
Abstract
A total of 22 Japanese patients with hypophosphatasia were included in a study analysing the relationship between mutations in the tissue-nonspecific alkaline phosphatase (TNSALP) gene and the severity of the phenotype in Japanese patients with hypophosphatasia. The enzymatic activity of some of the identified mutant TNSALP proteins was also examined using corresponding expression vectors. Eighteen mutations, including 6 novel ones, were identified in the patients. Among them, the common mutations were F310L and T1559del. Of note, five patients with F310L mutation in one of the alleles exhibited a relatively mild phenotype without respiratory complications despite its perinatal onset. In contrast, the T1559del mutation is associated with perinatal lethal and infantile forms when not found in patients with the F310L mutation. The genotype-phenotype relationship was, to some extent, consistent with the enzymatic activity of the mutant ALP proteins; mutations K207E and G409C found in a surviving case of infantile hypophosphatasia, as well as F310L, retained some residual activities, whereas T1559del caused a complete loss of activity. Conclusion:In Japanese patients, the common mutations F310L and T1559del are associated with the relatively mild and lethal forms of hypophosphatasia, respectively. Our results may enhance the importance of genotyping patients with hypophosphatasia to predict their prognosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypophosphatasia (OMIM 146300, 241500 and 241510) is characterised by hypomineralisation of bone associated with the impaired activity of tissue-nonspecific alkaline phosphatase (TNSALP). Hypophosphatasia has diverse phenotypes and is usually classified into five subtypes based on the age of onset and clinical features; perinatal, infantile, childhood and adult type, and odontohypophosphatasia [24]. The severity of the disease is generally well correlated with the onset of the disease, except for odontohypophosphatasia where only the teeth are affected. Patients with the perinatal form of hypophosphatasia almost always die around birth due to impaired development of the lungs and the severe hypomineralisation of their bones [19]. However, the classification of these subgroups is not definite and there is diversity in the phenotype creating a so-called spectrum. For example, we have previously reported a patient who achieved long-term survival without respiratory failure despite fetal onset [17]. The presence of such a case may depend on the development of a diagnosis procedure or on the specificity of the genotype. Elucidation of the molecular heterogeneity underlying hypophosphatasia may contribute to our understanding of the clinical heterogeneity observed and the improvement of treatment, especially in patients with severe forms of the disease. The perinatal form is called a lethal form and the second most severe form, the infantile form, is still associated with high mortality because of the impairment of respiratory function and hypercalcaemia [2, 9]. To date, more than 100 mutations have been reported in the TNSALP gene in patients with hypophosphatasia, mainly in Caucasians and Asians (Japanese) [11, 12, 15, 20, 21]; however, the mutations are scattered throughout the whole coding region and only a few have been recognised to occur frequently in the gene. Moreover, although the relationship between the clinical features and the mutation of the corresponding gene has also been analysed, only a few reports detected the positive correlation between the mutations and severity [4, 6]. The mutations responsible for mild hypophosphatasia may not cause a complete loss of TNSALP function, suggested by several reconstruction experiments in which mutated TNSALP activity was examined [26]. On the other hand, mutations found in the severe form of the disease do not tend to be associated with restricted positions, although the three-dimensional model study showed that most of the severe missense mutations were localised in crucial domains such as the active site [26].
Although hypophosphatasia is usually inherited in an autosomal recessive manner, autosomal dominant inheritance is also recognised in some families with the mild form of the disease. Interestingly, the mutations in those families have been reported to show the dominant negative effect on the wild-type TNSALP activity, as one of the remarkable results of the recent progress in molecular biology [7, 13].
In the present report, we describe 22 Japanese patients with various types of hypophosphatasia, whose TNSALP genes were analysed. The enzymatic activity of the mutant TNSALP proteins was also investigated with reconstruction experiments. The results suggest some correlation between genotype and phenotype in Japanese patients with hypophosphatasia.
Subjects and methods
Subjects
A total of 22 unrelated Japanese patients with hypophosphatasia were included in the study after informed consent was obtained.
Sequence analysis of the tissue-nonspecific alkaline phosphatase gene
Sequencing of the TNSALP gene was performed following the extraction of genomic DNA from peripheral mononuclear cells of the patients. In some patients, DNA from the parents was also analysed. The primers described in the previous report were utilized for the PCR [3, 23]. In some cases, total RNA was also extracted from peripheral mononuclear cells and served as the template for reverse transcription (RT)-PCR using random hexamer and reverse polymerase (Super Script II; Invitrogen, Carlsbad, Calif.). The fragments amplified by PCR or RT-PCR were sequenced directly or after cloning into pT7-Blue vector (Novagen, Madison, Wi.) using a Model 377 sequencer (Perkin-Elmer, Norwalk, Conn.). The nucleotide and amino acid numbers are designated according to the Nomenclature Working Group [1].
Mutagenesis and reconstruction experiments
The human TNSALP expression vector (pSV2Aalp) was generously provided by Dr. P.S. Henthorn [5]. TNSALP cDNAs carrying the following mutations were generated by PCR-mediated mutagenesis or the Kunkel method following the manufacturer’s protocol (Gene Editor; Promega); T1559del, F310L, F311L, F311del, K207E, L282P and G409C. The wild-type and mutated TNSALP cDNAs were then cloned into pcDNA3.0 vector (Invitrogen). The expression vectors of the wild-type and mutated TNSALP were introduced into COS-7 cells using the FuGENETM6 Reagent (Roche) and the activity of TNSALP was measured by the Lowry method using p-nitrophenylphosphate as a substrate 2 days after the transfection [8]. The expression of each plasmid containing wild-type or mutant cDNA of TNSALP was confirmed by RT-PCR analysis using RNA samples obtained from the identical transfectants.
Results
Clinical forms and genotypes of patients
The patients with hypophosphatasia subjected to the mutation analysis of the TNSALP gene are summarised in Table 1 with respect to sex, age at the time of analysis, clinical forms, amino acid changes, serum levels of TNSALP at diagnosis and the outcome. Table 2 contains the changes of nucleotides and amino acids in mutations found in the study. In our study, numbers of male and female patients were 18 and four respectively, although hypophosphatasia is an autosomal trait. Classification of the patients into corresponding clinical forms was performed based on the onset of the disease and the severity of the clinical features including bone abnormalities and respiratory problems. Interestingly, six patients (numbers 1–6 in Table 1) have survived and had relatively high serum TNSALP levels for hypophosphatasia (>50 IU/l) despite the perinatal onset of the disease. In contrast, ten patients (numbers 7–16) were classified as the classical perinatal form; nine of them died and the remaining one is currently on mechanical ventilation with very low levels of TNSALP (<30 IU/l). Three patients (numbers 17–19) were diagnosed with infantile hypophosphatasia. All of these three patients were diagnosed before 6 months of age based on their severe bony hypomineralisation and hypercalcaemia. Two of them died from respiratory failure or sudden death before they reached 1 year of age. Three patients (numbers 20–22) were diagnosed with childhood hypophosphatasia and are still alive.
The mutation analyses revealed 40 alleles with missense mutations or deletion of a nucleotide, although we were not able to find mutations in the remaining four alleles. Two of the most frequent mutations, Phe to Leu conversion at codon 310 (F310L) and deletion of T at positions 1559 (T1559del) were detected in five and 15 patients, respectively. On the allele basis, the F310L and the T1559del mutations were found in 5 and 18 alleles respectively in 44 alleles of the patients.
The F310L is found in patients with the relatively mild form of hypophosphatasias
The F310L mutation was found in patients 1–5 who have survived despite the perinatal onset of their disease. The profile of patient 1 has been previously described elsewhere [17]. The patient, a female, exhibited deformity of the long bones and the characteristic bone spurs in the fibulae noted at birth [14]. At the age of 1 year, she underwent surgery to correct the bilateral leg deformity. Although she experienced premature tooth loss at the age of 2, she reached the age of 9 years without any other complications and the deformity of the long bones improved (Fig. 1A). However, she was short (120 cm, −1.8 SD at the age of 9 years) with bilateral short fibulae. The serum TNSALP level remained at approximately 100 IU/l, ranging from 70 to 159 IU/l.

A X-ray film of the knees of patient 1 at the age of 9 years. The curvature of the femur and the tibia has improved significantly since birth. Note the slight irregularity of metaphysis. B X-ray film of the knees of patient 2 taken at the age of 18 months. Note the bending of the lower leg bones. There is no sign of impaired mineralisation or metaphyseal irregularity
Patient 2 was a male baby, previously described in [3]. He had skin dimples of the lower legs and a skeletal survey resembled that of patient 1 except for the bone spurs (Fig. 1B). He was diagnosed with hypophosphatasia on the basis of a low level of serum TNSALP activity (56 IU/l) and the characteristic bone findings. His parents had also low TNSALP activity levels (father 32 IU/l, mother 68 IU/l).
Patient 3 was a female baby born in the 36th gestational week with a relative low birth weight (2380 g). She was the second baby of unrelated parents. Her brother was healthy and his serum TNSALP level was 224 IU/l. An ultrasound scan in the 33rd gestational week revealed a shortning of her left femur. She had no respiratory problems, but the deformity and shortness of the extremities were confirmed by skeletal survey (not shown); however, she had neither skin dimples of the lower legs nor bone spurs. The patient was diagnosed as having hypophosphatasia on the basis of a low level of serum TNSALP activity (89 IU/l), elevated urine levels of phosphoethanolamine (1419 µmol/mg creatinine) and bone findings. The serum TNSALP level increased thereafter, but remained low, ranging from 149 to 238 IU/l.
Of note, five of patients 1–6 had the F310L mutation in one of the alleles. In addition, the other patient with this form ( number 6) had a mutation at a similar position of the gene (F311L). On the other hand, the mutations F310L and F311L were not found in the other patients with more severe phenotypes.
T1559del is the most frequent mutation in Japanese patients with hypophosphatasia
The T1559del mutation was found in 15 patients and in 18 alleles, and was the most frequent mutation detected in our patients (Table 1) as reported by Orimo et al. [16]. Three patients with a homozygous T1559del mutation (numbers 7, 14, 15) were all diagnosed with the classical perinatal form of hypophosphatasia and died shortly after birth. Including these patients, nine out of ten cases of perinatal hypophosphatasia have the T1559del mutation in at least one of the alleles. However, this mutation was also found in compound heterozygotes of other forms of the disease.
Enzymatic activities of the mutant tissue-nonspecific alkaline phosphatase proteins
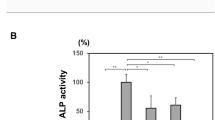
Enzymatic activities of some mutant TNSALP proteins were determined in the reconstruction experiments. Interestingly, the F310L mutant, which was associated with the non-lethal neonatal hypophosphatasia, has been shown to retain around 70% of enzymatic activity compared with the wild-type protein as previously reported [3] (Fig. 2). On the other hand, the F311L and F310del mutations led to an almost complete loss of enzymatic activitiy.

Enzymatic activities of mutant TNSALP proteins where codon F310 or F311 was mutated and found in the infantile form of hypophosphatasia. COS7 cells were transfected with the expression vectors coding the wild-type (WT) ALP, F310L, F311L, delF310, K207E, L282P, G409C or T1559del mutant TNSALP. The TNSALP activities of the cell lysates harvested 2 days after transfection were measured according to Lowry et al. [8] and standardised to the amount of protein. The TNSALP activity of the cells transfected with wild-type TNSALP expression vector was designated as 100%
We had three patients with infantile hypophosphatasia (numbers 17, 18, 19, Table 1). The enzymatic activities of the mutant proteins found in these patients (K207E, L282P. G409C, T1559del) were also evaluated (Fig. 2). K207E, L282P and G409C mutants retained some enzymatic activity corresponding to 43.0±10%, 9.7±1.7% and 18.5±1.2% of the activity of wild-type protein, respectively, whereas the T1559del mutant almost completely lost its activity. Interestingly, the single surviving patient with infantile hypophosphatasia (number 18 in Table 1) has the K207E and G409C mutations, both of the mutants retaining substantial enzymatic activity.
Discussion
Currently, 127 mutations of the TNSALP gene in patients with hypophosphatasia have been described in the TNSALP gene mutations database (http://www.sesep.uvsq.fr/Database.html). Most of the identified mutations are missense mutations, the remainder being several deletion and insertion mutations of one to four nucleotides. The deletion of the whole or a large part of the gene seems rare. Mutations are scattered in each exon with comparable mutation rates (13.0%–34.8% per number of coding nucleotides of each exon). In the present investigation, 18 mutations, including 6 novel ones, were identified in the 22 Japanese patients with hypophophatasia (Table 1). Interestingly, mutations common to the other races were relatively rare in the Japanese patients. In addition, the most and the second most frequent mutations in Japanese patients, T1559del (41%) and F310L (11%) respectively, have not been reported in other races. However, the frequency of the mutations may be biased because we were able to investigate more patients with severe forms of the disease. In our experience, de novo mutations of the TNSALP gene seem rare because the parents of the patients with hypophosphatasia were found to be carriers of one of the mutations found in their children when DNA analysis was performed (data not shown). Thus, it is likely that these common mutations occurred in an ancient Japanese patient with hypophosphatasia and has been maintained in the population. In Europe, the E174K mutation is reported to be frequent (31%) in mild hypophosphatasia [6].
Patients with the classical perinatal form of hypophosphatasia almost always die in utero or in the neonatal period. Therefore, the perinatal form is a synonym for the lethal form. However, here we have reported six patients with hypophosphatasia of fetal onset who manifested the bending of the long bones and/or characteristic bone spurs, but had neither respiratory failure nor apparent hypomineralisation. Most importantly, the prognosis of these patients was rather good, i.e., they achieved long-term survival without life-threatening complications. Obviously the phenotype of these particular patients was benign. Since these patients survive longer despite their fetal onset, we should pay great attention to their care and to genetic counselling. Interestingly, there have been a couple of reports dealing with a benign prenatal form of hypophosphatasia; one of the cases had a D361V mutation [10, 18]. Indeed, routine fetal sonography can greatly contribute to the detection of mild forms of skeletal dysplasia in utero, but this particularly benign form of hypophosphatasia found in Japanese patients is considered to be associated with a specific F310L mutation whose product retained approximately 70% of its enzymatic activity. The relatively high activity of the mutation may contribute to this relatively mild form. However, one patient with similar mild phenotype had the F311L mutation where the enzymatic activity was low. The discrepancy between the phenotype and genotype still remains unclear.
As shown in Table 1, three patients with infantile hypophosphatasia (numbers 17, 18, 19) were also included in the current study. One of them (number 18) was the previously reported patient [17] and is still alive with characteristic skeletal changes, i.e. tongue-like lucent lesions of the metaphyses. In contrast, two of them (numbers 17, 19) died of respiratory failure in infancy. Thus, the prognosis of infantile hypophosphatasia is still poor. In an attempt to improve the poor prognosis, marrow cell transplantation has been performed in infantile hypophosphatasia although more studies on its effect are required before the treatment becomes standard [25]. In our study, both alleles from the surviving case carry the mutants with residual enzymatic activities.
In four patients (numbers 9, 13, 21, 22), including two with childhood hypophosphatasia, mutations were found in only one of the alleles of the TNSALP gene. The information concerning their parents was not available and thus the possibility of dominant inheritance cannot be excluded. However, deletion of the whole or a part of the gene and mutations affecting the expression level are also to be considered. At least secondary hypophosphatasaemia caused by a form of cleidocranial dysplasia (OMIM 119600) and zinc deficiency can be excluded because one allele had the mutation of the gene [22].
In conclusion, we have investigated the relationship between the genotype and phenotype in 22 Japanese patients with hypophosphatasia. Two of the most common mutations were T1559del and F310L and the latter was associated with a unique subtype of hypophosphatasia in which long-term survival was achieved despite the fetal onset of the disease. In the reconstruction experiments, some of the mutant TNSALP proteins associated with infantile hypophosphatasia as well as with the F310L mutation have been revealed to retain residual enzymatic activities. These results enhance the significance of the identification of mutations and the evaluation of the enzymatic activity of the mutant proteins to predict the prognosis of the patients with hypophosphatasia.
Abbreviations
- TNSALP :
-
tissue-nonspecific alkaline phosphatase
References
Antonarakis SE, the Nomenclature Working Group (1998) Recommendations for a nomenclature system for human gene mutations. Hum Mutat 11: 1–3
Barcia JP, Strife CF, Langman CB (1997) Infantile hypophosphatasia: treatment options to control hypercalcemia, hypercalciuria, and chronic bone demineralization. J Pediatr 130: 825–828
Cai G, Michigami T, Yamamoto T, Yasui N, Satomura K, Yamagata M, Shima M, Nakajima S, Mushiake S, Okada S, Ozono K (1998) Analysis of localization of mutated tissue-nonspecific alkaline phosphatase proteins associated with neonatal hypophosphatasia using green fluorescent protein chimeras. J Clin Endocrinol Metab 83: 3936–3942
Henthorn PS, Whyte MP (1992) Missense mutations of the tissue-nonspecific alkaline phosphatase gene in hypophosphatasia. Clin Chem 38: 2501–2505
Henthorn PS, Raducha M, Fedde KN, Lafferty MA, Whyte MP (1992) Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypophosphatasia. Proc Natl Acad Sci USA 89: 9924–9928
Herasse M, Spentchian M, Taillandier A, Mornet E (2002) Evidence of a founder effect for the tissue-nonspecific alkaline phosphatase (TNSALP) gene E174K mutation in hypophosphatasia patients. Eur J Hum Genet 10: 666–668
Lia-Baldini AS, Muller F, Taillandier A, Gibrat JF, Mouchard M, Robin B, Simon-Bouy B, Serre JL, Aylsworth AS, Bieth E, Delanote S, Freisinger P, Hu JC, Krohn HP, Nunes ME, Mornet E (2001) A molecular approach to dominance in hypophosphatasia. Hum Genet 109: 99–108
Lowry OH, Roberts NR, Wu ML, Hixon WS, Crawford EJ (1954) The quantitative histochemistry of brain. II. Enzyme measurements. J Biol Chem 207: 19–37
Mochizuki H, Saito M, Michigami T, Ohashi H, Koda N, Yamaguchi S, Ozono K (2000) Severe hypercalcemia and respiratory insufficiency associated with infantile hypophosphatasia caused by two novel mutations of the TNSALP gene. Eur J Pediatr 159: 375–379
Moore CA, Curry CJR, Henthorn PS, Smith JA, Smith JC, O’Lague P, Coburn SP, Weaver DD, Whyte MP(1999) Mild autosomal dominant hypophosphatasia: in utero presentation in two families. Am J Med Genet 86: 410–415
Mornet E (2000) Hypophosphatasia: the mutations in the tissue-nonspecific alkaline phosphatase gene. Hum Mutat 15: 309–315
Mornet E, Taillandier A, Peyramaure S, Kaper F, Muller F, Brenner R, Bussiere P, Freisinger P, Godard J, Le Merrer M, Oury JF, Plauchu H, Puddu R, Rival JM, Superti-Furga A, Touraine RL, Serre JL, Simon-Bouy B (1998) Identification of fifteen novel mutations in the tissue-nonspecific alkaline phosphatase (TNSALP) gene in European patients with severe hypophosphatasia. Eur J Hum Genet 6: 308–314
Müller HL, Yamazaki M, Michigami T, Kageyama T, Schönau E, Schneider P, Ozono K (2000) Asp361Val mutant of alkaline phosphatase found in patients with dominantly inherited hypophosphatasia inhibits the activity of the wild-type enzyme. J Clin Endocrinol Metab 85: 743–747
Oestreich AE, Bofinger MK (1989) Prominent transverse (Bowdler) bone spurs as a diagnostic clue in a case of neonatal hypophosphatasia without metaphyseal irregularity. Pediatr Radiol 19: 341–342
Orimo H, Hayashi Z, Watanabe A, Hirayama T, Hirayama T, Shimada T (1994) Novel missense and frameshift mutations in the tissue-nonspecific alkaline phosphatase gene in a Japanese patient with hypophosphatasia. Hum Mol Genet 3: 1683–1684
Orimo H, Goseki-Sone M, Inoue M, Tsubakio Y, Sakiyama T, Shimada T (2002) Importance of deletion of T at nucleotide 1559 in the tissue-nonspecific alkaline phosphatase gene in Japanese patients with hypophosphatasia. J Bone Miner Metab 20: 28–33
Ozono K, Yamagata M, Michigami T, Nakajima S, Sakai N, Cai G, Satomura K, Yasui N, Okada S, Nakayama M (1996) Identification of novel missense mutations (Phe310Leu and Gly439Arg) in a neonatal case of hypophosphatasia. J Clin Endocrinol Metab 81: 4458–4461
Pauli RM, Modaff P Sipes SL, Whyte MP(1999) Mild hypophosphatasia mimicking severe osteogenesis imperfecta in utero: bent but not broken. Am J Med Genet 86: 434–438
Sergi C, Mornet E, Troeger J, Voigtlaender T (2001) Perinatal hypophosphatasia: radiology, pathology and molecular biology studies in a family harboring a splicing mutation (648+1A) and a novel missense mutation (N400S) in the tissue-nonspecific alkaline phosphatase (TNSALP) gene. Am J Med Genet 103: 235–240
Spentchian M, Merrien Y, Herasse M, Dobbie Z, Glaser D, Holder SE, Ivarsson SA, Kostiner D, Mansour S, Norman A, Roth J, Stipoljev F, Taillemite JL, van der Smagt JJ, Serre JL, Simon-Bouy B, Taillandier A, Mornet E (2003) Severe hypophosphatasia: characterization of fifteen novel mutations in the ALPL gene. Hum Mutat 22: 105–106
Taillandier A, Zurutuza L, Muller F, Simon-Bouy B, Serre JL, Bird L, Brenner R, Boute O, Cousin J, Gaillard D, Heidemann PH, Steinmann B, Wallot M, Mornet E (1999) Characterization of eleven novel mutations (M45L, R119H, 544delG, G145V, H154Y, C184Y, D289V, 862+5A, 1172delC, R411X, E459K) in the tissue-nonspecific alkaline phosphatase (TNSALP) gene in patients with severe hypophosphatasia. Hum Mutat 13: 171–172
Unger S, Mornet E, Mundlos S, Blaser S, Cole DE (2002) Severe cleidocranial dysplasia can mimic hypophosphatasia. Eur J Pediatr 161: 623–626
Weiss MJ, Henthorn PS, Lafferty MA, Slaughter C, Raducha M, Harris H (1986) Isolation and characterization of a cDNA encoding a human liver/bone/kidney-type alkaline phosphatase. Proc Natl Acad Sci USA 83: 7182–7186
Whyte MP (1994) Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev 15: 439–461
Whyte MP, Kurtzberg J, McAlister WH, Mumm S, Podgornik MN, Coburn SP, Ryan LM, Miller CR, Gottesman GS, Smith AK, Douville J, Waters-Pick B, Armstrong RD, Martin PL (2003) Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res 18: 624–636
Zurutuza L, Muller F, Gibrat JF, Taillandier A, Simon-Bouy B, Serre JL, Mornet E (1999) Correlations of genotype and phenotype in hypophosphatasia. Hum Mol Genet 8: 1039–1046
Acknowledgements
We thank Dr. P. S. Henthorn of the University of Pennsylvania School of Veterinary Medicine for generously providing the TNSALP expression vector. We also thank the physicians listed below for referring patients with hypophosphatasia for TNSALP gene analysis: Drs. Kennichi Satomura and Hidehiko Kawabata, Osaka Medical Centre and Research Institute for Maternal and Child Health; Dr. Norio Miharu, Hiroshima University; Dr. Tsuyoshi Oikawa, Jikei Medical University; Dr. Yu Ikeda, Shiga Medical University; Dr. Yasushi Uchida, Nagahama Municipal Hospital; Dr. Go Hasegawa, Nikko Memorial Hospital; Drs. Hiroyuki Tanaka, Masaru Inoue, Okayama University School of Medicine; Dr. Toru Ogiwara, Osaka Medical School; Drs. Katshuhiko Tachibana, Masayuki Adachi, Tetshusi Ueno, Hiroshima Municipal Hospital; Dr. Hiroshi Mochizuki, Saitama Medical Centre; Dr. Oguchi, Hata Hospital; Dr. Shuichiro Akagi, Chifune Hospital; Dr. Yuka Sasamoto, St. Marianna University; Dr. Kusuki, Osaka Medical Centre; Dr. Ichiba, Osaka City University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Michigami, T., Uchihashi, T., Suzuki, A. et al. Common mutations F310L and T1559del in the tissue-nonspecific alkaline phosphatase gene are related to distinct phenotypes in Japanese patients with hypophosphatasia. Eur J Pediatr 164, 277–282 (2005). https://doi.org/10.1007/s00431-004-1612-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-004-1612-9