
Abstract
Mutations in the ALPL gene encoding tissue-nonspecific alkaline phosphatase (TNSALP) cause hypophosphatasia (HPP), a genetic disorder characterized by deficiency of serum ALP and hypomineralization of bone and teeth. Three missense mutations for glycine 426 (by standard nomenclature) of TNSALP have been reported: cysteine (p.G426C), serine (p.G426S), and aspartate (p.G426D). We expressed TNSALP mutants carrying each missense mutation in mammalian cells. All three TNSALP mutants appeared on the cell surface like the wild-type (WT) TNSALP, although the cells expressing each TNSALP mutant exhibited markedly reduced ALP activity. TNSALP (WT) was mainly present as a 140 kDa catalytically active dimeric form, whereas ~80 kDa monomers were the predominant molecular species in the cells expressing TNSALP (p.G426D) or TNSALP (p.G426S), suggesting that aspartate or serine at position 426 may hamper the subunit assembly essential for the enzymatic function of TNSALP. Alternatively, the subunits of TNSALP (p.G426C) were found to be aberrantly cross-linked by disulfide bonds, giving rise to a 200 kDa form lacking ALP activity. Taken together, our results reveal that the amino acid substitutions at position 426 of TNSALP differentially affect the structure and function of TNSALP, leading to understanding of the molecular and cellular basis of HPP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypophosphatasia (HPP) is an inborn metabolism error resulting from multiple mutations of the ALPL gene encoding tissue-nonspecific alkaline phosphatase (TNSALP) [1–6], which is anchored to the cell membrane via glycosylphosphatidylinositol (GPI) and hydrolyzes phosphomonoesters such as inorganic pyrophosphate, phosphoethanolamine, and pyridoxal phosphate [1, 2]. Generally, the loss-of-function of TNSALP causes the accumulation of inorganic pyrophosphate, a crystal poison of hydroxyapatite, leading to a reduction in bio-mineralization [1–6]. HPP is classified into five principal forms: (1) perinatal, (2) infantile, (3) childhood, (4) adult, and (5) odonto-HPP [1–4]. It is clinically broad-ranging, encompassing a wide range of symptoms from still birth in utero with severe skeletal hypomineralization in perinatal HPP to premature loss of deciduous teeth and dental caries without bone symptoms in odonto-HPP [1–4]. Severe forms (perinatal and infantile) of HPP are inherited in an autosomal recessive manner, while milder forms (childhood, adult, and odonto-HPP) manifest both autosomal recessive and autosomal dominant patterns [1–4].
TNSALP consists of two homologous subunits harnessed by noncovalent bonds [1]. TNSALP is biosynthesized as a ~70 kDa immature form with high-mannose-type oligosaccharides and becomes an ~80 kDa mature form with complex-type oligosaccharide chains during its transport from the ER to the cell membrane via the Golgi [1, 7]. A total of 322 mutations in the ALPL gene have been identified worldwide as of November in 2016 (http://www.sesep.uvsq.fr/03_hypo_mutations.php). Most of them are missense mutations (75%) with deletions, splicing mutations, nonsense mutations, and insertions: these loss-of-function mutations may affect several steps during the synthesis of TNSALP and its subsequent trafficking, resulting in diminished and altered expression of TNSALP on the cell surface [7–15]. Several TNSALP mutants failed to fold correctly or assemble properly, and hence accumulated in the ER [7, 9, 11, 13, 15] or Golgi [10], followed by ER-associated degradation [10, 11, 13]. Some TNSALP mutants appeared on the cell surface, although they exhibited a decreased and/or altered catalytic activity [16, 17]. In particular, TNSALP mutants related to the dominant form of HPP were present on the cell surface as a monomer completely lacking catalytic activity [18–20]. One particular frame-shift mutant, which is most prevalent in Japanese HPP patients, was no longer modified with GPI and became a soluble form [21].
Three different missense mutations have been reported at position 426 of TNSALP: p.G426C [22] and p.G426S (http://www.sesep.uvsq.fr/03_hypo_mutations.php) in infantile HPP and p.G426D [23] in childhood HPP. A 3D model of TNSALP revealed that glycine at position 426 was located at the top of the crown domain of TNSALP, a unique domain involved in collagen-binding, allosteric nature, heat stability, and subunit assembly of TNSALP [24, 25]. However, little is known about how each missense mutation affects TNSALP at the molecular level. In this report, we characterized three TNSALP mutants bearing an amino acid substitution at position 426 of TNSALP by expressing them in a heterogeneous mammalian expression system. Our data reveal that TNSALP (p.G426D) and TNSALP (p.G426S) exist mostly as monomers in transfected cells, suggesting that aspartate or serine at position 426 may compromise the subunit assembly of TNSALP. In marked contrast to TNSALP (p.G426D) and TNSALP (p.G426S), a unique cross-linked product via disulfide bonds was found in the cells expressing TNSALP (p.G426C).
Materials and methods
Plasmids and transfection
pSG5 vector (Stratagene, La Jolla, CA, USA) encoding TNSALP (WT) was created as described previously [7] and further used as a template for oligonucleotide-directed mutagenesis. The QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) was used to introduce mutations. The oligonucleotides used (mutated nucleotides are shown in bold) were: p.G426C sense, 5′-CAAGGTGGTGGGC- TGTGAACGAGAGAATGTC 3′ and p.G426C antisense, 5′-TGCCGGCGCTGTCAAGGACCTGGG-CATTGGT-3′; p.G426D sense 5′-CAAGGTGGTGGGCGATGAACGAGAGAATGTC-3′ and p.G426D antisense, 5′-GACATTCTCTCGTTCATCGCCCACC-ACCTTG-3; p.G426S sense, 5′-CAAGGTGGTGG-GCAGTGAACGAGAGAATGTC-3′ and p.G426S antisense, 5′-ACATTCTCTCGTTCACTGCCCACC-ACCTTG-3′ The full coding sequences of the TNSALP mutants were verified by DNA sequencing. TNSALP (p.G426C) was further ligated into pTRE2-hyg (Invitrogen, Carlsbad, CA, USA). COS-1 cells, inoculated at a density of 8 × 104 cells/35-mm dish 24 h before transfection, were transiently transfected with 0.5 μg of each plasmid using Lipofectamine Plus reagent (Invitrogen, Carlsbad, CA, USA). COS-1 cells were further cultured for 24 h after transfection and used as described previously [7]. For establishing cells, CHO K1 Tet-On cells (Invitrogen, Carlsbad, CA, USA) were transfected with 0.5 μg of pTRE2-hyg encoding TNSALP (p.G426C) using Lipofectamine Plus reagent. Two days after transfection, the cells were subjected to selection with hygromycin B (400 μg/ml) essentially according to the manufacturer’s protocol and screened by immunofluorescence for induction of TNSALP (p.G426C) in the presence of 1 μg/ml doxycycline (DOX), as described previously [26]. The established cells were cultured in the presence of 1 μg/ml doxycycline (DOX) for 24 h before use. CHO K1 Tet-On cells expressing TNSALP (WT) [20] or TNSALP (p.R450C, R433in nonstandard nomenclature) [16] were established as described previously.
Miscellaneous procedures
Conventional SDS-PAGE and modified SDS-PAGE for ALP were carried out essentially as described previously [19, 20]. SuperSep™ Ace (10% gel; Wako Pure Chemical, Osaka, Japan) was used throughout the experiment. Precision Plus Protein Standards and Precision Plus Protein Western C standards were obtained from BIO-RAD (Hercules, CA, USA). Unless otherwise stated, 5 μg of the cell homogenate was applied for western blotting. Cytochemical staining [9, 10], immunofluorescence [11], ALP assay [7, 11], western blotting [10, 13] and digestion using phosphatidylinositol-specific phospholipase C [20] were carried out as described previously. ALP activity was expressed as nmol of p-nitrophenyl formed per min at 37 °C. Anti-human TNSALP antibody was raised in rabbits [27].
Results
Expression of TNSALP mutants in COS-1 cells
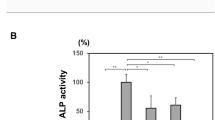
In order to investigate how three TNSALP mutations (p.G426C, pG426S, and p.G426D) affect the structure and/or function of TNSALP at the molecular level thereby causing HPP, we attempted to express each TNSALP mutant protein in COS-1 cells transiently as shown in Fig. 1. All three TNSALP mutants appeared on the cell surface like TNSALP (WT), as evidenced by immunofluorescence, although the intensity of ALP activity staining of each TNSALP mutant was significantly weaker than that of TNSALP (WT). Consistent with morphological observations, the cellular enzymatic activity of each TNSALP mutant was markedly diminished based on a quantitative ALP assay (Fig. 1b). These results indicate that the catalytic function, but not cell surface localization of the TNSALP mutants, was perturbed by each amino acid substitution linked to HPP.

Expression of TNSALP (p.G426C), TNSALP (p.G426D), and TNSALP (p.G426S) in COS-1 cells. a Cells were untransfected (U) or transfected with a plasmid encoding TNSALP (WT), TNSALP (p.G426C), TNSALP (p.G426D), or TNSALP (p.G426S), and processed for immunofluorescence using an anti-TNSALP antibody or cytochemically stained for ALP activity for 10 min at room temperature. WT, 426C, 426D, and 426S denote TNSALP (WT), TNSALP (p.G426C), TNSALP (p.G426D), and TNSALP (p.G426S), respectively. Each TNSALP mutant protein appeared on the cell surface like TNSALP (WT); however, they produced reduced ALP staining products compared with TNSALP (WT). In (b), cells were cultured as in (a), but were homogenized and assayed for their ALP activity (expressed as a percentage of the wild-type enzyme) using p-nitrophenylphosphate as a substrate. Data represent the average of two independent experiments. Consistent with cytochemical staining in (a), enzymatic activities of TNSALP mutants were markedly reduced compared with that of TNSALP (WT). Only trace ALP activity was found in the cells expressing TNSALP (p.G426D)
As the dimeric structure of TNSALP is held non-covalently, it dissociates into monomers in conventional SDS-PAGE. Western blotting using anti-TNSALP antibody showed that TNSALP (WT), TNSALP (p.G426D), and TNSALP (p.G426S) were very similar to one another (Fig. 2a); in addition to an immature form, a mature monomer form, as observed in TNSALP (WT), was also detected in the cells expressing TNSALP (p.G426D) or TNSALP (p.G426S) compatible with their cell surface appearance (Fig. 1a). Next, the same samples were analyzed by a modified SDS-PAGE for ALP (Fig. 2b). TNSALP (WT) retained its dimeric structure and catalytic activity when it was dissolved with SDS at a higher pH and incubated at 37 °C instead of 95 °C [19, 20]. TNSALP (WT) migrated as a 140 kDa form retaining ALP activity (lanes, 2, 7). Note that the mature monomer band was completely absent (lane 7), indicating all mature forms were efficiently assembled into the dimeric form in the cells expressing TNSALP (WT). Under the same conditions, however, virtually no 140 kDa form was detected in the cells expressing TNSALP (p.G426D) or TNSALP (p.G426S) by western blotting; both the immature and mature monomers were the predominant molecular species in transfected cells (lanes 9, 10). This suggests that aspartate or serine at position 426 markedly compromised the subunit assembly of TNSALP such that only a trace amount of the dimeric form was produced in the cells (lanes 4, 5). In addition, aspartate had a more profound effect on TNSALP than serine did at position 426 (Figs. 1b, 2b, lanes 4, 5).

Molecular species of TNSALP mutants analyzed by SDS-PAGE. The cells transfected as in (Fig. 1) were subjected to conventional SDS-PAGE under reducing or non-reducing conditions, followed by western blotting using the anti-TNSALP antibody (a). WT, 426C, 426D, and 426S denote TNSALP (WT), TNSALP (p.G426C), TNSALP (p.G426D), and TNSALP (p.G426S), respectively. M and IM indicate mature monomeric and immature monomeric TNSALP, respectively. Under reducing conditions, each TNSALP mutant resembled TNSALP (WT) with the immature and mature bands. However, TNSALP (p.G426C) formed a unique 200 kDa band cross-linked by disulfide bonding (lanes 3, 8). In (b), the same samples as in (a) were analyzed by a modified SDS-PAGE for ALP. The samples were adjusted to a final concentration of 50 mM in Tris–HCl buffer (pH 8.8) and incubated for 30 min at 37 °C in the presence of SDS, followed by electrophoresis with zinc ions. The gels were stained for ALP activity using 5-bromo-3-indolyl phosphate salt as a substrate or subjected to western blotting. D, M, and IM indicate dimeric, mature monomeric, and immature monomeric TNSALP, respectively. In contrast to TNSALP (WT), showing a 140 kDa dimeric functional form (lanes 2, 7) as a major band, both TNSALP (p.G426D) and TNSALP (p.G426S) consisted of monomers (lanes 9, 10). The 200 kDa band of TNSALP (p.G426C) exhibited no ALP activity (lanes 3, 8)
Some TNSALP mutants linked to mild forms of HPP are known to be transmitted in a dominant inheritance manner [28, 29]. TNSALP (p.P108L) [20], TNSALP (p.A116T) [26], TNSALP (p.N417S) [19], and TNSALP (p.G420S) [18] fall into this group. Previous studies demonstrated that these TNSALP mutants failed to assemble into the functional dimeric structure, although they were able to gain access to the cell surface as a monomer [18–20] or a heterogeneously aggregated form [20, 26]. Importantly, these TNSALP mutants exerted a dominant negative effect on TNSALP (WT) when each TNSALP mutant was co-expressed with TNSALP (WT) [18–20, 29]. It is of interest to see if TNSALP (p.G426D) and TNSALP (p.G426S) have a dominant negative nature. The transfection efficiency of each plasmid was found to be the same level as assessed by western blotting (Fig. 2a). Figure 3 shows a co-expression experiment in which TNSALP (p.G426D) or TNSALP (p.G426S) was expressed with TNSALP (WT). Neither TNSALP (p.G426D) nor TNSALP (p.G426S) blocked the enzymatic activity of TNSALP (WT), excluding the possibility that these mutants are transmitted dominantly.

Co-expression experiments. Cells were co-transfected with TNSALP (WT) and TNSALP (p.G423D) (a) or TNSALP (p.G426S) (b) in differing ratios (1:0, 0.9:0.1, 0.5:0.5, 0.1:0.9, 0:1, x-axis). Cells were homogenized and assayed for ALP activity (expressed as a percentage of the wild-type enzyme, Y-axis). A dotted line represents the recessive model, where each TNSALP mutant does not exert an inhibitory effect on TNSALP (WT). Data represent the average of two independent experiments. Neither TNSALP (p.G426D) nor TNSALP (p.G426S) impaired the enzymatic activity of TNSALP (WT)
In a marked contrast to TNSALP (p.G426D) or TNSALP (p.G426S), a 200 kDa form was found in the cells expressing TNSALP (p.G426C) (Fig. 2a, lane 8). As this high-molecular mass band completely disappeared under reducing conditions (lanes 3, 8), it is highly likely that this 200 kDa TNSALP (p.G426C) was composed of the monomers cross-linked by disulfide bonds. Furthermore, ALP activity staining revealed that the 200 kDa form completely lost ALP activity (Fig. 2b, lanes 3, 8). However, a weak ALP staining product was also located at the position on the gel corresponding to the 140 kDa TNSALP (WT) (lanes 2, 3). Although this dimeric form of TNSALP (p.G426C) was negligible on the western blot, it seems reasonable that a significant ALP staining product was amplified and accumulated due to its catalytic nature.
Establishing Tet-On CHO cells expressing TNSALP (p.G426C)
There was a possibility that the 200 kDa disulfide-bonded band may have been an artificial form resulting from transient expression, in which a huge amount of the TNSALP mutant was produced; its appearance does not necessarily reflect in vivo state. To circumvent this, we established Tet-On cells expressing TNSALP (p.G426C) in response to doxycycline (DOX). The advantage of this induction system was reported previously [16, 17]. TNSALP (p.G426C) appeared on the cell surface (Fig. 4a) and exhibited weak ALP staining along the contours of CHO cells (Fig. 4b) in the presence but not absence of DOX. In agreement with the cytochemical staining, the cells expressing TNSALP (p.G426C) had a small but significant enzymatic activity (Fig. 3c). Moreover, the 200 kDa disulfide-bonded form lacking ALP activity was prominent in the established cells expressing TNSALP (p.G426C), but not TNSALP (WT) (Fig. 5, lanes 4, 5), and was found to be released into the medium upon digestion with phosphatidylinositol-specific phospholipase C (data not shown). A weak staining product was again localized at the same position as the 140 kDa TNSALP (WT) (lanes 7, 8), implying that TNSALP (p.G426C) is naturally able to form a small amount of the 140 kDa functional form. Thus, we demonstrated that TNSALP (p.G426D) gave rise to the unique 200 kDa disulfide-bonded form using both the transient expression system and the established cell line. Nanoscale liquid chromatography coupled with tandem mass spectrometry (nano LC–MS/MS) showed that no peptide other than TNSALP was detected in purified 200 kDa GPI-anchorless TNSALP (p.G426C) (data not shown), demonstrating that this disulfide-bonded form solely consisted of the monomers of TNSALP. Previously, we reported that TNSALP (p.G450C, equivalent to p.R433C in nonstandard nomenclature), which was reported in severe HPP [16], formed an inactive disulfide-bonded dimeric enzyme based on sucrose-density-gradient centrifugation [16]. Indeed, TNSALP (p.G426C) was found to be larger than TNSALP (p.R450C) (Lanes 5, 6, and 11, 12). However, unexpectedly, even TNSALP (p.R450C) migrated slower than the authentic 140 kDa TNSALP (WT) (lanes 10, 12), as judged by the modified SDS-PAGE for ALP.

Established Tet-On CHO-K1 cells expressing TNSALP (p.G426C). Established cells harboring a plasmid encoding TNSALP (WT) or TNSALP (p.G426C) were cultured without (-DOX) or with (+DOX) doxycycline for 24 h, followed by immunofluorescence using the anti-TNSALP antibody (a) or cytochemical staining (b). WT and 426C denote TNSALP (WT) and TNSALP (p.G426C), respectively. TNSALP (p.G426C) appeared on the cell surface, but showed only weak ALP staining products compared with TNSALP (WT). In (c), established cells were cultured without (-D) or with (+D) doxycycline for 24 h, homogenized and then assayed for ALP activity (expressed as a percentage of the wild-type enzyme, Y-axis). WT and 426C denote TNSALP (WT) and TNSALP (p.G426C), respectively. Data represent the average of two independent experiments. In agreement with b, TNSALP (p.G426C) showed markedly reduced enzymatic activity

Analysis of TNSALP (p.G426C) and TNSALP (p.R450C) by SDS-PAGE. Established Tet-On CHO cells harboring a plasmid encoding TNSALP (WT), TNSALP (p.G426C), or TNSALP (p.R450C) were cultured in the presence of DOX for 24 h, homogenized and then subjected to conventional or modified SDS-PAGE for ALP. WT, 426C, and 450C denote TNSALP (WT), TNSALP (p.G426C), and TNSALP (p.R450C), respectively. D, M, and IM indicate dimeric, mature monomeric, and immature monomeric TNSALP, respectively. TNSALP (p.G426C) migrated slower than TNSALP (p.R450C) did in the two SDS-PAGE systems (lanes 5, 6 and 11, 12), possibly reflecting a difference in the numbers of the subunits cross-linked by disulfide bonds. M denotes molecular markers
Discussion
HPP is an inborn error of metabolism and caused by multiple loss-of-function mutations in the ALPL gene encoding TNSALP. More than 75% of the mutations are missense. With the knowledge of the 3D structure of TNSALP, these natural mutations give us an opportunity to elucidate the effects of amino acid substitutions on TNSALP at the molecular level, thus leading to the understanding of the molecular and cellular basis of HPP [24]. Glycine at position 426 is localized at the top of the substructure of TNSALP, the crown domain, and is unique to mammalian ALPs [24]. The crown domain, which consists of 65 amino acid residues including G426 of each subunit, is involved in collagen-binding, allosteric nature, heat stability, and subunit assembly of TNSALP [24, 25]. Three different missense mutations have been reported at position 426: p.G426C and p.G426S in infantile HPP, and p.G426D in childhood HPP. In this report, we characterized these three TNSALP mutants by expressing them in mammalian cells to gain insight to the molecular mechanism, whereby these mutations lead to the loss-of-function of TNSALP. Both TNSALP (p.G426D) and TNSALP (p.G426S) appeared on the cell surface like TNSALP (WT) as assessed by immunofluorescence; however, their ALP activities were markedly reduced compared with that of TNSALP (WT) (Fig. 1). By an advantage of the modified SDS-PAGE for ALP, we demonstrated that TNSALP (WT) was mostly present as a dimeric functional enzyme, while the monomers of TNSALP (p.G426D) and TNSALP (p.G426S) were only observed by western blotting (Fig. 2). Therefore, these results demonstrate that aspartate or serine at position 426 markedly reduces the probability of the subunit assembly of TNSALP. It is worth noting that an ALP staining product on the gel corresponding to the authentic 140 kDa TNSALP (WT) was detected even in cells expressing TNSALP (p.G426D) or TNSALP (p.G426S). Thus, both TNSALP mutants were able to assemble into the dimeric structure albeit to a quite lesser extent. The bulky side chain and/or negative charge of aspartate at position 426 may impede the dimer formation more severely than the hydroxyl group of serine, leading to an assumption that only glycine accommodates well in this crucial spot of the subunit interface of TNSALP. Regardless of their apparent resemblance to some dominantly transmitted molecular phenotypes, both TNSALP (p.G426D) and TNSALP (p.G426S) may be inherited in a recessive manner as suggested by co-expression studies (Fig. 3).
Substitution of glycine with cysteine at position 426 resulted in the formation of the 200 kDa disulfide-bonded molecular species (Fig. 2). Presumably, the additional cysteine residue on the crown domain may elicit the aberrant disulfide-bonded crosslinking of TNSALP with only a small amount of the dimeric functional TNSALP. There is a precedent to TNSALP (p.G426C). We previously reported that TNSALP (p.R450C) formed a disulfide-bonded molecular species [16]. As estimated by sucrose-density-gradient centrifugation, we tentatively assumed that TNSALP (p.R450C) was an inter-chain cross-linked dimer via the cysteine residue at position 450 [16]. However, TNSALP (p.R450C) was found to migrate more slowly than TNSALP (WT) in the modified SDS-PAGE for ALP (Fig. 5). Furthermore, it was evident that TNSALP (p.G426C) was larger than TNSALP (p.R450C). According to the estimated molecular size based in SDS-PAGE, we speculate that TNSALP (p.G426C) consists of three subunits cross-linked by disulfide bonds. Further study is necessary to build the molecular models of these aberrant TNSALPs.
Finally, it is worth noting that both basic and clinical researches on HPP have recently culminated in a treatment of HPP: enzyme-replacement therapy using a genetically engineered soluble form of TNSALP has been successfully used for HPP patients [30].
Abbreviations
- ALP:
-
Alkaline phosphatase
- TNSALP:
-
Tissue-nonspecific alkaline phosphatase
- HPP:
-
Hypophosphatasia
- TNSALP (p.G426C):
-
TNSALP with a glycine to cysteine substitution at position 426
- TNSALP (p.G426S):
-
TNSALP with a glycine to serine substitution at position 426
- TNSALP (p.G426D):
-
TNSALP with a glycine to aspartate substitution at position 426
- TNSALP (WT):
-
Wild-type TNSALP
- GPI:
-
Glycosylphosphatidylinositol
- ER:
-
Endoplasmic reticulum
- DOX:
-
Doxycycline
References
Millán JL (2006) Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. Wiley, Weinheim
Whyte MP (2002) The metabolic and molecular bases of inherited diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW (eds) vol. IV 8th edn. McGraw-Hill, New York, pp. 5319–5329
Mornet E (2007) Hypophosphatasia. Orphanet J Rare Dis 2:40
Whyte MP (2010) Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann NY Acad Sci 1192:190–200
Millán JL (2013) The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int 93:299–306
Mornet E (2013) Genetics of hypophosphatasia. Clin Rev Bone Miner Metab 11:71–77. doi:10.1007/s12018-013-9140-7
Shibata H, Fukushi M, Igarashi A, Misumi Y, Ikehara Y, Ohashi Y, Oda K (1998) Defective intracellular transport of tissue-nonspecific alkaline phosphatase with an Ala162Thr mutation associated with lethal hypophosphatasia. J Biochem (Tokyo) 123:968–977
Cai G, Michigami T, Yamamoto T, Yasui N, Satomura K, Yamagata M, Shima M, Nakajima S, Mushiake S, Okada S, Ozono K (1998) Analysis of localization of mutated tissue nonspecific alkaline phosphatase proteins associated with neonatal hypophosphatasia using green fluorescent protein chimeras. J Clin Endocrinol Metab 83:3936–3942
Fukushi-Irie M, Ito M, Amaya Y, Amizuka N, Ozawa H, Omura S, Ikehara Y, Oda K (2000) Possible interference between tissue-non-specific alkaline phosphatase with an Arg54-Cys substitution and a counterpart with an Asp277-Ala substitution found in a compound heterozygote associated with severe hypophosphatasia. Biochem J 348:633–642
Ito M, Amizuka N, Ozawa H, Oda K (2002) Retention at the cis-Golgi and delayed degradation of tissue-non-specifi. Biochem J. doi:10.1042/bj3610473
Ishida Y, Komaru K, Ito M, Amaya Y, Kohno S, Oda K (2003) Tissue-nonspecific alkaline phosphatase with an Asp289-Val mutation fails to reach the cell surface and undergoes proteasome-mediated degradation. J Biochem (Tokyo) 134:63–70
Brun-Heath Lia-Baldini AS, Maillard S, Taillandier A, Utsch B, Nunes ME, Serre JL, Mornet E (2007) Delayed transport of tissue-nonspecific alkaline phosphatase with missense mutations causing hypophosphatasia. Eur J Med Genet 50:367–378
Satou Y, Al-Shawafi HA, Sultana S, Makita S, Sohda M, Oda K (2012) Disulfide bonds are critical for tissue-nonspecific alkaline phosphatase function revealed by analysis of mutant proteins bearing a C201-Y or C489-S substitution associated with severe hypophosphatasia. Biochim Biophys Acta 1822:581–588
Yang H, Wang L, Geng J, Yu T, Yao R, Shen Y, Yin L, Ying D, Huang R, Zhou Y, Chen H, Liu L, Mo X, Shen Y, Fu Q, Yu Y (2013) Characterization of six missense mutations in the tissue-nonspecific alkaline phosphatase (TNSALP) gene in Chinese children with hypophosphatasia. Cell Physiol Biochem 32:635–644
Komaru K, Satou Y, Al-Shawafi HA, Numa-Kinjoh N, Sohda M, Oda K (2016) Glycosylation-deficient mutations in tissue-nonspecific alkaline phosphatase impair its structure and function and are linked to infantile hypophosphatasia. FEBS J 283:1168–1179
Nasu M, Ito M, Ishida Y, Numa N, Komaru K, Nomura S, Oda K (2006) Aberrant interchain disulfide bridge of tissue-nonspecific alkaline phosphatase with an Arg433Cys substitution associated with severe hypophosphatasia. FEBS J 273:5612–5624
Numa N, Ishida Y, Nasu M, Sohda M, Misumi Y, Noda T, Oda K (2008) Molecular basis of perinatal hypophosphatasia with tissue-nonspecific alkaline phosphatase bearing a conservative replacement of valine by alanine at position 406 structural importance of the crown domain. FEBS J 275:2727–2737
Makita S, Al-Shawafi HA, Sultana S, Sohda M, Nomura S, Oda K (2012) A dimerization defect caused by a glycine substitution at position 420 by serine in tissue nonspecific alkaline phosphatase associated with perinatal hypophosphatasia. FEBS J 279:4327–4337
Sultana S, Al-Shawafi HA, Makita S, Sohda M, Amizuka N, Takagi R, Oda K (2013) An asparagine at position 417 of tissue-nonspecific alkaline phosphatase is essential for its structure and function as revealed by analysis of the N417S mutation associated with severe hypophosphatasia. Mol Genet Metab 109:282–288
Numa-Kinjoh N, Komaru K, Ishida Y, Sohda M, Oda K (2015) Molecular phenotype of tissue-nonspecific alkaline phosphatase with a proline (108) to leucine substitution associated with dominant odontohypophosphatasia. Mol Genet Metab 115:180–185
Komaru K, Ishida Y, Amaya Y, Goseki-Sone M, Orimo H, Oda K (2005) Novel aggregate formation of a frame-shift mutant protein of tissue- nonspecific alkaline phosphatase is ascribed to three cysteine residues in the C-terminal extension, retarded secretion and proteasomal degradation. FEBS J 272:1704–1717
Mochizuki H, Saito M, Michigami T, Ohashi H, Koda N, Yamaguchi S, Ozono K (2000) Severe hypercalcaemia and respiratory insufficiency associated with infantile hypophosphatasia caused by two novel mutations of the tissue-nonspecific alkaline phosphatase gene. Eur J Pediatr 159:375–379
Mumm S, Jones J, Finnegan P, Henthorn PS, Podgornik MN, Whyte MP (2002) Denaturing gradient gel electrophoresis analysis of the tissue nonspecific alkaline phosphatase isoenzyme gene in hypophosphatasia. Mol Genet Metab 75:143–153
Mornet E, Stura E, Lia-Baldini AS, Stigbrand T, Ménez A, Le Du MH (2001) Structural evidence for a functional role of human tissue nonspecific alkaline phosphatase in bone mineralization. J Biol Chem 276:31171–31178
Bossi M, Hoylaerts MF, Millán JL (1993) Modifications in a flexible surface modulate the isozyme-specific properties of mammalian alkaline phosphatase. J Biol 268:25409–25416
Ishida Y, Komaru K, Oda K (2011) Molecular characterization of tissue-nonspecific alkaline phosphatase with an Ala to Thr substitution at position 116 associated with dominantly inherited hypophosphatasia. Biochim Biophys Acta 1812:326–332
Oda K, Amaya Y, Fukushi-Irie M, Kinameri Y, Ohsuye K, Kubota I, Fujimura S, Kobayashi J (1999) A general method for rapid purification of soluble versions of glycosylphosphatidylinositol-anchored proteins expressed in insect cells: an application for human tissue-nonspecific alkaline phosphatase. J Biochem (Tokyo) 126:694–699
Fauvert D, Brun-Heath I, Lia-Baldini AS, Bellazi L, Taillandier A, Serre JL, de Mazancourt P, Mornet E (2009) Mildform forms of hypophosphatasia most result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet 10:51
Lia-Baldini AS, Muller F, Taillandier A, Gibrat JF, Mouchard M, Robin B, Simon-Bouy B, Serre JL, Aylsworth AS, Bieth E, Delanote S, Freisinger P, Hu JC, Krohn HP, Nunes ME, Mornet E (2001) A molecular approach to dominance in hypophosphatasia. Hum Genet 109:99–108
Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millán JL, Skrinar AM, Crine P, Landy H (2012) Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 366:904–913
Acknowledgements
The authors thank Miyako Okamura for her technical assistance. This work was supported in part by a Grand-in-Aid for Scientific Research (to K.O., No. 24592794), from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Al-Shawafi, H.A., Komaru, K. & Oda, K. Molecular defect of tissue-nonspecific alkaline phosphatase bearing a substitution at position 426 associated with hypophosphatasia. Mol Cell Biochem 427, 169–176 (2017). https://doi.org/10.1007/s11010-016-2908-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2908-6