:1542–1547
Ohali M, Shalev H, Schlesinger M, Katz Y, Kachko L, Carmi R, Sofer S, Landau D (1998) Hypocomplementemic autosomal recessive hemolytic uremic syndrome with decreased factor H. Pediatr Nephrol 12:619–624
Orth D, Wurzner R (2006) What makes an enterohemorrhagic Escherichia coli? Clin Infect Dis 43:1168–1169
Perez-Caballero D, Gonzalez-Rubio C, Gallardo ME, Vera M, Lopez-Trascasa M, Rodriguez de Cordoba S, Sanchez-Corral P (2001) Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet 68:478–484
Pradel N, Livrelli V, De Champs C, Palcoux J-B, Reynaud A, Scheutz F, Sirot J, Joly B, Forestier C (2000) Prevalence and characterization of shiga toxin-producing Escherichia coli isolated from cattle, food, and children during a one-year prospective study in France. J Clin Microbiol 38:1023–1031
Remuzzi G, Ruggenenti P, Codazzi D, Noris M, Caprioli J, Locatelli G, Gridelli B (2002) Combined kidney and liver transplantation for familial haemolytic uraemic syndrome. Lancet 359:1671–1672
Remuzzi G, Ruggenenti P, Colledan M, Gridelli B, Bertani A, Bettinaglio P, Bucchioni S, Sonzogni A, Bonanomi E, Sonzogni V, Platt JL, Perico N, Noris M (2005) Hemolytic uremic syndrome: a fatal outcome after kidney and liver transplantation performed to correct factor h gene mutation. Am J Transplant 5:1146–1150
Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, Tielemans CL, Goodship JA, Goodship TH (2001) Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet 68:485–490
Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, Muslumanogglu MH, Kavukcu S, Filler G, Pirson Y, Wen LS, Atkinson JP, Goodship THJ (2003) Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. PNAS 100:12966–12971
Safdar N, Said A, Gangnon RE, Maki DG (2002) Risk of hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 enteritis: a meta-analysis. JAMA 288:996–1001
Saland JM, Emre SH, Shneider BL, Benchimol C, Ames S, Bromberg JS, Remuzzi G, Strain L, Goodship TH (2006) Favorable long-term outcome after liver-kidney transplant for recurrent hemolytic uremic syndrome associated with a factor H mutation. Am J Transplant 6:1948–1952
Sanchez-Corral P, Gonzalez-Rubio C, Rodriguez de Cordoba S, Lopez-Trascasa M (2004) Functional analysis in serum from atypical hemolytic uremic syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol 41:81–84
Sanchez-Corral P, Perez-Caballero D, Huarte O, Simckes AM, Goicoechea E, Lopez-Trascasa M, de Cordoba SR (2002) Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet 71:1285–1295
Saunders RE, Goodship TH, Zipfel PF, Perkins SJ (2006) An interactive web database of factor H-associated hemolytic uremic syndrome mutations: insights into the structural consequences of disease-associated mutations. Hum Mutat 27:21–30
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, Boudailliez B, Bouissou F, Deschenes G, Gie S, Tsimaratos M, Fischbach M, Morin D, Nivet H, Alberti C, Loirat C (2007) Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 18:2392–2400
Sheoran AS, Chapman S, Singh P, Donohue-Rolfe A, Tzipori S (2003) Stx2-specific human monoclonal antibodies protect mice against lethal infection with Escherichia coli expressing Stx2 variants. Infect Immun 71:3125–3130
Trachtman H, Cnaan A, Christen E, Gibbs K, Zhao S, Acheson DWK, Weiss R, Kaskel FJ, Spitzer A, Hirschman GH (2003) Effect of an oral shiga toxin-binding agent on diarrhea-associated hemolytic uremic syndrome in children: a randomized controlled trial. JAMA 290:1337–1344
Veyradier A, Obert B, Haddad E, Cloarec S, Nivet H, Foulard M, Lesure F, Delattre P, Lakhdari M, Meyer D, Girma J-P, Loriat C (2003) Severe deficiency of the specific von Willebrand factor-cleaving protease (ADAMTS 13) activity in a subgroup of children with atypical hemolytic uremic syndrome. J Pediatr 142:310–317
Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, Goodship JA (1998) Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 53:836–844
Waters AM, Kerecuk L, Luk D, Haq MR, Fitzpatrick MM, Gilbert RD, Inward C, Jones C, Pichon B, Reid C, Slack MPE, Van’t Hoff W, Dillon MJ, Taylor CM, Tullus K (2007) Hemolytic uremic syndrome associated with invasive pneumococcal disease: The United Kingdom experience. J Pediatr 151:140–144
Werber D, Behnke SC, Fruth A, Merle R, Menzler S, Glaser S, Kreienbrock L, Prager R, Tschape H, Roggentin P, Bockemuhl J, Ammon A (2007) Shiga toxin-producing Escherichia coli infection in Germany: different risk factors for different age groups. Am J Epidemiol 165:425–434
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johnson, S., Taylor, C.M. What’s new in haemolytic uraemic syndrome?. Eur J Pediatr 167, 965–971 (2008). https://doi.org/10.1007/s00431-008-0745-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-008-0745-7