
Abstract
Cytomegaloviruses (CMVs), members of the β-subfamily of the herpesvirus family, have co-speciated with their respective mammalian hosts resulting in a mutual virus–host adaptation reflected by sets of ‘private’ viral genes that a particular CMV species does not share with other CMVs and that define the host-species specificity of CMVs. Nonetheless, based on “biological convergence” in evolution, fundamental rules in viral pathogenesis and immune control are functionally analogous between different virus–host pairs. Therefore, the mouse model of infection with murine CMV (mCMV) has revealed generally valid principles of CMV–host interactions. Specifically, the mouse model has paved the way to cellular immunotherapy of CMV disease in immunocompromised recipients of hematopoietic cell transplantation (HCT). Precisely in the context of HCT, however, current view assumes that there exists a major difference between hCMV and mCMV regarding “latent virus reservoirs” in that only hCMV establishes latency in hematopoietic lineage cells (HLCs), whereas mCMV establishes latency in endothelial cells. This would imply that only hCMV can reactivate from transplanted HLCs of a latently infected donor. In addition, as viral transcriptional activity during latency is discussed as a driver of clonal T-cell expansion over lifetime, a phenomenon known as “memory inflation”, it is important to know if hCMV and mCMV establish latency in the same cell type(s) for imprinting the immune system. Here, we review the currently available evidence to propose that the alleged difference in latent virus reservoirs between hCMV and mCMV may rather relate to a difference in the focus of research. While studies on hCMV latency in HLCs likely described a non-canonical, transient type-2 latency, studies in the mouse model focussed on canonical, lifelong type-1 latency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Like all members of the herpesvirus family, cytomegaloviruses (CMVs) are retained in their respective mammalian hosts after immune-mediated clearance of productive infection in a dormant state referred to as “latency”. By definition, infectious virions are absent in latency, so that the host is no longer infectious, but the virus genome is maintained for a lifetime in a replicatively dormant state from which the productive cycle can be reactivated, resulting in recurrent infection with a risk of recrudescent disease [1]. Recurrence of latent human cytomegalovirus (hCMV) is a major clinical concern in immunocompromised recipients of hematopoietic cell transplantation (HCT), where interstitial pneumonia is a frequent organ manifestation, as well as of solid organ transplantation (SOT), where it can cause graft failure and also spread to host organs, depending on the degree of immunosuppression (for selected clinical reviews, see [2,3,4,5,6,7]). To predict the risk of transmitting latent CMV from a latently infected transplant donor to a recipient in HCT and SOTs, knowledge of cellular reservoirs of latent virus genomes is a fundamental need. Surprisingly, despite decades of research, “latent virus reservoirs” are still a matter of debate and current evidence suggests a difference between hCMV latency in human cells and the mouse model of latent infection with murine CMV (mCMV). Here, we discuss our view on the interpretation of data on cellular sites of latent infection, comparing clinical CMV and the mouse model.
The dilemma of how to verify CMV latency on the organismal level
For decades of research on herpesviral latency in general, the strict definition of latency as it originally was proposed for neuronal latency of alpha-herpesviruses by Roizman and Sears [1] was disputed with the argument that available assays for infectious virions may not be sensitive enough to rigorously exclude a low-level productive “persistent” infection. Some authors still do not clearly distinguish between the terms “latency” and “persistence”. Whereas the existence of a latent state of the viral genome on the cellular level is meanwhile undoubted and characterized in molecular depth (see below), latency on the organismal level is more difficult to verify. The distinction between latency at all sites and persistence at particular sites is a relevant issue, as, under conditions of immunosuppression, very low numbers of infectious virus can expand in numbers by replication and can spread in the body, thereby mimicking virus recurrence with no preceding event of reactivation from latency. Obstacles include the following:
Limited assay sensitivity
Roizman’s strict definition of latency would demand that not a single infectious virion is present. However, as shown in the mouse model, one infectious unit of mCMV defined by plaque formation on permissive embryonic fibroblasts, that is a plaque-forming unit (PFU), represents ~ 500 viral genomes, that is the genome-to-infectivity ratio is ~ 500:1 [8]. This order of magnitude was subsequently confirmed for all tested strains of mCMV, including recombinant viruses generated by bacterial artificial chromosome (BAC) mutagenesis ([9], and references therein). This does not mean that the vast majority of viral genomes is defective, as the method of “centrifugal enhancement of infectivity” ([10], and references therein) combined with the detection of immediate-early (IE) transcripts in the indicator fibroblast cell cultures reduced the genome-to-infectivity ratio to a range of 2–9 [8]. Many in vivo studies on latency in the mouse model relied on latency defined just by arbitrary time after virus application with no evidence at all or on absence of infectivity based on the conventional, quite insensitive PFU assay. In our view, many of these studies are, thus, inconclusive. As the clearance kinetics of productive infection can vary substantially depending on the host organ as well as on the precise experimental conditions, including mouse genetics and immune status, a time when latency is established cannot be predicted. Instead, the status of CMV latency must be verified in each study situation.
Sampling error
Evidently, the absence of infectivity in humans on the whole-body level cannot be verified by taking biopsies of all tissue sites, and even within a particular organ of interest, a biopsy specimen may miss the site at which virus replicates focally. In the mouse model, it is in principle possible to test an organ homogenate in toto, but analysis is mostly restricted to known key organs of viral replication. Whole-body imaging using reporter viruses, which nicely revealed sites of full-blown acute infection by recombinant virus mCMVluc expressing luciferase for bioluminescence imaging [11], might offer a solution, provided that the sensitivity is not a limitation for conclusions on latency versus low-level persistence.
Neutralizing antiviral antibodies
After clearance of acute, productive infection, that is at a time when latency is thought to be established, the host has usually developed neutralizing antibodies that intercept virions as soon as they are released from a cell, which has been shown in the mouse model of virus reactivation from latency, where neutralizing antibodies prevented virus spread and kept reactivated virus focal in the organ in which the reactivation event had occurred ([12, 13], for more detail, see Krmpotić et al. in this issue of MMIM [14]). In addition to virus neutralization in situ, antibodies present in organ homogenates might also interfere in vitro in the infectivity assay, thus further reducing assay sensitivity. An option to overcome this problem is the use of constitutively or conditionally B-cell-deficient mice or, even better, of the IgMi mouse strain that still has B cells with membrane-bound IgM as B-cell receptor for an antigen-presentation function but unable to secrete IgM antibodies and also unable to go through class switch recombination [15]. As with any genetically modified mouse model [16], a putative interference with the establishment of latency is a caveat that needs to be considered for the interpretation of data.
Viral genome degradation kinetics and subclinical virus reactivation
The presence of viral genome after clearance of productive primary infection, operationally defined as latency, is not necessarily genome derived from a latent virus reservoir. One has to rule out the possibilities that viral genome or PCR-amplifiable fragments thereof originate from the acute infection and remain detectable for a period of time after resolution of infectivity, depending on catabolic half-life. A particular problem is posed by episodes of subclinical virus reactivation that reload viral DNA from a productive source.
In our view, the currently best evidence for CMV latency in an organ is the presence of reactivatable viral genomes in the absence of essential viral transcripts of the productive cycle, as determined by highly sensitive, quantitative RT-PCRs (qRT-PCRs) ([17, 18], reviewed in [19]), a criterion that is not reliant on sensitivity of the infectivity assay and not influenced by antiviral antibodies. We advocate the view that molecular CMV latency can already be established in a particular organ of interest, while productive infection persists at other sites. In the mouse model, there exists ample evidence for organ-specific kinetics in the clearance of infectivity, with a temporal ranking of fast clearance, and, thus, rapid establishment of latency, in spleen and liver, followed by lungs, and last in the salivary glands. Furthermore, when antiviral antibodies prevent spread of reactivated virus, recurrent productive infection stays confined to the organ in which the reactivation event occurred, while, at the same time, other organs remain latently infected [12].
Importantly, latent virus load in organs was found not to reflect the duration of productive infection. Specifically, although virus replication persists long-term in salivary gland tissue, latent viral load was found to be low compared to the lungs where productive infection was resolved much faster [12, 20]. An explanation may be that the acinar glandular epithelial cell, the cellular site of high and prolonged non-cytopathogenic virus production in salivary gland tissue [21], is not a cellular site of latency, because termination of virus production in this cell type, supposed to be cytokine/lymphokine-mediated in an antigen-unspecific fashion, is associated with cell death and, thus, with loss of the viral genome [22].
Importance of latent virus reservoirs for the theoretical risk of CMV reactivation in transplant recipients
A reactivation risk exists if transplant donor (D) or recipient (R) or both are latently infected, as indicated in practice by “seropositivity” for CMV-specific antibodies, specifically IgG in the absence of IgM to distinguish between past and acute infection. Serological screening, however, may miss cases of very recent infection or fail to detect low amounts of antibodies because of issues of assay sensitivity and false-negative detection. This may explain reported cases of CMV DNAemia in seronegative recipients who received a transplant from a seronegative donor (D−R−) [23]. A more direct evidence for latency could be the detection of viral DNA in the absence of infectious virus. Testing this for the transplant is an option, though prone to high sampling error, but unrealistic for organ biopsies from the recipient. Therefore, the serological status serves as a surrogate evidence for latent infection in clinical routine. The serological status of donor and recipient defines risk constellations D+R−, D−R+, and D+R+, respectively. Depending on the latent virus reservoirs, reactivation to productive infection can initiate from latently infected hematopoietic lineage cells (HLCs) and/or diverse tissue cell types (TCs) constituting organs. Obviously, in a D+R− constellation in HCT, reactivation can initiate only from donor-derived HLCs, provided that these cells are, indeed, a reservoir of latent CMV. Other constellations are a bit more complicated, in particular since, in HCT, some recipient HLCs may resist conditioning hematoablative treatment and since, in SOT, transplanted organs may harbor also differentiated HLCs as blood cells in the vasculature, some of which can be perfusion resistant by attachment to blood vessel endothelia. Table 1 summarizes the theoretical risk of virus reactivation from latently infected donor or recipient HLCs or TCs in HCT and SOT under the idealized assumption that HLCs and TCs are both latent virus reservoirs and comparable in numbers of cells actually harboring latent viral DNA (Table 1).
Observed risk of hCMV reactivation in HCT and SOTs
For estimating the contribution of HLCs and TCs to CMV reactivation in HCT and SOTs, it is instructive to consider clinical experience on the association between viral DNA load in blood or urine and CMV disease after CMV reactivation in D+R− and D−R+ recipients [23]. In clinical routine, transplantation of donor HLCs or of donor organs that are productively infected with hCMV is at best an accident. Therefore, infection of transplant recipients in the constellation D+R− usually results from reactivation of latent virus transmitted with the transplant, that is primarily TCs in SOTs and HLCs in HCT (recall Table 1).
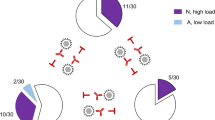
In renal as well as in liver SOT, reactivated viral load was high in D+R− recipients and low in D−R+ recipients, and symptomatic cases were observed only in high-load D+R− recipients. The speculation that low risk in the D−R+ constellation results exclusively from immunity in the recipients is rendered unlikely by the observation of cases of symptomatic infection in the constellation D+R+, although reduced load in this group compared to group D+R− indicates some effect of pre-existing immunity in SOT recipients (Table 2, simplified based on [23]).
In HCT, the situation is just the mirror image. Here, viral load was highest in D−R+ recipients and low in D+R− recipients, and symptomatic cases were observed primarily in high-load D−R+ recipients. The speculation that low risk of reactivation in the D+R− constellation results exclusively from transferred donor immunity is rendered unlikely by the observation of symptomatic cases with high load in the constellation D+R+. Reduced load in this group compared to group D−R+, however, indicates an at least moderate effect of pre-existing immunity in HCT donors (Table 2), an effect that predictably depends on the level of stem cell purification.
Therefore, risk of hCMV reactivation and disease is associated with D+ status in SOTs and R+ status in HCT. Differences in clinical regimens, including underlying primary disease, graft manipulation, type, degree and duration of iatrogenic immunosuppression, as well as therapeutic measures, complicate the interpretation. Nonetheless, a straightforward explanation might be that latently infected cells are frequent in D+ as well as R+ organs but rare in D+ HLCs. Altogether, risk in HCT results primarily from reactivation of the recipient’s virus from her/his own organs, whereas risk in SOTs results primarily from reactivation of the donor’s virus from the organ transplant. Thus, as a common trait, in both cases, reactivation initiates from organs rather than from HLCs, thus, suggesting the existence of latency in non-hematopoietic cell types.
The paradox of hCMV latency in human HLCs
It is widely accepted that CD34+ stem/progenitor HLCs [24,25,26,27], myeloid lineage-committed CD33+ progenitor cells co-expressing CD14 or CD15 along with dendritic cell (DC) markers [28], and CD14+ blood monocytes [29, 30] represent cellular sites of hCMV latency (for more recent reviews, see [31] and the review by Elder and Sinclair in this issue of MMIM [32]). A number of studies followed to define the latent state in molecular terms, which led to the identification of “CMV latency-associated transcripts” (CLTs) ([33, 34], reviewed in [35]). Notably, CLTs are mainly derived from the enhancer-driven major immediate–early (MIE) region, transcribed in sense or anti-sense direction [35]. In a cell culture model of latently infected granulocyte–macrophage progenitors [36], CLTs were found to be present only in 3–5% of cells carrying latent viral genome in a copy number of 1–8/cell. While cells carrying viral DNA in virtual absence of CLTs at the time of analysis might actually express CLTs sporadically, this finding showed that continuous presence of CLTs is not required for maintenance of the viral genome. Regarding the order of magnitude of the frequency of CD33+ myeloid lineage progenitors expressing CLTs in naturally infected individuals, Hahn et al. gave an estimate ranging from 0.001 to 0.01% [28]. More recent studies strongly suggest that certain CLTs, as exemplified by latency-associated cmvIL-10 (LAcmvIL-10), manipulate the microenvironment of latently infected cells, putatively contributing to maintenance of the viral genome by dampening the immune response (reviewed in [37]). Notably, recent definition of the transcriptional landscape during hCMV latency in vivo did not reveal a highly restricted latency-associated viral gene expression program but a quantitative rather than qualitative difference to lytic gene expression [38].
From a clinician’s point of view, the problem in patients is not latency as such, but reactivation to productive, cytopathogenic, and thus tissue-destructing recurrent infection leading to recrudescent disease. The early studies on latency in HLCs related virus reactivation to cell differentiation from a viral replication non-permissive myeloid lineage stem/progenitor to a permissive mature cell type, specifically macrophages and myeloid DCs [28, 30, 39]. This differentiation-dependent shift in permissivity involves viral chromatin remodeling (reviewed in [40]) and is likely driven by pro-inflammatory cytokines [28], for instance, under conditions of allogeneic stimulation [41]. The current state of reactivation regulation involving MIE gene expression repressor US28 and its proposed derepression by an US28 inhibitor is discussed by Elder and Sinclair in this issue of MMIM [32].
From all these findings on a hematopoietic reservoir of latent viral genomes of hCMV, a picture emerges proposing a continuous and enduring bone marrow (BM) source of latent viral genomes, consisting of latently infected CD34+ stem cells and CD33+ myeloid progenitors. These give rise to still latently infected CD14+ blood monocytes exported to the blood, where the latent viral genomes can eventually reactivate to productive infection upon inflammatory cytokine-driven differentiation to macrophages and myeloid DCs. With regard to hCMV latency in the CD34+ stem cells, however, their role as an efficient source delivering latent viral genomes to downstream lineage-committed progenitors might be limited by the inhibition of their proliferation as reported by Sindre and colleagues [27]. Along the same line of argument, clinical isolates of hCMV that show tropism for hematopoietic stem/progenitor cells inhibit hematopoiesis in long-term BM-cell cultures, thus explaining the clinical problem of CMV-associated myelosuppression [42]. All in all, the picture is less clear than currently assumed.
The proposed lifelong hematopoietic latency of hCMV is also difficult to reconcile with the relatively low risk of virus reactivation in recipients of D+R− HCT as discussed above (Table 2) and also not compatible with the routine clinical practice of reactivation monitoring in HCT recipients. At transplantation centers worldwide, HCT recipients are longitudinally monitored post-transplantation by quantitative PCR (qPCR) for hCMV reactivation as defined by the onset of hCMV DNAemia. Detection even of trace amounts of viral DNA in blood cells or plasma is the medical indication for starting with “pre-emptive” antiviral therapy, which means medication with antivirals prior to clinically defined CMV organ disease, interstitial pneumonia in particular. In the follow-up, clearance of viral DNA from blood is used to monitor success of the antiviral therapy [5]. Conversion from CMV DNA-negative to -positive and back implies that hematopoietic lineage progeny in peripheral blood are either not cellular sites of latency—as defined by harboring viral DNA for lifetime—or that latent genome copy number and/or frequency of latently infected blood cells are below the detection limit and are subject to sampling error in the routine diagnostic qPCR monitoring. Alternatively, and in agreement with the overwhelming evidence of a latent state of hCMV in HLCs, such a hematopoietic reservoir might not last for lifetime, but may be a transient stage.
Can the mouse model reproduce hematopoietic lineage latency?
Reproducing the clinical correlate is the acid test for the validity of any CMV animal model [43]. An advantage of model CMVs in general is the experimentally defined time of infection. This allows longitudinal studies on the clearance of productive infection as well as a comparative quantitation of viral DNA load in BM, in peripheral blood, and in host organs. In contrast, in “seropositive” individuals carrying hCMV DNA in HLCs, time, dose, and route of primary infection are unknown parameters, because primary infection of an immunocompetent person is associated at best with mild and unspecific “feverish” symptoms and is, thus, rarely diagnosed. As a consequence, it is difficult to distinguish between (1) viral DNA still present in peripheral blood after clearance of productive infection depending on catabolic half-life of viral DNA within cells as well as the half-life of the cell type itself, (2) replenishment of viral DNA in the blood based on subclinical episodes of virus reactivation at any tissue site in the body, and (3) genuinely latent viral DNA continuously delivered to the blood with progeny of latently infected BM-resident hematopoietic stem and progenitor cells.
These alternatives were tested in the BALB/c mouse model of viral latency after experimental syngeneic HCT, a regimen that involves hematoablative conditioning of recipients by total-body γ-irradiation, followed by intravenous infusion of tibial and femoral donor BM cells and intraplantar/footpad infection with mCMV (Fig. 1a) (model reviewed in [44,45,46]).

Kinetics of viral genome clearance in BM cells and blood leucocytes after HCT. a Sketch of the HCT mouse model. The flash symbol indicates hematoablative treatment by total-body γ-irradiation. b Clearance of infectious virus in organs, based on data from [47]. Gray boxes indicate data range. c Clearance of viral DNA (vDNA), illustrated schematically based on data from [47]. Cone symbols indicate decline of vDNA load over time for BM cells (BMC yellow) and peripheral blood cells (PBC red)
If the prediction from the human correlate applies, viral DNA should not be cleared from the BM compartment. As latently infected stem and progenitor cells are expected to pass the latent viral genome to all their more differentiated clonal progeny, mCMV DNA may be found amplified in peripheral blood cells compared to BM cells in absolute terms. However, assuming that the progeny clone size and export rate are similar for latently infected and uninfected bone marrow stem and progenitor cells, the proportion of latently infected cells in both compartments should remain constant over time. In the actual experiment [47], productive viral replication in organs of the infected model HCT recipients was slowly cleared with different kinetics in spleen, lungs, and salivary glands by 2, 3, and 4 months after HCT, respectively (Fig. 1b). In parallel to the resolution of productive infection, viral DNA load in BM cells and blood leucocytes also declined with time after HCT (Fig. 1c) and was cleared in BM after 5 months as defined by the absence of detectable viral DNA in 107 BM cells, which equals the cell yield from a complete femur plus tibia. At the time when viral DNA was cleared from the BM, 105 blood cells still contained about 100 copies of viral DNA. Notably, throughout the time course, the frequency of cells carrying viral genome was about 1000-fold higher in peripheral blood compared to BM. As the BM compartment is vascularized, this difference might possibly just reflect the proportion of blood leucocytes circulating in BM capillaries, so that the available data do not provide evidence for the proposed existence of a lifelong hematopoietic reservoir of latent mCMV in the BM.
Although this experiment, dating back to 1994, did not include a phenotyping of viral DNA-carrying peripheral blood leucocyte subsets with markers that are state-of-the-art technology of today, a rough cell sorting was performed to localize viral DNA [47]. At 3 months after HCT and acute infection, when ~ 1 cell in 104 blood leucocytes harbored viral DNA, cells were sorted based on physical properties of size (forward scatter) and granularity (sideward scatter), followed by cytofluorometric detection of lead subset markers. The bulk of viral DNA localized to a high-granularity population of cells co-expressing the myeloid lineage marker CD11b and the granulocyte marker Gr-1 (covering Ly-6C and Ly6G), which primarily represent neutrophilic granulocytes. With today’s technology, this finding should be confirmed using Ly6G as a more specific marker expressed exclusively by neutrophils, whereas Ly6C is expressed to different levels also by monocyte subsets. Nonetheless, in accordance with the diagnostic antigenemia test for productive/reactivated hCMV infection, the high-granularity Gr-1+CD11b+mCMV-DNA+ cells likely are polymorphonuclear neutrophils that have taken up virus derived during productive infection from infected tissue cells [48, 49]. With less than 10 genome copies per 104 cells, viral DNA also localized to Gr-1−CD11b+ cells, a phenotype attributed to patrolling monocytes, a recognized cellular site of hematopoietic hCMV latency (see above). These findings on hematopoietic lineage cell types that harbor viral genomes in peripheral blood are in accordance with reports on the detection of mCMV DNA in monocytes/macrophages and polymorphonuclear leucocytes in mice defined as being latently infected based on the absence of productive infection [50, 51]. Subsequent studies in the HCT model confirmed the message of Fig. 1 in that they consistently demonstrated decline of mCMV DNA over time, more rapidly in BM compared to peripheral blood, to levels below the detection limit of qPCR, whereas a high latent genome burden was maintained lifelong in organs of the very same individual mice, with the lungs having been in the focus of interest [8, 17, 18, 52]. In accordance with a vanishing hematopoietic source of mCMV latency, single-cell detection of latent reporter virus reactivation in explanted lung tissue slices did not reveal CD11b+CX3CR1+ HLCs as cellular sites of virus reactivation [53].
The most important conclusion from the longitudinal studies after HCT in the mouse model is that HLCs are not latent virus reservoirs for lifetime and, thus, do not fulfill the more strict Roizman’s definition of herpesvirus latency. These data prompted us to define type-1 “canonical latency” as lifelong presence of reactivation-competent viral DNA in organs after clearance of transient type-2 “non-canonical latency” from BM and peripheral blood (discussed in [44]). Actually, the transient nature of mCMV type-2 latency in HLCs was the reason why it was not pursued further, unlike, in its human counterpart, so that molecular state of the latent viral genome is a blank area for mCMV latency in HLCs. Instead, clearance of mCMV DNA from blood was even used regularly as an additional condition for defining “organ latency”, since this excludes detection of viral DNA present in perfusion-resistent leucocytes that stick to the vasculature.
To our knowledge, a longitudinal comparative analysis, ideally performed over lifetime, of latent hCMV DNA load in BM, peripheral blood and organs of seropositive volunteers is missing. Admittedly, such an ambition is unrealistic, first because the time of primary infection is usually undefined in humans and second because there is no easy access to repeated tissue biopsies for longitudinal analysis in latently infected but otherwise healthy volunteers. As a consequence, it remains to be seen if latency of hCMV in HLCs is transient or fulfills the definition of lifelong “canonical latency”.
Is CMV latency transmittable by HCT?
The risk of transmission of latent virus by HCT performed with HLCs derived from the BM of latently infected donors was tested in the mouse model [54]. In this work, latency was established in immunocompromised and infected female (XX, sex-determining region of Y gene sry−) recipients after hematopoietic reconstitution by HCT using male (XY, gene sry+) donors, so that, in the HCT recipients, transplanted HLCs were XY, whereas all cells constituting tissues, including BM stromal cells, were XX. These latently infected BM chimeras were then used as donors in a consecutive HCT into immunocompromised, uninfected XX recipients to test if latently infected XY HLCs present in BM transmit latent virus. The result was unequivocally negative, consistent with the previous conclusion that HLCs are not sites of lifelong “canonical” latency of mCMV.
This seems to contrast with data in clinical HCT with D+ HLC donors, where viral DNA became detectable in R− recipients, even though the risk was low ([23], and Table 2). However, as discussed in greater detail above, the personal infection history of HCT donors, specifically the period of time that has passed after acute primary infection, possibly in early childhood decades ago, is unknown. Thus, transmitted viral genomes may have derived from HLCs latently infected only transiently. In addition, there is a practical issue that needs to be considered: when BM cells are isolated from femoral or tibial BM of mice, BM-derived HLCs are only minimally contaminated by blood leucocytes circulating in the BM capillaries, whereas such a contamination can be significant after BM puncture and aspiration in clinical HCT, so that transmitted latent virus may be derived from co-transplanted latently infected blood monocytes rather than from latently infected stem/progenitor cells in the BM compartment as the source.
Experimental “proof of concept” for latent hCMV infection of HLCs has been provided by a humanized mouse model [55]. In this model, NOD-scid/IL2Rγ-chainnull (NSG) mice were humanized for HLCs by engraftment of human CD34+ (huCD34+) cord blood cells. These interspecies chimeras were shown to carry latent, reactivatable hCMV DNA in human HLCs that had colonized murine host tissues at 4 weeks after primary infection with neonatal normal human dermal fibroblasts (NHDF) that were infected with low-passage clinical hCMV isolate TRpM1A. Acute infection of the humanized mice through infected NHDF was crucial, since hCMV DNA was not detected at any site in huCD34+ cell-engrafted mock-infected mice.
While this showed hCMV latency in human HLCs, it also implies, however, that latent hCMV DNA was not transmitted with the huCD34+ graft, although, unfortunately, the CMV-specific serostatus of the huCD34+ cell donors was not specified in this study. To close this gap, a so far underappreciated follow-up study modelled a D+R− HCT constellation (see Tables 1, 2) by engrafting NSG mice with hemopoietin-mobilized human peripheral blood stem cells (PBSC) from latently infected donors (56). Notably, thus humanized-recipient mice demonstrated hCMV reactivation in diverse organs. While this is so far the best evidence for transmittable hCMV latency in human hematopoietic stem/progenitor cells, the question remains if latency in HLCs of the PBSC transplantation donors represented lifelong type-1 “canonical latency” or transient type-2 “non-canonical” latency. The clinical finding of an only up to 20% post-transplantation incidence of primary infection in recipients of a D+R− HCT (referenced in [56]) might be a hint to transmission occurring only when donors are in the stage of transient type-2 latency in HLCs.
In conclusion, in our view, the main “difference” between clinical CMV and the mouse model might be that clinical studies refer to the latent virus reservoir(s) during transient type-2 latency, whereas most studies in the mouse model refer to the latent virus reservoir(s) during lifelong type-1 latency.
Link between “memory inflation” and latent virus reservoirs
Lifelong imprinting of the immune system is a hallmark of CMV infections and the theme of this Special Issue of MMIM. Specifically, latent mCMV infection is associated with a viral-epitope selective accumulation of functional KLRG1+CD62L−CD8+ short-lived effector-(memory) cells (SLECs) in latently infected extra-lymphoid tissues [57,58,59,60,61], a phenomenon coined with the catch phrase “memory inflation” ([59], for reviews, see [62,63,64]) and contributions to this special issue of MMIM [65,66,67]. These cells were shown to be involved in the immune surveillance of latency by sensing viral epitope presented as a result of limited transcription events during latency [68]. As memory inflation is driven by latency-associated antigen/antigenic peptide presentation, memory inflation in BM chimeras can help to decide if latency is established in donor BM-derived HLCs or in recipient-derived TCs, or possibly in both. Using genetically different but, regarding the underlying idea, related types of BM chimeras, two groups independently showed that recipient-genotype TCs but not donor-genotype HLCs are the main drivers of memory inflation [69, 70].
In accordance with this understanding, it was found that recruitment of naïve CD8 T cells contributes little to memory inflation [61, 71], which is reasonably explained by the fact that priming of naïve T cells requires antigen presentation by professional APCs, that is in particular DCs, which are HLCs of the myelopoietic differentiation lineage. To speculate, a minor contribution of naïve CD8+ cells to fueling memory inflation [61, 65] may date to the period of infection when virus is still latent in HLCs, as discussed above as the transient “non-canonical” type-2 latency.
All in all, memory inflation not only predicts but also demands viral latency in a non-hematopoietic TC type.
Evidence for CMV latency in non-hematopoietic tissue cells
Focus of hCMV latency research on latent infection of human HLCs (see above), favored by easy access to clinical samples, has distracted attention from a possible latency in human TCs, disfavored by difficult access to biopsies from organs of latently infected but otherwise healthy volunteers. Based on cell separation followed by virus reactivation in co-culture with permissive cells, an early report on CMV latency in the mouse model localized latent, reactivatable mCMV in the spleen to a stromal/reticular cell type not expressing MHC class-II (MHC-II) antigens [72]. Although the authors were very careful with their interpretation, comparison with infected cells detected during acute infection of the spleen [72] somehow suggested a “sinusoidal lining cell” or an endothelial cell (EC) as candidates, a view that needs to be revised in part today as spleens of mice are asinusoidal, so that human CD34−CD8α+ sinusoid-lining littoral cells (LCs) have no direct correspondence in mice [73], whereas a murine counterpart of human CD34+CD8α− splenic vascular endothelial cells (SVECs) remains in discussion. A more recent study in sex chromosome BM chimeras with XY HLCs and XX TCs has positively identified recipient-genotype, and, thus, non-hematopoietic lineage-derived, MHC-II−CD11b−CD11c− but L-SIGN+CD31+CD146+ long-lived liver sinusoidal ECs (LSECs) as one cellular site of latent, reactivatable mCMV ([74], reviewed in [62]). As views haunt in the literature saying that EC also originate from a hematopoietic lineage progenitor, it may be important to note that in the sex-mismatched BM chimeras donor-derived, and thus hematopoietic lineage-derived, cells expressing the EC marker CD146 constitutively expressed also MHC-II, unlike the recipient-derived LSECs, and did not harbor latent viral genome [74].
In the lungs, latent viral genomes were found to co-purify with CD31+ cells (own unpublished data), suggesting that the cellular site of mCMV latency in the lungs is a capillary EC. This is of relevance, as it implies that recognition of antigen in latently infected lungs for driving memory inflation takes place in the vascular bed of the lungs, which is bordered by ECs, rather than in trans-endothelial lung parenchyma. This view is supported by the recent finding of a systemic hematogenous maintenance of memory inflation [75].
And what about hCMV? A link between vascular disease, atherosclerosis in particular, and CMV is controversially discussed. Based on PCR and in situ hybridization, it has been proposed that the human aorta may be a site of hCMV latency [76,77,78]. For testing the hypothesis of hCMV latency in blood vessel ECs, Reeves and colleagues have tried to detect latent hCMV DNA in ECs as well as in smooth muscle cells isolated from the saphenous veins of seropositive persons—with negative results [79]. Is this a final decision against latent hCMV infection of human ECs? We hesitate to already accept this for several reasons: (1) the great saphenous vein is a large superficial vein of the leg and becomes accessible for studies as surplus tissue, as it is routinely used for autotransplantation in coronary artery bypass surgery. Can we be sure that veins of an extremity are representative and are reached in acute infection for the establishment of a latent infection? We also do not know if any data exist on the risk of transmission of latent hCMV in coronary artery bypass surgery, as compared to the risk in kidney or liver SOTs (recall Table 2). Lack of information likely relates to the fact that blood vessel autotransplantation does not involve iatrogenic immunosuppression, so that transmission of latent CMV cannot be recognized by virus recurrence in the transplant recipients; (2) ECs are not a single-cell type but come in different flavors [80,81,82], which may or may not all qualify for latent infection; (3) as the frequency of latently infected cells is generally very low, sampling error may be an issue, and finally (4) enrichment for ECs by a period of growth in cell culture [79] may actually have selected against latently infected ECs.
All in all, we believe that the question of hCMV latency in TCs, with ECs being only one candidate, is not yet settled. The high risk of transmitting latent hCMV in D+R− kidney-SOT and liver-SOT (Table 2) calls for a non-hematopoietic lineage source of latent hCMV in organs.
Virus strain variance in cell-type tropism: a neglected issue in the search for latent virus reservoirs
So far, the search for the cellular site(s) of hCMV latency did not take account of the increasingly recognized variance in clinical strains/isolates that often impacts on cell type tropism ([83, 84], for a review, see [85]). Obviously, for establishing molecular latency in a cell type, virus must be able to enter the cell in the first place, and its genome must be replicatively silenced but maintain the capacity to reactivate to productive infection. Regarding the infection of HLCs, an early study by the group of Beverly Torok-Storb has noted phenotypic differences among short-term propagated clinical isolates of hCMV ([42], discussed in [46]). Specifically, only 8 out of 20 isolates were found to infect hematopoietic, myeloid lineage-committed progenitors in long-term BM-cell cultures, associated with myelosuppression. The remaining 12 isolates infected BM stromal cells. Notably, among the eight isolates with tropism for the hematopoietic progenitors, four had lost tropism for stromal cells. This gives us a new perception of the relatively low incidence of hCMV reactivation following D+R− HCT (see above, and Table 2). A donor graft latent with a strain that infects a TC type but not HLCs will of course not transmit latent virus to an HCT recipient, but will transmit to an SOT recipient. As suggested by the important study of Simmons et al. [42], TC-tropic strains are in the majority. This might contribute to the high incidence of hCMV reactivation following D+R− SOTs.
Short résumé
The search for latent CMV reservoirs is by no means a closed chapter. From reviewing the literature, we got the impression that the proposed difference in latent virus reservoirs between hCMV and mCMV may relate to a difference in the focus of the search. Studies on hCMV latency in HLCs likely described the non-canonical, transient type-2 latency, whereas studies in the mouse model focussed on canonical, lifelong type-1 latency (Fig. 2).

Graphical abstract. Key features of lifelong (type-1) and transient (type 2) latency. TC non-hematopoietic stromal and/or parenchymal tissue cell(s), which includes endothelial cells. HLC hematopoietic lineage cell(s). APC professional antigen-presenting cell(s) of the myelopoietic lineage; red circles circularized latent vDNA localizing to the cell nucleus. Red dot antigenic peptide presented during episodes of limited transcriptional activity by MHC class-I molecules of the latently infected cells to the T-cell receptors of effector memory and naïve T cells during type-1 and type 2 latency, respectively. Red symbols indicate durable and vanishing vDNA load in TC and HLC/APC, respectively. Antigenic peptide presentation by latently infected, non-hematopoietic TC drives memory inflation. Antigenic peptide presentation by latently infected HLC/APC primes naïve T cells
References
Roizman B, Sears AE (1987) An inquiry into the mechanisms of herpes simplex virus latency. Annu Rev Microbiol 41:543–571. https://doi.org/10.1146/annurev.mi.41.100187.002551
Riddell SR (1995) Pathogenesis of cytomegalovirus pneumonia in immunocompromised hosts. Semin Respir Infect 10:199–208
Ho M (2008) The history of cytomegalovirus and its diseases. Med Microbiol Immunol 197:65–73. https://doi.org/10.1007/s00430-007-0066-x
Boppana SB, Britt WJ (2013) Synopsis of clinical aspects of human cytomegalovirus disease. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II. Caister Academic Press, Norfolk, pp 1–25
Seo S, Boeck M (2013) Clinical cytomegalovirus research: hematopoietic cell transplantation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II. Caister Academic Press, Norfolk, pp 337–353
Emery VC, Milne RS, Griffiths PD (2013) Clinical cytomegalovirus research: liver and kidney transplantation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II. Caister Academic Press, Norfolk, pp 301–311
Avery RK (2013) Clinical cytomegalovirus research: thoracic organ transplantation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II. Caister Academic Press, Norfolk, pp 286–300
Kurz S, Steffens HP, Mayer A, Harris JR, Reddehase MJ (1997) Latency versus persistence or intermittent recurrences: evidence for a latent state of murine cytomegalovirus in the lungs. J Virol 71:2980–2987
Lemmermann NA, Podlech J, Seckert CK, Kropp KA, Grzimek NK, Reddehase MJ, Holtappels R (2010) CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model. In: Kabelitz D, Kaufmann SHE (eds) Methods in microbiology. Immunology of infection, vol 37. Academic Press, London, pp 369–420
Podlech J, Holtappels R, Grzimek NKA, Reddehase MJ (2002) Animal models: murine cytomegalovirus. In: Kaufmann SHE, Kabelitz D (eds) Methods in microbiology. Immunology of infection, vol 32. Academic Press, London, pp 493–525
Klenovsek K, Weisel F, Schneider A, Appelt U, Jonjic S, Messerle M, Bradel-Tretheway B, Winkler TH, Mach M (2007) Protection from CMV infection in immunodeficient hosts by adoptive transfer of memory B cells. Blood 110:3472–3479. https://doi.org/10.1182/blood-2007-06-095414
Reddehase MJ, Balthesen M, Rapp M, Jonjić S, Pavić I, Koszinowski UH (1994) The conditions of primary infection define the load of latent viral genome in organs and the risk of recurrent cytomegalovirus disease. J Exp Med 179:185–193
Jonjić S, Pavić I, Polić B, Crnković I, Lucin P, Koszinowski UH (1994) Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J Exp Med 179:1713–1717
Krmpotić A, Podlech J, Reddehase MJ, Britt WJ, Jonjić S (2019) Role of antibodies in confining cytomegalovirus after reactivation from latency: three decades’ résumé. Med Microbiol Immunol. https://doi.org/10.1007/s00430-019-00600-1
Waisman A, Croxford AL, Demircik F (2008) New tools to study the role of B cells in cytomegalovirus infections. Med Microbiol Immunol 197:145–149. https://doi.org/10.1007/s00430-008-0088-z
Benedict CA, Crozat K, Degli-Esposti M, Dalod M (2013) Host genetic models in cytomegalovirus immunology. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II. Caister Academic Press, Norfolk, pp 259–285
Kurz SK, Rapp M, Steffens HP, Grzimek NK, Schmalz S, Reddehase MJ (1999) Focal transcriptional activity of murine cytomegalovirus during latency in the lungs. J Virol 73:482–494
Kurz SK, Reddehase MJ (1999) Patchwork pattern of transcriptional reactivation in the lungs indicates sequential checkpoints in the transition from murine cytomegalovirus latency to recurrence. J Virol 73:8612–8622
Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK (2008) Murine model of cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol 325:315–331
Balthesen M, Messerle M, Reddehase MJ (1993) Lungs are a major organ site of cytomegalovirus latency and recurrence. J Virol 67:5360–5366
Jonjić S, Mutter W, Weiland F, Reddehase MJ, Koszinowski UH (1989) Site-restricted persistent cytomegalovirus infection after selective long-term depletion of CD4 + T lymphocytes. J Exp Med 169:1199–1212
Henson D, Strano AJ. Mouse cytomegalovirus (1972) Necrosis of infected and morphologically normal submaxillary gland acinar cells during termination of chronic infection. Am J Pathol 68:183–202
Emery VC (1998) Relative importance of cytomegalovirus load as a risk factor for cytomegalovirus disease in the immunocompromised host. Monogr Virol 21:288–301
Maciejewski JP, Bruening EE, Donahue RE, Mocarski ES, Young NS, St Jeor SC (1992) Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 80:170–178
von Laer D, Meyer-Koenig U, Serr A, Finke J, Kanz L, Fauser AA, Neumann-Haefelin D, Brugger W, Hufert FT (1995) Detection of cytomegalovirus DNA in CD34 + cells from blood and bone marrow. Blood 86:4086–4090
Mendelson M, Monard S, Sissons P, Sinclair J (1996) Detection of endogenous human cytomegalovirus in CD34 + bone marrow progenitors. J Gen Virol 77:3099–3102. https://doi.org/10.1099/0022-1317-77-12-3099
Sindre H, Tjøonnfjord GE, Rollag H, Ranneberg-Nilsen T, Veiby OP, Beck S, Degré M, Hestdal K (1996) Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 88:4526–4533
Hahn G, Jores R, Mocarski ES (1998) Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci USA 95:3937–3942
Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair JH (1991) Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol 72:2059–2064. https://doi.org/10.1099/0022-1317-72-9-2059
Söderberg-Nauclér C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA (2001) Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol 75:7543–7554. https://doi.org/10.1128/JVI.75.16.7543-7554.2001
Goodrum F (2016) Human cytomegalovirus latency: approaching the Gordian Knot. Annu Rev Virol 3:333–357. https://doi.org/10.1146/annurev-virology-110615-042422
Elder E, Sinclair J (2019) HCMV latency: what regulates the regulators? Med Microbiol Immunol. https://doi.org/10.1007/s00430-019-00581-1
Kondo K, Kaneshima H, Mocarski ES (1994) Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci USA 91:11879–11883
Kondo K, Xu J, Mocarski ES (1996) Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc Natl Acad Sci USA 93:11137–11142
Slobedman B, Avdic S, Abendroth A (2013) Transcription associated with human cytomegalovirus latency. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol I. Caister Academic Press, Norfolk, pp 347–362
Slobedman B, Mocarski ES (1999) Quantitative analysis of latent human cytomegalovirus. J Virol 73:4806–4812
Poole E, Sinclair J (2015) Sleepless latency of human cytomegalovirus. Med Microbiol Immunol 204:421–429. https://doi.org/10.1007/s00430-015-0401-6
Shnayder M, Nachshon A, Krishna B, Poole E, Boshkov A, Binyamin A, Maza I, Sinclair J, Schwartz M, Stern-Ginossar N (2018) Defining the transcriptional landscape during cytomegalovirus latency with single-cell RNA sequencing. MBio 9:e00013–e18. https://doi.org/10.1128/mBio.00013-18
Taylor-Wiedeman J, Sissons P, Sinclair J (1994) Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J Virol 68:1597–1604
Reeves M, Sinclair J (2013) Epigenetic regulation of human cytomegalovirus gene expression: impact on latency and reactivation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol I. Caister Academic Press, Norfolk, pp 330–346
Söderberg-Nauclér C, Fish KN, Nelson JA (1997) Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126
Simmons P, Kaushansky K, Torok-Storb B (1990) Mechanisms of Cytomegalovirus-mediated myelosuppression: perturbation of stromal cell function versus direct infection of myeloid cells. Proc Natl Acad Sci USA 87:1386–1390. https://doi.org/10.1073/pnas.87.4.1386
Reddehase MJ, Lemmermann NAW (2018) Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: predictions for medical translation that survived the “test of time”. Viruses 10:e693. https://doi.org/10.3390/v10120693
Reddehase MJ, Podlech J, Grzimek NK (2002) Mouse models of cytomegalovirus latency: overview. J Clin Virol 25:S23–S36
Holtappels R, Ebert S, Podlech J, Fink A, Böhm V, Lemmermann NAW, Freitag K, Renzaho A, Thomas D, Reddehase MJ (2013) Murine model for cytoimmunotherapy of CMV disease after hematopoietic cell transplantation. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol II. Caister Academic Press, Norfolk, pp 353–381
Reddehase MJ (2016) Mutual interference between cytomegalovirus and reconstitution of protective immunity after hematopoietic cell transplantation. Front Immunol 7:294. https://doi.org/10.3389/fimmu.2016.00294
Balthesen M, Šuša M, Lučin P, Reddehase MJ (1994) Cytomegalovirus DNA detected in blood leukocytes after resolution of productive infection does not originate from latently infected hematopoietic stem cells in the bone marrow. Croat Med J 35:19–25
Revello MG, Percivalle E, Zavattoni M, Parea M, Grossi P, Gerna G (1989) Detection of human cytomegalovirus immediate early antigen in leukocytes as a marker of viremia in immunocompromised patients. J Med Virol 29:88–93
Kas-Deelen AM, The TH, Blom N, van der Strate BW, De Maar EF, Smit J, van Son WJ, Harmsen MC (2001) Uptake of pp65 in in vitro generated pp65-positive polymorphonuclear cells mediated by phagocytosis and cell fusion? Intervirology 44:8–13. https://doi.org/10.1159/000050024
Mitchell BM, Leung A, Stevens JG (1996) Murine cytomegalovirus DNA in peripheral blood of latently infected mice is detectable only in monocytes and polymorphonuclear leukocytes. Virology 223:198–207. https://doi.org/10.1006/viro.1996.0468
Pollock JL, Presti RM, Paetzold S, Virgin HW 4th (1997) Latent murine cytomegalovirus infection in macrophages. Virology 227:168–179. https://doi.org/10.1006/viro.1996.8303
Grzimek NK, Dreis D, Schmalz S, Reddehase MJ (2001) Random, asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J Virol 75:2692–2705. https://doi.org/10.1128/JVI.75.6.2692-2705.2001
Marquardt A, Halle S, Seckert CK, Lemmermann NA, Veres TZ, Braun A, Maus UA, Förster R, Reddehase MJ, Messerle M, Busche A (2011) Single cell detection of latent cytomegalovirus reactivation in host tissue. J Gen Virol 92:1279–1291. https://doi.org/10.1099/vir.0.029827-0
Seckert CK, Renzaho A, Reddehase MJ, Grzimek NK (2008) Hematopoietic stem cell transplantation with latently infected donors does not transmit virus to immunocompromised recipients in the murine model of cytomegalovirus infection. Med Microbiol Immunol 197:251–259. https://doi.org/10.1007/s00430-008-0094-1
Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, Othieno FA, Streblow DN, Garcia JV, Fleming WH, Nelson JA (2010) Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 8:284–291. https://doi.org/10.1016/j.chom.2010.08.001
Hakki M, Goldman DC, Streblow DN, Hamlin KL, Krekylwich CN, Fleming WH, Nelson JA (2014) HCMV infection of humanized mice after transplantation of G-CSF-mobilized peripheral blood stem cells from HCMV-seropositive donors. Biol Blood Marrow Transpl 20:132–135. https://doi.org/10.1016/j.bbmt.2013.10.019
Holtappels R, Pahl-Seibert MF, Thomas D, Reddehase MJ (2000) Enrichment of immediate-early 1 (m123/pp89) peptide-specific CD8 T cells in a pulmonary CD62L(lo) memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. J Virol 74:11495–11503
Holtappels R, Thomas D, Podlech J, Reddehase MJ (2002) Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J Virol 76:151–164
Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, Klenerman P (2003) Memory inflation: continuous accumulation of antiviral CD8 + T cells over time. J Immunol 170:2022–2029 (Corrigendum J Immunol 171:3895)
Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB (2006) Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol 177:450–458
Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB (2008) Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity 29:650–659. https://doi.org/10.1016/j.immuni.2008.07.017
Seckert CK, Griessl M, Büttner JK, Scheller S, Simon CO, Kropp KA, Renzaho A, Kühnapfel B, Grzimek NK, Reddehase MJ (2012) Viral latency drives ‘memory inflation’: a unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med Microbiol Immunol 201:551–566. https://doi.org/10.1007/s00430-012-0273-y
Seckert CK, Griessl M, Buttner JK, Freitag K, Lemmermann N, Hummel M, Liu XF, Abecassis M, Angulo A, Messerle M, Cook CH, Reddehase M (2013) Immune surveillance of cytomegalovirus latency and reactivation in murine models: link to memory inflation. In: Reddehase MJ (ed) Cytomegaloviruses, vol 1. Caister Academic PressNorfolk, UK, pp 374–416
Klenerman P, Oxenius A (2016) T cell responses to cytomegalovirus. Nat Rev Immunol 16:367–377. https://doi.org/10.1038/nri.2016.38
Welten SPM, Baumann NS, Oxenius A (2019) Fuel and brake of memory T cell inflation. Med Microbiol Immunol. https://doi.org/10.1007/s00430-019-00587-9
Cicin-Sain L (2019) Cytomegalovirus memory inflation and immune protection. Med Microbiol Immunol. https://doi.org/10.1007/s00430-019-00607-8
Renzaho A, Schmiedeke JK, Griessl M, Kühnapfel B, Seckert CK, Lemmermann NAW (2019) Transcripts expressed in cytomegalovirus latency coding for an antigenic IE/E phase peptide that drives “memory inlation”. Med Microbiol Immunol. https://doi.org/10.1007/s00430-019-00615-8
Simon CO, Holtappels R, Tervo HM, Böhm V, Däubner T, Oehrlein-Karpi SA, Kühnapfel B, Renzaho A, Strand D, Podlech J, Reddehase MJ, Grzimek NK (2006) CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J Virol 80:10436–10456. https://doi.org/10.1128/JVI.01248-06
Seckert CK, Schader SI, Ebert S, Thomas D, Freitag K, Renzaho A, Podlech J, Reddehase MJ, Holtappels R (2011) Antigen-presenting cells of haematopoietic origin prime cytomegalovirus-specific CD8 T-cells but are not sufficient for driving memory inflation during viral latency. J Gen Virol 92:1994–2005. https://doi.org/10.1099/vir.0.031815-0
Torti N, Walton SM, Brocker T, Rülicke T, Oxenius A (2011) Non-hematopoietic cells in lymph nodes drive memory CD8 T cell inflation during murine cytomegalovirus infection. PLoS Pathog 7:e1002313. https://doi.org/10.1371/journal.ppat.1002313
Loewendorf AI, Arens R, Purton JF, Surh CD, Benedict CA (2011) Dissecting the requirements for maintenance of the CMV-specific memory T-cell pool. Viral Immunol 24:351–355. https://doi.org/10.1089/vim.2010.0140
Mercer JA, Wiley CA, Spector DH (1988) Pathogenesis of murine cytomegalovirus infection: identification of infected cells in the spleen during acute and latent infections. J Virol 62:987–997
Qiu J, Salama ME, Hu CS, Li Y, Wang X, Hoffman R (2018) The characteristics of vessel lining cells in normal spleens and their role in the pathobiology of myelofibrosis. Blood Adv 2:1130–1145. https://doi.org/10.1182/bloodadvances.2017015073
Seckert CK, Renzaho A, Tervo HM, Krause C, Deegen P, Kühnapfel B, Reddehase MJ, Grzimek NK (2009) Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J Virol 83:8869–8884. https://doi.org/10.1128/JVI.00870-09
Smith CJ, Turula H, Snyder CM (2014) Systemic hematogenous maintenance of memory inflation by MCMV infection. PLoS Pathog 10:e1004233. https://doi.org/10.1371/journal.ppat.1004233
Hendrix MG, Dormans PH, Kitslaar P, Bosman F, Bruggeman CA (1989) The presence of cytomegalovirus nucleic acids in arterial walls of atherosclerotic and nonatherosclerotic patients. Am J Pathol 134:1151–1157
Pampou SY, Gnedoy SN, Bystrevskaya VB, Smirnov VN, Chazov EI, Melnick JL, DeBakey ME (2000) Cytomegalovirus genome and the immediate-early antigen in cells of different layers of human aorta. Virchows Arch 436:539–552
Chen R, Xiong S, Yang Y, Fu W, Wang Y, Ge J (2003) The relationship between human cytomegalovirus infection and atherosclerosis development. Mol Cell Biochem 249:91–96
Reeves MB, Coleman H, Chadderton J, Goddard M, Sissons JG, Sinclair JH (2004) Vascular endothelial and smooth muscle cells are unlikely to be major sites of latency of human cytomegalovirus in vivo. J Gen Virol 85:3337–3341. https://doi.org/10.1099/vir.0.80285-0
Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO (2003) Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci USA 100:10623–10628. https://doi.org/10.1073/pnas.1434429100
Conway EM, Carmeliet P (2004) The diversity of endothelial cells: a challenge for therapeutic angiogenesis. Genome Biol 5:207. https://doi.org/10.1186/gb-2004-5-2-207
Jarvis MA, Nelson JA (2007) Human cytomegalovirus tropism for endothelial cells: not all endothelial cells are created equal. J Virol 81:2095–2101. https://doi.org/10.1128/JVI.01422-06
Wilkinson GW, Davison AJ, Tomasec P, Fielding CA, Aicheler R, Murrell I, Seirafian S, Wang EC, Weekes M, Lehner PJ, Wilkie GS, Stanton RJ (2015) Human cytomegalovirus: taking the strain. Med Microbiol Immunol 204:273–284. https://doi.org/10.1007/s00430-015-0411-4
Renzette N, Pokalyuk C, Gibson L, Bhattacharjee B, Schleiss MR, Hamprecht K, Yamamoto AY, Mussi-Pinhata MM, Britt WJ, Jensen JD, Kowalik TF (2015) Limits and patterns of cytomegalovirus genomic diversity in humans. Proc Natl Acad Sci USA 112:E4120–E4128. https://doi.org/10.1073/pnas.1501880112
Adler B, Sinzger C (2013) Cytomegalovirus interstrain variance in cell type tropism. In: Reddehase MJ (ed) Cytomegaloviruses: from molecular pathogenesis to intervention, vol I. Caister Academic Press, Norfolk, pp 297–321
Acknowledgements
The authors are supported by the Deutsche Forschungsgemeinschaft SFB1292, individual project TP11.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by: Sebastian Voigt.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Special Issue on Immunological Imprinting during Chronic Viral Infection.
Rights and permissions
About this article

Cite this article
Reddehase, M.J., Lemmermann, N.A.W. Cellular reservoirs of latent cytomegaloviruses. Med Microbiol Immunol 208, 391–403 (2019). https://doi.org/10.1007/s00430-019-00592-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-019-00592-y