
Abstract
Peptides are able to cross the blood–brain barrier (BBB) through various mechanisms, opening new diagnostic and therapeutic avenues. However, their BBB transport data are scattered in the literature over different disciplines, using different methodologies reporting different influx or efflux aspects. Therefore, a comprehensive BBB peptide database (Brainpeps) was constructed to collect the BBB data available in the literature. Brainpeps currently contains BBB transport information with positive as well as negative results. The database is a useful tool to prioritize peptide choices for evaluating different BBB responses or studying quantitative structure–property (BBB behaviour) relationships of peptides. Because a multitude of methods have been used to assess the BBB behaviour of compounds, we classified these methods and their responses. Moreover, the relationships between the different BBB transport methods have been clarified and visualized.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
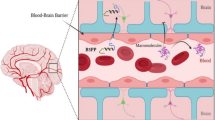
The blood–brain barrier (BBB) was discovered 125 years ago by Ehrlich and later supported by Goldmann based on the observation that blood-borne coloured substances did not leak into the brain, whereas intracerebroventricularly injected dyes did not exit the brain (Ehrlich 1885; Goldmann 1913). The barrier is mainly formed by brain capillary endothelial cells, joined together by tight junctions and surrounded by a basal membrane, next to other cell types as pericytes, astrocytes and neuronal cells (see Fig. 1). This physical barrier separates blood (systemic circulation) from brain (central nervous system), protecting the brain by regulation of influx and efflux. The transport of most foreign molecules is blocked, while essential substances can pass the BBB. This means that many drugs, capable of treating central nervous system (CNS) disorders are denied access to the regions where they would be effective. Owing to the presence of tight junctions, paracellular transport is quasi-impossible, but molecules are able to cross the BBB by passive transmembrane diffusion, depending on their lipophilicity. Additionally, several specific transport systems are present at the BBB, transporting compounds by a carrier-, receptor- or adsorptive-mediated transfer mechanism. In contrast, efflux transporters, such as P-glycoprotein (Pgp) are described as well, pumping compounds back into the blood. Because selective and sufficient entry of drugs and their modification tools is important in CNS drug design, while still limiting the access of toxic compounds, knowledge about the BBB behaviour of drugs is highly researched (Chu et al. 2006; Engelhardt 2006). Moreover, the addition of physical techniques to temporarily and spatial-selectively open the BBB, can even further extend the clinical possibilities (Campbell et al. 2011; Liu et al. 2010; Madsen and Hirschberg 2010; Rossi 2011).

BBB morphology (M metabolites)
Peptides are a promising group of therapeutic and diagnostic drugs, which are expected to play a significant role in the next decennia alleviating the current rising attrition rate of small molecules. These physiologically active molecules exert a variety of direct or indirect CNS effects, often showing hormesis, so that peptide drug development is a promising, but extraordinarily difficult challenge. Earlier, it was believed that peptides were not able to penetrate the BBB, but several studies disproved this criticism (Banks and Kastin 1996; Van Dorpe et al. 2010, 2011). Although the sequence and other structural and genomic information can be retrieved from general protein/peptide databases like Swiss-Prot, experimental data on the BBB transport characteristics of peptides are, however, very scattered in the literature and hence, a need for a more formal approach was felt. Therefore, we built up a database: the ‘Blood–brain barrier peptide database (Brainpeps)’, analogous to existing non-ribosomal and antimicrobial (plant) peptide databases (Caboche et al. 2008; Hammami et al. 2009; Maupetit et al. 2009; Vanhee et al. 2010; Wang et al. 2009). The multi-disciplinary complexity of the BBB research has led to several techniques, each resulting in a specific parameter describing a specific aspect of brain penetration. The different BBB techniques were already overviewed in literature (Nag 2003; Pardridge 1999a), however, to the best of our knowledge, no coherent overview of all the responses is currently available. As the attrition rate of candidate CNS drugs is significantly higher than in other therapeutic areas (Alavijeh et al. 2005), a thorough understanding of the different BBB-penetration aspects is a key aspect in developing CNS drugs. This manuscript thus also focuses on the different methods and responses currently available to study BBB transport of peptides.
Database model
The construction of the Brainpeps database is based on the conceptual design shown in Fig. 2. The level of detail shown in this figure is limited in order to focus only on the important conceptual decisions. The model used for this design is related both to entity-relationship (ER)-modelling (Chen 1976) and unified modelling language (UML)-modelling (Booch et al. 1999). The design shows six entity types (visualized in Fig. 2 as rectangles with right angles). These entity types are ‘peptide’, ‘origin’, ‘experiment’, ‘response’, ‘method’ and ‘publication’. The rectangle shows the name of the entity type, followed by its attributes. Each entity type can have an unlimited number of instantiations in the real world, called entities. For example: the entity ‘Oxytocin’ is an instantiation of the entity type ‘peptide’. In the physical database, each entity type translates to a table, where each attribute translates to a table column and each entity is represented by a table row. Attributes displayed in bold font are mandatory (i.e., missing values are not allowed), while optional attributes are displayed in regular font. The primary key of an entity is a combination of attributes that uniquely and irreducibly describes each entity and is visualized as one or more underlined attributes (Codd 1970). So is each single peptide uniquely identified by the attribute PID (Peptide IDentification). Other attributes represent the Simplified Molecular Input Line Entry Specification (SMILES)-string, the peptide name, the peptide chemical sequence and the International Union of Pure and Applied Chemistry (IUPAC) name. Other entity types are described below.

Database architecture
Next to the entity types, the diagram shows relationship types between entity types. Two kinds of relationship types are distinguished in Fig. 2. The first kind is called a one-to-many relationship type and is symbolized by an arrow between two entity types, marked by two cardinalities. Basically, this type models that one entity (marked by cardinality 1) of the first entity type corresponds to multiple entities (marked by cardinality n) of the second entity type, hence the name one-to-many. For example: the arrow between peptide and experiment means that a peptide can occur in multiple experiments, but each experiment is related to exactly one peptide. In the physical database, one-to-many relationship types do not require an additional table. They are implemented by copying the primary key of the entity type with cardinality 1 in the entity type with cardinality n. The copy of the primary key then serves as a reference and is called the foreign key (denoted FK). For example, the primary key of peptide (PID) is copied in experiment to serve as a reference (FK1). Note that arrows in Fig. 2 always point from a foreign key to a primary key. The second kind of relationship type is called many-to-many relationship. This kind of relationship requires an additional table, visualized by rectangles with rounded angles, and basically implemented by two one-to-many relationship types. There are two many-to-many relationship types in the conceptual design of the Brainpeps database: ‘modification’ and ‘peptide-origin’.
Having explained the introduced notation as used in Fig. 2, a brief discussion about the database schema is given. The central entity type is experiment. Each experiment is characterized by applying a method (MID) to a peptide (PID) in order to measure a particular response (RID). The result of each experiment is exactly one value. Further on, each experiment is conducted within the context of exactly one publication (PBID). Having the database model as it is, database users can investigate experiments by focussing on specific responses measured, by specific methods used or by specific peptides that are the subject of experiments. Peptide modifications that are deliberately applied to peptides, such as modifications for measuring purposes like direct or indirect radio-iodination, can be stored by using the relationship type ‘modification’. Finally, the origin from peptides is characterized by a combination of organism and tissue. The relationship between peptides and origins (OID) is stored by using the relationship type ‘peptide-origin’.
Information collection
For loading information in the database, literature data was gathered by using the search engines ScienceDirect, PubMed and Web of Knowledge. ‘Brain’, ‘blood–brain barrier’ and ‘peptides’, each separately, as well as ‘transport, ‘transfer’, ‘permeation’ and ‘permeability’, using the Boolean operation “AND”, covering the period 1990–2010.
Database availability
The database is available at the following website: http://brainpeps.ugent.be/. Researchers can search the database by peptide nomenclature (e.g. peptide name, 3-letter code, 1-letter code), BBB method (e.g. multiple time regression, efflux), BBB response value (e.g. K in, k out) and literature (author, year, journal). In addition, there are tools to contact us and propose to submit data to the database.
BBB-penetration mechanisms
Brain penetration is the exposure of the peptide (active drug or drug delivery excipient) to the therapeutic target in the brain. This is function of two main processes limiting the brain penetration: permeation and distribution (Liu et al. 2004). The first process describes the rate of entry, whereas the second process represents the relative extent of material that entered the brain (Reichel 2009). The results of brain penetration experiments define compounds in two classes: CNS+ and CNS−, for high and poor penetrators, respectively, although this is often linked to the permeability or distribution component only. For example, compounds with a low log BB (<0) are sometimes defined as poor brain penetrators (CNS−) versus CNS+ compounds having a log BB >0 (Mensch et al. 2010). Similarly, compounds with K p,brain ≪ 1 are designated as poor CNS distributors versus K p,brain > 1 as good CNS distributors (Peremans et al. 2008). Another classification was obtained by defining limits of the effective permeability coefficient obtained with the parallel artificial membrane permeability assay (PAMPA): CNS+ for P e > 4 × 10−6 cm/s are classified as being highly BBB permeable, CNS− when P e < 2 × 10−6 cm/s for low BBB-permeable compounds and CNS+/− when P e ranges from 2 to 4 × 10−6 cm/s for compounds with an intermediate BBB permeability (Di et al. 2003). When using Caco-2 cell lines, the following two classes were distinguished: P app < 2×10−6 cm/s: low permeability and P app > 20×10−6 cm/s: high permeability. Similarly, the immobilized artificial membrane (IAM) capacity factor was used: CNS+ when k IAM/Mw × 10−10 > 0.85 and CNS− when this ratio is lower than 0.84 (Yoon et al. 2006).
Permeation mechanisms
The permeation mechanisms are focused on the blood–brain endothelial barrier, including transcellular passive diffusion as well as active uptake, pinocytosis and paracellular permeation, but also efflux activity and metabolization (phases I and II) within the endothelial cells structurally modifying the compounds before they reach the target brain tissue (Di et al. 2003). BBB permeation describes velocity aspects of peptide flux between blood and brain tissue as obtained from theoretical calculations, in vitro and/or in vivo experiments.
Efflux transporters present at the BBB are P-glycoprotein (Pgp), breast cancer resistance protein (BCRP) and multidrug resistance associated protein-1 (MRP-1). The efflux potential is expressed by the efflux ratio (ER), defined as the ratio of the permeability in the efflux direction divided by the permeability in the influx direction. It is most often assessed by an in vitro cell monolayer permeability assay. Generally, ER > 2 indicates significant efflux and is uncommon in current CNS drugs: the majority of the current CNS drugs shows an ER < 2 (Feng et al. 2008).
Overall, brain permeation is described by the following global responses:
-
P app: the apparent permeability coefficient. Several in vitro system are applied to obtain P app using cell cultures as well as artificial membranes.
-
PS: the permeability–surface area coefficient, defined as the product of permeability P and capillary surface area S (Bickel 2005). This response represents the uptake clearance from blood to brain (Pardridge 2004). The measurement of the PS product is obtained by in vitro as well as by in vivo techniques, using a tissue culture or animal models, respectively.
-
K in: the unidirectional influx rate constant, describing the kinetics of the permeability. By measuring the brain and plasma concentration after intravenous administration and constructing the concentration–time profile, the unidirectional influx rate constant is estimated (Gjedde 1981; Pan et al. 1998a).
-
t 1/2eq,in and t 1/2eq (Liu et al. 2005): the (intrinsic) equilibration half-life. This parameter is derived from the BB concentration profile as being the time to reach 50% of the concentration plateau (Liu et al. 2005). The intrinsic half-life is an extension, whereby the plasma elimination constant rate is neglected, i.e. the plasma concentration is considered constant (Liu et al. 2005).
More specifically, mechanistic aspects are investigated by following methods:
-
Efflux potential in vitro Pgp methods (e.g. Caco-2, knock-out mice).
-
Uptake transporters:
-
in vitro cell layer transport assays using cells transfected with transporter genes, cell uptake assays (incl. oocyte uptake method), inverted vesicle assay, ATPase assay for ABC transporters, calcein-AM assay for Pgp-inhibitors;
-
in vivo experiments using genetically modified animals. For example, the in vivo Pgp efflux ratio is obtained by comparison of Pgp deficient (mdr1a−/−) mice to Pgp competent (mdr1a+/+) mice.
-
Distribution mechanisms
Once in the blood stream, the drug distributes across the body. Several brain distribution mechanisms limit the access of the compound to its target brain cells: metabolic clearance, plasma protein binding, nonspecific binding to proteins and lipids in brain tissue and clearance of the compound from the brain extracellular fluid (ECF) into the blood and cerebrospinal fluid (CSF).
Metabolic clearance (systemic clearance ClS = ClH + ClR + minors) predominantly includes hepatic clearance (ClH): a high rate of liver metabolization (e.g. t 1/2 of a few minutes) will greatly reduce the plasma concentration and thus limit the access of the peptide to the brain. This is also the case for kidney clearance (renal clearance ClR) (Hansen et al. 2002). Species differences are to be taken into consideration, as indicated in Table 1.
When the compound is highly bound to plasma proteins and the on/off kinetics are moderate to slow, then little free drug is available to penetrate the brain tissue. However, it has been shown that release of free drug from bound is higher in vivo than in vitro plasma assays (Pardridge 2004), as demonstrated by their much higher dissociation constant (K d) values in vivo compared to in vitro with bovine serum albumin (BSA) and human α1 acid glycoprotein (hAAG) (Pardridge 1995).
Generally, although data are not consistent, it is the unbound free drug in the brain that is responsible for activity, while the nonspecific binding is no longer contributing to its activity. Measurement of unbound drug in brain for current CNS drugs ranges widely from 0.07 to 52% (Maurer et al. 2005), indicating other factors, like target distribution in the brain, play a role in functional activity.
Brain distribution is described by following responses:
-
B/P: the blood–brain equilibrium distribution defined as the brain-to-plasma concentration ratio at steady-state. It is calculated from in vivo kinetic experiments as \( \frac{{{\text{AUC}}_{\text{brain}} }}{{{\text{AUC}}_{\text{plasma}} }}, \) quantified as log BB.
-
K p: the brain-to-plasma partition coefficient, determined from the total brain and plasma concentration.
-
K p,free: the brain-to-plasma partition coefficient determined from the unbound brain and plasma concentration. This is a better parameter to quantitate the extent of brain equilibrium, because it is independent of plasma and brain tissue binding (Liu et al. 2005).
Responses: definitions and calculations
As demonstrated in Table 2, the different BBB transport techniques can be associated with several responses. Moreover, the response calculation could be carried out in different ways. A certain research group mostly uses the same BBB techniques to study the BBB transport and calculates the responses as they are used to do. For these two reasons, we collected the different calculations used to characterize the BBB transport behaviour as well as the associated methodologies. This way, it is possible to link the BBB techniques with the responses and vice versa in order to present a clearer overview.
Most drugs are transported into the brain by passive diffusion, which is affected by different factors: (1) lipophilicity, (2) ionization, (3) molecular weight and (4) plasma protein binding (Dash and Elmquist 2003). From these four factors, lipophilicity comprises the measure of drug–membrane partitioning and is thus the most important physicochemical parameter for predicting and interpreting membrane permeability (Stewart and Chan 1998).
Lipophilicity
Several groups have correlated the lipophilicity of the compounds with their brain permeability and demonstrated a linear relationship (Banks and Kastin 1985; Cornford 1985; Levin 1980).
The lipophilicity (Liu et al. 2006a) is expressed as log P, log D or delta log P. The partition coefficient (P) (Ecker and Noe 2004; Kumar et al. 2007; Liu et al. 2006a) for test compounds is defined as the concentration ratio of test compound found in the organic phase such as octanol (C org [nl−3]) and that in the aqueous phase (C wat [nl−3]), quantified as log P:
For ionic compounds, the distribution coefficient (D) is used, quantified as log D (Bickel 2005). This coefficient represents the ratio of the sum of the concentrations of all species of the compound, ionic and neutral, in octanol to the corresponding concentration sum in water and is dependent on the pH:
Δlog P is defined as the difference between the log P value of the 1 − octanol/water system and that of the cyclohexane/water system (Ecker and Noe 2004; Eddy et al. 1997):
The classic way to determine the lipophilicity is called the shake-flask method and consists of mixing equal volumes of organic and aqueous solution. Then, the layers are separated and the test compound is added to the aqueous phase and mixed with the organic phase again. Following this, the solution is centrifuged and the content in the aqueous phase is determined using high-performance liquid chromatography (HPLC). The content in the organic phase is also determined with HPLC after lyophilizing and reconstitution in methanol (Ducarme et al. 1998). The octanol/buffer coefficient is finally calculated as the ratio of the concentration in the octanol layer to that in the buffer layer.
IAM capacity factor
As the shake-flask method is time-consuming and less susceptible for automation, (Giaginis and Tsantili-Kakoulidou 2008), immobilized artificial membrane (IAM) chromatography was introduced as a more rapid, efficient, reliable and accurate measurement of lipophilicity (Stewart and Chan 1998). IAM columns contain a monolayer of phosphatidylcholine covalently immobilized on a silica support, which mimics very closely the lipid membrane bilayer (Ducarme et al. 1998; Ecker and Noe 2004; Reichel and Begley 1998). Given the chemical and physical similarity between these IAM columns and the biological cell membranes, these chromatographic columns are mainly used for predicting biomembrane transport properties (Cucullot et al. 2006; Lazaro et al. 2005). The compounds on these columns are analysed in an HPLC system with a physiological buffer (mostly phosphate buffered saline) as eluent and characterized by their capacity factor, expressed as log k IAM:
where t r [t] is the retention time of the test compound and t 0 [t] is the retention time of a reference compound that is not retained by the column (e.g. citrate) and is also called dead time. The obtained capacity factor is then correlated to brain penetration, but is only applicable when the drug is transported by diffusion.
Pidgeon (Pidgeon and Venkataram 1989) developed the IAM column and suggested that k IAM always gives a better correlation for predicting biomembrane transport compared to the capacity factor in reversed-phase HPLC (RP-HPLC) and the octanol–water partition coefficient. For polar and ionizable compounds, it was observed that k IAM is superior over the octanol–water partition coefficient, distribution coefficient and the capacity factor determined by RP-HPLC (Dash and Elmquist 2003).
Surface activity
Another way to predict the blood–brain barrier penetration via passive diffusion can be achieved by constructing surface activity profiles. The BBB consists of a lipid bilayer, characterized as a highly anisotropic system with two distinct regions: (1) the hydrophilic head group–water interface and (2) the hydrophobic region (Fischer et al. 1998). Therefore, conventional investigations that derive the diffusion ability from octanol–water partition coefficients or HPLC assays often fail to predict the membrane permeation for larger compounds because of the limited anisotropy in these systems. Thus, the surface activity was proposed as an alternative to correlate with the BBB penetration (Fischer et al. 1998; Gerebtzoff and Seelig 2006).
The surface activity is determined by molecular properties such as the number of lipophilic groups, the number of charged group and their extent of ionization and the molecular size, which also indicate the ability of the compound to cross the BBB (Seelig et al. 1994).
The surface pressure profile represents the change in surface activity as a function of compound concentration at the air–water interface. This parameter is evaluated in a single experiment measuring the Gibbs adsorption isotherm. The isotherm is a quantitative measure for the tendency of a compound to move to the air–water interface. Because the dielectric constant of air (ε = 1) and the hydrocarbon region of the lipid membrane (ε = 2) are similar, the air–water interface provides a good model for orientation of the compound in the lipid–water interface.
The Gibbs adsorption isotherm can be written as:
where C [nl−3] is the concentration of the compound in the bulk solution, RT [ml2 t 2 n −1] is the thermal energy, N A [n−1] is the Avogadro constant and A s [l2] is the surface area of the surface active compound at the interface.
represents the surface excess concentration (i.e. the concentration at the surface in excess of the bulk concentration), which increases linearly with C at low concentrations and reaches a limiting value denoted Г∞:
This equation may also be written as:
known as the Szyszkowski equation, where K aw is the air–water partition coefficient (Fischer et al. 1998).
To measure the surface activity, a filter paper is equilibrated in buffer until a steady surface tension was reached. The surface tension of the buffer (γ 0) is set to zero and the surface pressure of the compound (π [force length−1 = mt−2]) is recorded by adding small aliquots of the compound stock solution with a microsyringe until equilibrium is reached, typically being between 15 and 20 min. The measurements are performed in a multi-channel microtensiometer, designed for high throughput using small sample volumes and are based on the maximum pull force. Because of the adsorption of a compound at the air–water interface, the surface pressure is defined as the difference between the surface tension of pure buffer and the surface tension of the buffer solution containing the test compound (Petereit et al. 2010):
The permeability
Permeability is defined as the ability, expressed as rate, of a test compound to move into brain, with units of distance per time, whereas brain clearance represents the ability of the brain to remove the test compound from blood after a single passage across the BBB, with units of volume per time (Bonate 1995; de Boer and Breimer 1996; Franke et al. 2000). These two parameters are described in two different sections (“Permeability coefficient” and “The clearance”). Since the development of the first in vitro BBB model (Joo and Karnushi 1973), several co-culture systems were developed and further refined (Cecchelli et al. 1999; Malina et al. 2009; Shayan et al. 2011). The brain endothelial cells were isolated from different species (Lundquist and Renftel 2002; Walker and Coleman 1995). In addition, the use of immortal and non-cerebral cell lines was evaluated as an alternative in vitro system to predict the BBB transport behaviour in vivo (Cecchelli et al. 2006; Garberg et al. 2005; Hakkarainen et al. 2010; Neuhaus et al. 2010; Roux and Couraud 2005).
Next to the in vitro permeability (“The in vitro permeability”), this parameter is also determined with in vivo techniques (“The in vivo permeability”). For the clearance, also two different parameters exist: for influx (“The in vitro influx clearance” and “The in vivo influx clearance”) and efflux (“The efflux clearance”).
Permeability coefficient
The in vitro permeability
This parameter can be obtained by several cell systems using different cell types, as stated above, which all mimic the in vivo BBB as closely as possible. The ideal in vitro BBB model should address the following criteria (Cucullot et al. 2006; Reichel et al. 2003):
-
show restricted paracellular transport reflected by a high transendothelial electrical resistance (TEER) and low permeability of sucrose;
-
express BBB characteristics: morphology, markers and specific transporters;
-
be reproducible and predictable.
-
1.
Isolated brain microvessel endothelial cells (BMEC)
The cells are mostly isolated from the bovine, porcine, mouse, rat and even human brain (Culot et al. 2008; Lundquist and Renftel 2002; Malina et al. 2009; Walker and Coleman 1995) and are quite similar to the in vivo system. Several techniques were applied for the isolation and purification of the cells, involving mechanical and enzymatic dissociation, filtration and centrifugation procedures (Cecchelli et al. 2006, 1999; Grant et al. 1998). The advantages of the isolated BMECs consist of the close in vivo resemblance, the possible cryopreservation and the study of molecular interactions of endothelial functions (Cecchelli et al. 2007; Gumbleton and Audus 2001; Lundquist and Renftel 2002; Pardridge 1999b). However, these cells are not suitable for studying transendothelial transport or transcytosis because there is no separation between the luminal and abluminal compartments (de Boer and Breimer 1996; Eddy et al. 1997). Moreover, because the luminal compartment cannot be accessed, only the abluminal side can be studied (de Boer and Breimer 1996; Lundquist and Renftel 2002; Nicolazzo et al. 2006). In addition, difficulties can be observed during isolation, including limited viability of the cells, metabolic deficiency induction during isolation and differences in isolation selectivity regarding the co-isolation of contaminating cell types (Cecchelli et al. 2006; de Boer and Breimer 1996; Grant et al. 1998; Reichel et al. 2003).
-
2.
BMEC culture model
For quantitative permeability studies, BMEC culture systems are developed, differing in species origin, cell type, isolation procedure, culture conditions and characterization (Deli et al. 2005; Gaillard et al. 2001). Although several species are used to isolate endothelial cells, bovine and porcine are the two species receiving most attention in BBB culture models due to the limited yield of BMECs in murine species and the ethical issues related to the use of human material (Gumbleton and Audus 2001; Reichel et al. 2003). After isolation, the cells form a confluent monolayer on a rat-tail collagen-coated filter or microporous membrane. The resulting monolayer can be used to investigate transendothelial transport, in contrast to isolated cells (Eddy et al. 1997; Hansen et al. 2002; Nicolazzo et al. 2006).
Prior to the experiment, the tightness is evaluated by determination of the TEER and the permeability of high and low integrity markers (e.g. diazepam and sucrose) in order to assure the quality of the culture system. In addition, the permeability of the collagen-coated membrane or filter only is investigated in a control experiment to assure free permeability of the compound (Cecchelli et al. 1999; Malina et al. 2009). The permeability is measured in a diffusion apparatus consisting of a donor and receptor chamber, in between the cultured membrane or filter is inserted. A receptor chamber (abluminal or basolateral side) is filled with culture medium and at time = 0 min, the donor chamber (luminal or apical side) is filled with the test compound dissolved in the culture medium. Both chambers are stirred and after predetermined time points, samples from the receptor chamber are collected and replaced with fresh medium. In order to obtain the initial and final concentrations, samples are taken from the donor chamber in parallel at the beginning and end of the experiment. The amount of test compound in the acceptor chamber is analysed by HPLC or radioactivity and plotted as a function of time in order to calculate the apparent permeability:
where slope [mt−1] is the slope obtained from the acceptor chamber concentration profile, C DO [nl−3] is the initial concentration of test compound in the donor chamber and A [l2] is the membrane surface area (Cucullot et al. 2006; Franke et al. 2000; Hansen et al. 2002).
Another research group (Usansky and Sinko 2003) divided the obtained monolayer permeability from apical to basolateral BMEC side in a passive diffusion and a carrier-mediated transfer term. The monolayer permeability (P m [lt−1]) is then kinetically described as follows:
where J max [nt−1l−2] is the apparent maximal flux, K m is the apparent Michaelis–Menten constant [nl−3], C a is the compound concentration at the luminal side [nl−3], P p [lt−1] is the passive diffusive permeability and \( P_{\text{c}}^{\prime } \) [lt−1] is the apparent net carrier-mediated permeability. The intrinsic carrier-mediated permeability (P c) is defined as:
Some other research groups (Bickel 2005; Cecchelli et al. 1999; Franke et al. 2000; Lundquist et al. 2002) defined the permeability as an endothelial permeability, taking the filter-permeability into account:
where P endoth [lt−1] is the permeability of the endothelial cells, P total [lt−1] is the total permeability and P filter [lt−1] is the permeability of the filter which needs to be corrected for.
Although cell culture systems are expensive, time-consuming and deal with a limited lifespan of the cells, the cultivated cells can be frozen and provide large cell quantities (Cecchelli et al. 2006, 1999; Plattner et al. 2010). Furthermore, the purity as well as the homogeneity of the cell cultures are not easily assured because of the presence of contaminating cells like pericytes, leading to a large variability and low reproducibility (Deli et al. 2005; Plattner et al. 2010). Moreover, during subsequent cultivation, loss of BBB properties occurs, including suppression of transporters, therefore only low passage cells can be used (Prieto et al. 2004; Terasaki et al. 2003).
-
3.
Co-culture
As the BBB consists not only of BMECs, but also other cell types, such as astrocytes and pericytes, known to induce some fundamental BBB characteristics, several research groups developed co-culture models or cell-conditioned media, combining these cell types with BMECs (Gaillard et al. 2001; Nakagawa et al. 2009). This way, a higher TEER and lower permeability was achieved compared to BMEC cultures alone, resembling more the in vivo situation (Shayan et al. 2011). In addition, increased tight junction formation as well as increased expression of specific BBB markers, including transporters, was obtained (Cucullot et al. 2006). Different culture setups exists: non-contact (cells at the bottom) or contact (opposite side of membrane) to take into account the close anatomical association of BMECs and the other cell types (Audus et al. 1999; Malina et al. 2009). Most co-culture systems use rat astroctyes, while pericytes were found to further improve the BBB tightness. Therefore, a triple co-culture model was developed, comprising BMECs, astrocytes and pericytes, mimicking the in vivo BBB anatomy and integrity much more than the other co-culture systems (Nakagawa et al. 2009).
In the mono-culture systems, the test compound is added to the apical compartment at time 0 min. At designated times, the concentration of the test compound in the basolateral compartment together with a sample of the initial solution is measured. The apparent permeability is calculated as described with Eq. 10 (Gaillard et al. 2001; Van Dorpe et al. 2010).
-
4.
Immortalized cell line
In order to avoid the time-consuming isolation and maintenance of primary cell cultures, immortalizing primary BMECs was attempted. Many species were used to produce immortalized cell lines, but the rat model is the most frequently used (Reichel et al. 2003). Given that most in vivo BBB studies have been performed with rodents, this rat model is important to establish in vitro–in vivo correlations (Roux and Couraud 2005). Primary cultured cells were transduced with an immortalizing gene, such as SV40, polyoma virus large T-antigen or adenovirus E1A, by transfection of plasmid DNA or by infection using retroviral vectors. In contrast, conditionally immortalized have been established by using transgenic rodents, which better maintain the in vivo functions compared to immortalized cells (Roux and Couraud 2005; Terasaki et al. 2003). This has resulted in the generation of a number of immortalized cell lines (for an overview see Table 3). For the permeability study, the cell line was cultured on Transwell Clear inserts. Prior to the preparation of the analysis, the Transwell inserts are removed to empty wells within a tissue culture plate and the apical and basolateral chambers are filled with buffer. After a pre-incubation period, transport of the probes in the apical to basolateral direction across the monolayer is initiated by adding buffer, containing test compound, to the Transwell apical chamber. At predetermined times, the concentration of the test compound in the basolateral compartment together with a sample of the initial solution is measured. The apparent permeability is calculated with Eq. 10.
Although the common major drawback of the immortalized cell lines includes the lack of tightness, leading to the formation of a leaky monolayer (Reichel 2009; Smith et al. 2007), these systems proved to be useful for mechanistic and biochemical studies requiring large amounts of biological material due to the expression of relevant BBB transporters (Omidi et al. 2003; Reichel et al. 2003). In addition, no time-consuming cell isolation procedures are required, making these systems less labour intensive (Lundquist and Renftel 2002). Moreover, immortal cell lines can be cryopreserved and grown after recovery without loss of phenotype (Smith et al. 2007).
-
5.
Parallel artificial membrane permeation assay (PAMPA)
A cost-effective high-throughput alternative to in vitro cell methods, PAMPA was first introduced by Kansy (Kansy et al. 1998) and has been widely used in pharmaceutical industry to predict gastrointestinal absorption (Carrara et al. 2007; Dagenais et al. 2009; Ottaviani et al. 2008). The assay involves the use of non-biological artificial membranes and thus only focuses on the prediction of passive transcellular permeability. In order to be able to predict the BBB penetration, the lipid composition was modified using porcine brain lipids (Di et al. 2003; Mensch et al. 2010). PAMPA consists of hydrophobic filters coated with phospholipids in an organic solvent solution (Carrara et al. 2007; Ecker and Noe 2004; Fujikawa et al. 2005).
In order to determine the permeability (P), the acceptor filter is mounted on top of the donor plate to form a sandwich, in which two compartments are separated by the coated membrane filter (Di et al. 2003; Mensch et al. 2010). The test compound diffuses from the donor chamber through the membrane into the acceptor chamber because of the concentration gradient. After incubation of the “sandwich”, the sandwich is dissembled, the concentration of the test compound in acceptor chamber, donor chamber and reference well determined and the permeability calculated. The artificial membrane permeability [lt−1] is calculated as:
where V D [l3] is the donor volume, V A [l3] is the volume of the acceptor compartment, A [l2] is the accessible filter area and t [t] is the incubation time, C A and C D are concentrations of the compound in acceptor and donor well at time t and 0, respectively (Carrara et al. 2007; Feng et al. 2008; Mensch et al. 2010). Although the PAMPA-assay is accurate, low cost, reproducible and consumes only a limited amount of test compound, this system completely lacks pores and active transporter systems due to the artificial nature of the membrane (Verma et al. 2007). In addition, a long incubation time is required and adsorption by the assay plate occurs, attributed to the high tightness of the artificial membrane (Uchida et al. 2009).
-
6.
Non-cerebral cell lines
Despite the morphological and biological differences (membrane lipids, enzymes and transporters) between the BBB and non-cerebral peripheral epithelial cell lines (Caco-2 and MDCK), these cell lines possess sufficient restrictive paracellular transport (Lundquist and Renftel 2002; Nicolazzo et al. 2006; Reichel et al. 2003). These cells were initially used to predict intestinal absorption, but were also proven to estimate BBB permeability (Garberg et al. 2005; Nicolazzo et al. 2006). Caco-2 cells are derived from a human colon carcinoma cell line, expressing transporters such as the peptide transporter-1 (PEPT1), monocarboxylic transporters (MCT) and the P-glycoprotein (Pgp) efflux transporter. Thus, the Caco-2 cell monolayers can be used to study passive transport (transcellular and paracellular) as well as active transport (Fujikawa et al. 2005; Siissalo et al. 2009). Madin–Darby canine kidney (MDCK) cells are epithelial cells derived from dog kidney, mimicking some BBB properties (Navarro et al. 2011; Wang et al. 2005). These cells have also been used as a model for the BBB permeability, resulting in similar P app values as determined with BMEC (Garberg et al. 2005).
To study the transport across the cultured cells, the apical side (for apical to basolateral transport) or the basolateral side (for basolateral to apical transport) of the cell monolayer receives a test compound solution. After shaking, the amount of test compound in the receiver chamber, basolateral side (for apical to basolateral transport) or apical side (for basolateral to apical transport) is determined.
The apparent permeability (P app [lt−1]) is calculated according to Fick’s law of diffusion:
where \( \frac{{{\text{d}}Q}}{{{\text{d}}t}} \) [nt−1] is the transport rate of the test compound to appear in the receiver chamber, A [l2] is the surface area of the cell monolayers and C 0 [nl−3] is the initial concentration of test compound in the donor chamber (Garberg et al. 2005). The ratio of dQ and dt is also called the flux (J) (Yazdanian et al. 1998). Both cell lines are easily grown and the permeability assay can be automated, but only qualitative predictions of BBB permeability are drawn. Moreover, only low amounts of Pgp are expressed. Therefore, MDCK cells were transfected with the human multidrug resistance gene (MDR-1) leading to the overexpression of Pgp. In addition, the use of Caco-2 cells is limited due to the significant permeability differences between intestine and BBB cells (Cucullot et al. 2006; Gumbleton and Audus 2001; Navarro et al. 2011; Nicolazzo et al. 2006; Reichel 2009).
The in vivo permeability
Although, the use of in vitro systems revealed several advantages such as low amount of compound needed, high-throughput, mechanistic studies, extrapolation to the in vivo situation may be hampered because of the differences between the various in vitro techniques (de Boer and Breimer 1996; Usansky and Sinko 2003). These differences exist mainly due to the use of different model systems or improper and unclear experimental conditions (Franke et al. 2000).
In addition, currently no generally satisfying in vitro BBB model exists (Reichel 2009; Reichel et al. 2003; Walker and Coleman 1995). Nevertheless, the results still need to be correlated and validated with the in vivo data. Several animal-based in vivo models are available, but their applicability depends on aspects such as the sensitivity and selectivity of assays of drugs in the brain, fast and poor BBB penetrating compounds, the estimation of local concentrations in the brain or whole brain distribution, and the measurement of single time concentrations versus concentration time profiles (Lundquist et al. 2002; Nicolazzo et al. 2006).
-
1.
Indicator diffusion
Because the arterial concentration (C a) is not measured, a non-diffusible internal standard is co-injected with the test compound. This internal standard serves as a measure of the degree of dilution of the test compound within the cerebral circulation and thus reflects what C a would have been if no dilution occurred (Bonate 1995; Knudsen and Paulson 1999).
The test compound is injected in the artery, followed by immediate collection of several blood samples to construct time–concentration curves. After determining the extraction of the test compound as
the permeability coefficient [lt−1] could be calculated as (Crone 1965):
where \( \left( {\dot{Q}} \right) \) is the rate of blood flow per gram tissue and per time [l3m−1t−1] and the capillary surface area per gram tissue (A) [l2]. As the concentrations [nl−3] of the reference and test compound in the test solution are not equal, all concentrations are expressed relative to the concentrations in the injection solution (Crone 1965).
Although a single animal may be used for several time points, eliminating a major source of variation, this technique is technically difficult and the data are not easy to interpret (Bonate 1995).
-
2.
Mouse brain uptake assay (MBUA)
The MBUA is routinely used by Raub (Raub et al. 2006) and is more sensitive than BUI and indicator diffusion. In this test, a single intravenous dose of a test compound is administered, followed by blood and brain collection, typically at 5 min post-dose. The obtained brain and plasma concentrations are used to calculate an apparent permeability coefficient (P app). Brain concentrations are corrected for the plasma volume present in brain tissue, measured independently with an impermeable marker (e.g. inulin) (Garberg et al. 2005; Nicolazzo et al. 2006). Considering a two-compartment system, P app is estimated as originally described by Ohno et al. (1978):
where P app [lt−1] is the permeability coefficient, A [l2m−1] is the capillary surface area per gram brain, C br [Nm−1] and C pl [Nl−3] are the concentrations of test compound in brain and in plasma, respectively, and V e is the volume fraction of brain into which the test compound distributes.
It is assumed that metabolism, efflux out of the brain and tissue accumulation are negligible at the investigated time point (Garberg et al. 2005). However, permeability can only be measured under conditions that are independent of cerebral blood flow and for test compounds that are less permeable than glycerol (Ohno et al. 1978).
-
3.
Antinociception test
Peptides inducing an analgesic effect can be studied with the antinociception test. After administering the test compound intravenously or intracerebroventricularly into the animal, the tail flick withdrawal is evaluated by immersing the animal’s tail in hot water (55°C). The latency to withdrawal is measured at different time points in order to construct the time curve (Kleczkowska et al. 2010; Liu et al. 2006a). The dose producing 50% of the maximum response (i.e. ED50), derived from fitting the dose–response data, is multiplied with the ratio of peak response (PR) and total concentration as expressed as the AUC:
The ratio of PR and AUC normalizes for degradation and elimination.
Finally, the BBB permeability index (BBB-PI) is defined as the ratio of normalized ED50 after intravenous and intracerebroventricular injection, respectively (Fiori et al. 1997):
The clearance
While the permeability is defined as rate of a test compound to move into brain, brain clearance represents the volume blood that is completely removed by the brain after a single passage of the brain.
This parameter could be defined for influx transfer (determined in vitro as well as in vivo) and for efflux transport (only investigated in vivo), described separately below.
The in vitro influx clearance
After determining the concentration of the samples in acceptor and donor chamber, as explained above in “Permeability coefficient”, the clearance is calculated as (Cecchelli et al. 1999; Garberg et al. 2005; Nakagawa et al. 2009):
where C A and V A are the concentration and volume in acceptor chamber defining the amount present and C D is the concentration in donor chamber. This equation is applicable for all in vitro systems explained above: isolated and cultured BMEC, co-culture, immortalized cell lines and non-cerebral cell lines.
The in vivo influx clearance
The in vivo influx clearance is investigated with the brain uptake index method (BUI) or in situ brain perfusion technique.
-
1.
BUI
The brain uptake index method was developed by Oldendorf (1970), in which the test compound together with a freely diffusible reference is injected in the artery, followed by brain sampling after 5–15 s. An extraction of the test compound is determined, assuming that no metabolism and back-diffusion occurs of both compounds:
where E test/E ref is the ratio of the extracted concentration test and reference compound in brain and injectate, respectively. To a lesser extent, other related definitions of BUI were used, e.g. Isakovic et al. (2004), where BUI is defined as \( \left( {{\raise0.7ex\hbox{${E_{\text{test}} }$} \!\mathord{\left/ {\vphantom {{E_{\text{test}} } {E_{\text{ref}} }}}\right.\kern-\nulldelimiterspace} \!\lower0.7ex\hbox{${E_{\text{ref}} }$}}} \right) \times 100 \), or Bickel (2005), who defines BUI as \( \left( {\frac{{E_{\text{test}} - E_{\text{ref,vascular}} }}{{E_{\text{ref,permanent}} }}} \right) \times 100 \). From this BUI experiment, where E test is determined, the PS product [l3t−1] is calculated with the Renkin–Crone equation:
where CBF [l3t−1] is the cerebral blood flow (Bickel 2005; Isakovic et al. 2004).
This method may be used for small animals and can easily be modified to quantify efflux transfer, but is only applicable for highly permeable compounds (Bickel 2005; Bonate 1995; Cornford 1999).
-
2.
In situ brain perfusion
In order to measure the brain uptake also for slowly penetrating compounds and thus improve sensitivity, compared to the BUI technique, in situ brain perfusion was introduced. The higher sensitivity is ascribed to the prolonged experimental time (Begley 1999; Dash and Elmquist 2003). The method was developed by Takasato (Takasato et al. 1984) using rats and was adapted for mouse (Dagenais et al. 2000; Murakami et al. 2000) as already successfully applied to study brain uptake of peptides (Banks et al. 2002; Kastin and Akerstrom 1999; Pan et al. 2005; Zlokovic et al. 1990, 1994). The circulation to the brain is taken over via infusion into the heart, while the heart is stopped, such that no contribution of the blood flow occurs (Brandsch et al. 2008; Golden and Pollack 2003; Smith 2003; Urquhart and Kim 2009). The artery is catheterized, the heart ventricles severed and the brain is perfused with buffer, containing test compound and impermeable marker. Perfusion is terminated by decapitating the animal at a designated time point, typically 2–5 min.
The initial uptake clearance (Clup [l3t−1 m−1]) for short-time perfusion is determined as:
where C Brain [mm−1] is the brain tissue concentration, C Perfusion [ml−3] is the perfusion fluid concentration, T [t] is the perfusion time and V v [l3m−1] the brain vascular volume. The uptake clearance is then estimated from the slope of the initial linear portion of the C Brain/C Perfusate versus T plot (Liu et al. 2004).
The major advantage of this method consists of the possible manipulation of perfusion medium and time. In addition, metabolism and plasma protein binding are negligible, BBB integrity can be monitored with the impermeable marker and the method is also suitable for slowly penetrating compounds. However, the method is technically difficult and limited to the use of small animals as large volumes of perfusate would be needed to ensure adequate cerebral blood flow rates (Begley 1999; Smith 2003; Su and Sinko 2006).
The efflux clearance
The limited brain uptake of certain compounds can be ascribed to the presence of several efflux transporters at the BBB, making it important to investigate the efflux of test compounds (Brandsch et al. 2008; Golden and Pollack 2003; Urquhart and Kim 2009). The brain efflux index (BEI) method was developed by Kakee (Kakee et al. 1996) in order to quantify the efflux of a test compound across the BBB. A mixture of test and impermeable reference compound is injected into the brain parietal cortex area 2 (Par2), followed by brain collection at designated time points. From this injection site, diffusion of the impermeable marker into the rest of the CNS is limited (Hosoya et al. 2002). Because it is difficult to measure the effluxed portion, the amount associated with the brain is determined. The brain efflux index is thus defined as the percentage test compound remaining in the brain, normalized to the impermeable marker (Kusuhara et al. 2003):
where conc(test/ref) is the ratio of the concentration of test and reference compound remaining in the brain and injected solution, respectively.
By semilogarithmic fitting of the (100 − BEI)-values versus time, the apparent efflux rate constant from the brain (k eff) is obtained, which is necessary to determine the apparent brain efflux clearance (Cleff [l3t−1] (Isakovic et al. 2004; Moriki et al. 2005; Raybon and Boje 2001; Terasaki 1999):
where V br [l3] is the brain distribution volume of the test compound, determined with the brain slice uptake technique. Although it is very interesting to study efflux, this technique is not useful for screening. In addition, metabolism and damage from the needle, altering the BBB properties, could confound the results (Nicolazzo et al. 2006; Su and Sinko 2006).
Recovery
Because only the unbound fraction of a compound is available for BBB transport, the intracerebral microdialysis technique was applied to determine this fraction (Tsai 2003). The technique is based on the perfusion of a microdialysis probe, consisting of a semi-permeable membrane, with physiological buffer, whereby compounds that are small enough can diffuse through the membrane along the concentration gradient. The probe is stereotaxically implanted into the brain region of interest, which is an invasive procedure (Bickel 2005; Tunblad et al. 2003). After continuous blood and brain sampling, the concentration of the test compound that permeated into the brain is monitored over time. The free concentration of the compound in the brain ECF is reflected by the concentration measured in the dialysate and is used to construct the concentration–time curve. Generally, the transport properties are described by the extraction efficiency or the recovery (Tsai 2003):
where C perf [nl−3] is the concentration of the perfusate, C dial [nl−3] the concentration of the dialysate and C s [nl−3] the concentration of the sample.
Quantitative data are obtained after individual calibration of the probe, but depends on probe characteristics, elimination and metabolism (de Lange et al. 1999). Due to different diffusion properties of the compound in tissue, the recovery in vivo is different from that in vitro. Therefore, the internal reference technique was found to be useful as a calibration method for both, in vitro and in vivo experiments. An internal standard is added to the perfusion buffer, and then the concentration in the dialysate could be determined relative to the initial concentration in the perfusion medium (Hansen et al. 2002; Van Belle et al. 1993). As dialysis is performed in conscious animals, several advantages are associated to this technique: (1) continuous sampling; (2) measurement of concentration over different time points in a single animal allows the construction of a pharmacokinetic concentration–time profile; (3) reduction of number of animals and thus the inter-animal variation; (4) no influence of anaesthesia; and (5) no sample preparation before analysis is required due to exclusion of large molecules by the dialysis membrane (Dai and Elmquist 2003; de Lange et al. 1999; Hammarlund-Udenaes 2000). However, the method is limited by the need for probe calibration as well as by the sensitivity of the analytical technique used for the analysis of the dialysis samples (Bonate 1995; Davies et al. 2000; Stenken 1999). Moreover, microdialysis is invasive and may damage BBB integrity, probe implantation requires technical skills, only local concentrations are measured and low sample concentrations and volumes are obtained (Bickel 2005; Chaurasia et al. 2007; de Lange et al. 1999; Nicolazzo et al. 2006).
The probe can be used in the delivery mode, by addition of test compound to perfusion buffer, or in the sampling mode, in which perfusion buffer is devoid of test compound. This way, the recovery can be estimated either by gain or by loss as described below.
Recovery by gain
The calculation of the recovery by gain is only possible in vitro because a known test compound concentration (C s) is added to the sample solution surrounding the probe. The probe is perfused with buffer alone (C perf = 0) and the dialysate concentration (C dial) of the test compound is analysed. The relative recovery (Scheller and Kolb 1991) equals
Recovery by loss
Next to the determination of the recovery by gain, retrodialysis can be used to obtain the concentration recovery by loss in vitro as well as in vivo. In this assay, the test compound is added to the perfusion buffer (C perf [nl−3]), the unbound fraction present in the brain ECF diffuses into the probe and is collected for analysis. Before perfusion, neither the animal for in vivo assay nor the probe for in vitro assay is treated with test compound (C s = 0). The relative loss of test compound from the perfusion buffer into the surrounding medium or tissue through the membrane is calculated (Hansen et al. 2002; Tsai 2003; Tunblad et al. 2003):
The concentration of test compound in brain ECF is then defined as (Dai and Elmquist 2003):
Brain-to-plasma partition coefficient
The brain-to-plasma partition coefficient (K p) is the most widely used parameter to evaluate the extent of brain penetration or distribution (Liu et al. 2008; Reichel 2009), describing the ratio of total brain and plasma concentration at steady state:
The total concentrations in brain and plasma are defined as:
However, K p is of limited value because it depends on binding in plasma and brain, uptake and efflux transport at the BBB, metabolism in the brain and the bulk flow of CSF. It should be pointed out that it is the free concentration that exerts the CNS action, thus a large total brain concentration does not necessarily mean a high concentration available for action due to binding. If active transporters, brain metabolism and the bulk flow of CSF do not contribute significantly to brain compound disposition, then the free compound concentration in brain is equal to the free compound concentration in plasma and K p is expressed as the ratio of unbound fraction in plasma (f u,plasma) and unbound fraction in brain (f u,brain) at equilibrium (Gupta et al. 2006; Jeffrey and Summerfield 2010; Maurer et al. 2005):
where K p,uu is the ratio of the unbound concentration in brain and plasma at steady state\( \left( {K_{\text{p,uu}} = \frac{{C_{\text{u,brain}} }}{{C_{\text{u,plasma}} }}} \right) \) and K p,in is the intrinsic partition coefficient defined as the ratio of the unbound fractions in brain and plasma \( \left( {K_{\text{p,in}} = \frac{{f_{\text{u,brain}} }}{{f_{\text{u,plasma}} }}} \right) \).
K p,uu governs BBB properties independent of plasma and brain tissue binding, therefore represents a better parameter than K p to assess brain penetration, but is experimentally more difficult to obtain. K p,in is determined by nonspecific binding in brain and plasma (Becker and Liu 2006; Liu et al. 2005).
Several techniques can be used to determine the unbound fractions as discussed below.
-
1.
Equilibrium dialysis
The unbound fraction in plasma and brain homogenate, and thus K p,in, is determined with an in vitro or in vivo equilibrium dialysis method. When performing the in vitro analysis, plasma and diluted homogenized brain tissue are both spiked with test compound and loaded in the dialysis apparatus. At the start of the experiment, the receiver chamber is filled with buffer and during incubation the test compound can diffuse from donor to receiver chamber. After reaching equilibrium, samples are taken from the incubated receiver and donor chambers and the concentration investigated with an analytical technique, mostly HPLC.
Since brain is diluted prior to analysis, the dilution factor (D) is taken into account when calculating the unbound fraction (Kalvass and Maurer 2002; Summerfield et al. 2007):
Although the homogenates are readily available and can easily be stored until analysis, the main disadvantage of the technique consists of the necessity to homogenize brain before analysis. This may change the binding properties by unmasking binding sites that are not accessible in vivo (Liu et al. 2008; Read and Braggio 2010).
The dialysis method can also be applied to in vivo animals as described in “Recovery”. By placing the dialysis probe in blood and brain, it is possible to measure the unbound concentrations over time on each side of the BBB (Gupta et al. 2006; Jeffrey and Summerfield 2010).
-
2.
Ultrafiltration or ultracentrifugation technique
Ultrafiltration reveals tissue binding after adding plasma or brain homogenate, spiked with test compound, into the ultrafiltration tube. Test compounds are forced through the membrane using a pressure gradient. The solution is centrifuged at a speed of about 1,800g and the filtrate analysed to measure the free concentration. This concentration is then compared to the total concentration of plasma or brain homogenate. Separately, adsorption to the filtration membrane is measured (Berezhkovskiy 2008; Mano et al. 2002; Tsai 2003).
Ultracentrifugation, in contrast, uses a centrifugation tube with plasma or brain homogenate and test compound. The solution is ultracentrifuged at a speed of more than 400,000g, resulting in three layers according to the density of the substances present. The upper layer is used for the estimation of the total concentration, whereas the middle layer determines the unbound concentration. It is assumed that no protein contamination is present in the middle layer and binding equilibrium is not affected during ultracentrifugation. This technique is superior to ultrafiltration, because in the latter test compound may show adsorption to the membrane (Nakai et al. 2004).
-
3.
Brain slice method
Because the dialysis, ultracentrifugation and ultrafiltration techniques require tissue homogenization before analysis, as an alternative, the brain slice method could be applied. In the brain slice method, the brain tissue remains intact, being more physiologically relevant due to the conservation of cellular structures of the brain tissue (Di et al. 2011; Reichel 2009).
After decapitation of the animal, brain is collected and cut into slices. Following this, the slices are incubated with buffer containing test compound. At different time points, slices and samples of incubation buffer are taken, dried on a filter paper, homogenized and measured (Becker and Liu 2006; Friden et al. 2007). In contrast to the dialysis method, the free drug concentration is indirectly derived from the unbound brain distribution volume:
and thus
V i is the volume of buffer film remaining around the sampled slice due to incomplete absorption of buffer by the filter paper and is estimated from a separate experiment using inulin (Friden et al. 2009).
Compared to microdialysis in vivo, the brain slice method is applicable to a wide range of compounds, technically simple, more reproducible and yields a higher throughput. However, no extensive validation of this method is reported (Becker and Liu 2006; Di et al. 2011; Read and Braggio 2010).
The blood–brain equilibrium distribution
Next to the partition coefficient, described above, the brain equilibrium distribution could also be described with the BB parameter (Garberg et al. 2005; Liu et al. 2004). BB is simply defined as the ratio of the steady-state concentrations (or area under curve) in brain and blood, quantified as log BB (Alavijeh et al. 2005):
Once obtained, this parameter is applied in other calculations (e.g. P app) and in computational in silico predictions of the BBB permeability in terms of the physicochemical properties (Pardridge 2004; Subramanian and Kitchen 2003). Although, it is easier to determine log BB compared to kinetic data (e.g. log PS), the results may be misleading. A low log BB value may not necessarily mean a low penetration, because the parameter depends on binding to plasma and brain tissue as well as active transport (Alavijeh et al. 2005; Lanevskij et al. 2010; Liu et al. 2004). For this reason, it was suggested to replace log BB by log PS values (Martin 2004; Pardridge 2004).
Several in vivo methods, such as the intravenous injection technique, MBUA and microdialysis, can be performed to obtain the log BB (see “CSF concentration”, “The in vivo permeability” and “Recovery”, respectively) (Ecker and Noe 2004; Tsai 2003).
The unidirectional influx constant from plasma to brain
In order to quantify the rate of blood to brain transport, a transfer constant can be determined with the intravenous injection, in situ brain perfusion and microdialysis methods.
-
1.
Multiple time regression or intravenous injection technique
This technique represents the most widely applied approach to quantify the unidirectional transfer constant for slowly penetrating compounds (Bickel 1999). After intravenous injection of the test compound, arterial blood and brain are collected. The blood and brain collection is obtained at various times (between 1 and 60 min) after injection, each time point representing one animal (Banks et al. 2002; Kastin and Akerstrom 1999; Pan et al. 1998b; Pardridge 1995). As only the initial uptake is regarded, efflux from the brain into the blood can be ignored (Smith 2003). In order to avoid the dependence of a proper, specific compartmental model, a graphical approach was developed to study brain uptake. The steady-state unidirectional influx transfer constant is graphically derived from the following equation (Gjedde 1981):
where A m is the amount of radioactivity in brain at time t, C p is the amount of radioactivity in serum at time t, K in the unidirectional influx rate constant and V i the initial distribution volume, including endothelial binding and accumulation as well as intravascular distribution.
As the level of test compound in serum varies during the experimental period due to elimination, the exposure time (Exp.t) is defined as the integral of the serum radioactivity over time divided by the radioactivity at time t:
This integral of radioactivity over time is represented by the area under the curve (AUC) and its relationship with the experimental time was demonstrated by Kastin et al. (2001).
Finally, the brain/serum ratios (μl/g) are plotted versus exposure time and the slope of the linear portion of this relationship measures K in, while the intercept represents V i.
During this study, the BBB is intact, presenting all transporters, junctional proteins and enzymes at their physiological concentrations, the unique architecture of the blood vessels and perivascular cells is present and undisturbed, and cerebral metabolic pathways are not compromised (Nicolazzo et al. 2006; Smith 2003). In addition, saturation as well as regional studies can be performed with this method (Bonate 1995; Smith 2003). Moreover, this technique is technically easy, applicable to slowly penetrating compounds, gives information on pharmacokinetics in blood and brain and is sensitive (Bickel 1999; Nicolazzo et al. 2006). However, metabolism and distribution into tissues may confound the results, efflux is ignored, the method is labour intensive and time-consuming requiring a large number of animals and thus inter-animal variation may cause a large variance (Bonate 1995; Cecchelli et al. 2007; Smith 2003).
-
2.
Single time point analysis
The above-described experiment can also be carried out in order to obtain the influx transfer constant form a single time point analysis. A single brain is collected at one selected time point and several blood samples are collected at suitable time intervals from the catheterized artery (Mensch et al. 2009). The influx constant could be determined as follows (Bickel 1999):
where C br [nm−1] is the brain concentration, K in [l3m−1t−1] is the unidirectional influx constant and the integral expresses the plasma concentration over time. The brain tissue concentration is corrected for the intravascular content (V 0), determined by a vascular marker (e.g. BSA):
where V D [ml−3] is the apparent volume of distribution of the test compound in total brain at sampling time t and C p is the plasma concentration [nl−3].
Thus, when combining these two equations, the unidirectional influx constant could be determined as (Bickel 1999):
Although only a single animal is required for this study, reducing the inter-animal variation, it is however not frequently applied as replaced by the multiple time regression. The major drawback is the possible erroneous estimation of K in if there is a significant efflux during the experimental period (Patlak et al. 1983).
-
3.
External imaging detection methods
Detection in multiple time regression can also be carried out with the rapid, non-invasive imaging techniques, measuring real-time and local levels of blood and brain concentrations. Nevertheless, these methods have a high cost, are labour intensive, yield a low-throughput and are not able to separate parent and metabolite compounds (Bonate 1995; Cecchelli et al. 2007; Nicolazzo et al. 2006).
Dynamic magnetic resonance imaging (MRI) measurements register the concentration changes of a contrasting agent (e.g. gadolinium-diethylenetriaminepentaacetic acid) in brain tissue after an intravenous injection. Measurement is based on the magnetic properties of atoms and their ability to absorb energy at a specific radiofrequency, which is emitted after excitation (Dienel 2006). Determining the unidirectional influx rate by evaluation of the concentration–time curve of the contrast agent requires a compartmental kinetic model or graphical analysis analogous to Eq. 36 (Ewing et al. 2003; Taheri and Sood 2007):
where C tissue(t) [nl−3] is the tissue concentration of the indicator at time t per gram brain, C pa(τ) [nl−3] is the arterial plasma concentration over the duration of the experiment per unit plasma volume and V p [l3] is the tissue volume. Compared to the other imaging techniques, MRI has a limited sensitivity (Bickel 2005). However, nowadays this technique has improved sensitivity due to the use of high resolution MRI systems, which can now work at 15 T. In fact, this method remains the gold standard to assess BBB function and integrity (Albensi et al. 2000; Hoffmann et al. 2011).
As already described in the capillary depletion technique, it is important to consider whether the peptides do cross the blood–brain barrier or are bound to the endothelial cells. This binding might influence the permeability results. As already described in the capillary depletion technique, it is important to consider whether the peptides do cross the blood–brain barrier or are bound to the endothelial cells. This binding might influence the permeability results (Ermisch 1992; Ermisch et al. 1985, 1991; Kretzschmar et al. 1986). Observing the lack of consensus on nomenclature in quantitation of in vivo radioligand binding studies, Innis et al. (2007) proposed standard terminology in order to reduce confusion and improve clarity.
Positron emission tomography (PET) could also be applied to investigate the BBB permeability using a PET scanner with tracers, such as 82Rb or 68Ga-EDTA. A positron-emitting nuclide is injected, resulting in the annihilation of a positron with an electron. Following this, two gamma photons with an energy of 511 keV are emitted at 180° angle (Dienel 2006; Mueggler et al. 2009). Again a compartmental model is applied to determine the relationship between the transport rate constant and the tissue-plasma concentration as described in Eq. 41 (Logan 2000; Webb et al. 1989). Currently, this is the most sensitive imaging system (Dienel 2006).
Single photon emission tomography (SPECT) results in a lower gamma counting sensitivity and a more limited number of labelled tracers are available compared to PET. In order to solve the low-resolution problem in small animals, new SPECT systems were created (Beekman and Vastenhouw 2004). A gamma-emitting compound is injected in order to evaluate the unidirectional rate (see Eq. 41) (Bickel 2005; Dienel 2006).
-
4.
In situ brain perfusion
As already described above in detail (see “The in vivo influx clearance”), the in situ brain perfusion technique could be performed to obtain the in vivo clearance. Analogous to Eq. 36, the unidirectional influx rate (K in) is described as (Begley 1999):
where C br and C perf [nm−1] are the concentrations in brain and perfusate, respectively.
Since the concentration of the test compound in perfusion buffer is constant, the integral of concentration in perfusate equals C perf. Thus, the equation is rearranged and simplified to (Smith 2003):
where t is the perfusion time.
By plotting the brain/perfusate concentration ratio as a function of experimental time, the unidirectional influx transfer constant is estimated from the slope of the regression curve.
In addition, the single time point analysis can be described for perfusion as well (Begley 1999):
This method is technically more difficult than the intravenous injection technique, but permits the modification of perfusion buffer (Bickel 2005).
The half-life
The same definition applies to the calculation of the half-life from the brain elimination or efflux rate, independent of the method used, but the way to determine the efflux rate differs. The brain-to-plasma concentration ratio and the remaining activity in brain can be used as detailed below.
-
1.
Brain-to-plasma concentration ratio
The equilibration half-life (t 1/2eq) represents the time to reach 50% of the brain-to-plasma concentration ratio at equilibrium (i.e. 50% of the plateau level of the brain-to-plasma concentration time profile) and is calculated after injection of a single intravenous bolus as (Liu et al. 2005; Tunblad et al. 2003):
where k out [t−1] is the brain elimination rate constant and k el [t−1] is the plasma elimination rate constant. The efflux and elimination rate constants for brain and plasma are obtained as:
and
respectively.
In these equations, PS [l3t−1m−1] is the permeability–surface area product (explained later), f u,brain is the unbound fraction in brain tissue measured by microdialysis, V b [l3m−1] is the volume of brain tissue, Clp [l3t−1m−1] represents the systemic clearance and V p [l3m−1] is the plasma distribution volume.
Because Eq. 45 is only applicable for compounds where k out is larger than k el, the intrinsic equilibration half-life is introduced as the time to reach 50% of the brain-to-plasma concentration at equilibrium under the condition of constant plasma concentration (Liu et al. 2005):
Here, k out is calculated from the PS product and the unbound brain fraction as stated in Eq. 46.
Because these definitions are related to a pharmacokinetic compartmental model, the major drawback is that they are specifically valid for this model only. Moreover, microdialysis was necessary to obtain the free fractions of test compound.
-
2.
Efflux study
The animal receives an intracerebroventricular injection of radiolabelled test compound into the right lateral ventricle and the residual radioactivity in the brain is determined. By plotting the natural logarithm of this measured radioactivity remaining in the brain against time, the half-life of the compound is derived from the linear regression curve using Eq. 48.
In this study, k out is defined as the slope of the regression curve.
For time point zero, a control group is studied, in which the animal is killed by injecting an overdose of anaesthetics, followed by compound injection (Banks et al. 2002; Hsuchou et al. 2007).
Compartmental distribution
All in vivo methods lack the ability to discriminate between binding to the vasculature and complete penetration through the BBB. In order to distinguish between this endothelial binding and/or endocytosis by brain capillaries and actual transcytosis, the capillary depletion method or quantitative autoradiography is used (Bickel 1999; Triguero et al. 1990).
-
1.
Capillary depletion
After intravenous injection of the test compound, followed by subsequent blood and brain sampling, the brain is homogenized. Brain parenchyma and capillaries are separated by dextran density centrifugation (Banks et al. 2002; Chen et al. 2002; Pan et al. 2005; Price et al. 2007; Smith 2003). When the test compound is specifically bound to the brain endothelium, then the compound is not separated from the pellet (capillaries), while in the case of nonspecific binding, the test compound will distribute into the supernatant (parenchyma) during the brain centrifugation (Pardridge 1995).
The compartmental distribution (CD) is evaluated by plotting the tissue/serum ratio of brain cortex, parenchyma and capillaries as calculated by:
where N tissue [Nm−1] is the radioactivity per gram tissue (i.e. brain, parenchyma or capillaries) and N serum [Nl−3] is the radioactivity per volume serum.
This method was found to be rapid, sensitive and results in quantitative data (Triguero et al. 1990).
-
2.
Quantitative autoradiography
Although, the capillary depletion technique is much faster and has at least a comparable sensitivity, quantitative autoradiography (QAR) may visualize subcellular organelles within the endothelial cytoplasm, which are involved in trafficking (Triguero et al. 1990). The only difference with the above-described method consists of the brain activity measurement. This is performed on cryostat sections of the harvested brain tissue, which are dried and exposed to X-ray film (Bickel 2005). The film is developed and the distribution and quantitation of the radioactivity is measured (Dash and Elmquist 2003; Dienel 2006; Fenstermacher and Wei 1999).
CSF concentration
CSF can easily be sampled by cisternal puncture, performed through catheters inserted into the cistern magna or lumbar intrathecal space (Liu et al. 2006b; Okura et al. 2007). The CSF concentration reflects the unbound or free concentration in the brain when steady-state equilibration of freely diffusible test compound concentration is achieved throughout the brain (Maurer et al. 2005; Shen et al. 2004). When comparing to the BBB, different transporters are present at the blood–cerebrospinal fluid barrier (BCSFB) and the range of molecular size that can penetrate into the CSF is much larger (Saunders and Dziegielewska 1999). CSF concentration may be a poor indicator of brain ECF levels because only 10% of CSF arises from brain ECF, while 90% originates from the choroid plexus. Moreover, the CSF volume is limited and the method is only valid for slow uptake compounds (Hammarlund-Udenaes 2000; Read and Braggio 2010; Shen et al. 2004).
-
1.
Microdialysis
The spinal CSF concentration can be determined by subjecting the animal to microdialysis (see “Recovery”). In this case, the dialysis probe is inserted in the cisternal membrane to estimate the in vivo recovery as expressed in Eq. 28 (Okura et al. 2007):
The concentration of test compound in CSF is calculated based on the probe recovery (Okura et al. 2007):
-
2.
Cisternal puncture
As the microdialysis involves technical skills, the cisternal magna or lumbar intrathecal space can be catheterized for CSF sampling.
After administration of the test compound, arterial blood, cisternal CSF and blood are collected. CSF-to-plasma
brain-to-plasma
and CSF-to-brain
are then calculated.
The brain-to-CSF ratio is equivalent to the apparent distribution volume of the test compound normalized to the mass of the collected brain tissue (Liu et al. 2006b; Shen et al. 2004). The presence of the test compound in the CSF only reflects extracellular availability.
The determined CSF concentration and unbound plasma concentration are used to predict the unbound brain concentration indirectly (Liu et al. 2006b).
Derived parameters
Parameters derived from the ones described above, which are generally used, are mentioned here.
Surface parameters
The analysis of the Gibbs adsorption isotherm see "Surface activity") yields three physicochemical parameters:
-
1.
the surface area of the compound at the air–water interface (A D ).
-
2.
the minimum concentration to induce surface activity (C 0)
-
3.
the critical micelle concentration (CMC) at which the surface pressure reaches a limiting value
From the obtained data, a π-log C plot and a π versus C plot are constructed and several parameters are determined, such as C 0, CMC, C sat, K aw and Г∞. The parameters derived from the π-log C plot are:
-
C 0 is arbitrarily defined as the compound concentration at which the surface pressure is 0.1 mN/m [nl−3].
-
CMC is determined as the concentration at which the surface pressure becomes independent of the compound concentration [nl−3].
-
K aw is obtained by fitting the π/C curve using the determined Г∞.
-
C sat is determined as the concentration higher than the CMC leading to further aggregation and finally precipitation and thus creating a collapse of surface pressure [nl−3].
-
Г∞ is obtained from the slope of the linear part of the Gibbs adsorption isotherm.
-
The cross-sectional area, A s, can indirectly be determined from Г∞. A s was evaluated from the slope of the π versus ln C curve.
The correlation of the three parameters, described above A D , C 0 and CMC), with the BBB permeability was investigated. C 0 appears to correlate with the hydrophobicity of the test compounds, but not exactly parallel with the chromatographically determined hydrophobicity. CMC alone was not useful to predict the BBB permeability, but in combination with C 0 in a so-called C 0–CMC plot. From this diagram, three regions are divided:
-
1.
CNS− compounds constituted by very hydrophobic compounds.
-
2.
Less hydrophobic compounds which penetrate easily into the CNS.
-
3.
More hydrophilic compounds which become CNS+ at high concentrations.
At last, the A D parameter demonstrates that hydrophobic CNS− compounds have distinctly larger areas than the CNS+ compounds. Thus, in conclusion, CNS− compounds are not surface active, very hydrophobic with low C 0 and CMC values and large cross-sectional areas at the air–water interface or relatively hydrophilic with high C 0 and CMC values, especially when applied at low concentrations, whereas CNS+ compounds possess intermediate C 0 and CMC values and have cross-sectional areas which are smaller than that of a lipid molecule (Fischer et al. 1998; Gerebtzoff and Seelig 2006; Petereit et al. 2010).
In vitro transport rate
Transport rates are determined from in vitro cell systems (see “The in vitro permeability”) by plotting the cumulative amount of transported compound as a function of time (Madgula et al. 2007; Yazdanian et al. 1998). The efflux rate was calculated as (Madgula et al. 2007):
where P app(B–A) and P app(A–B) [l3t−1] are the apparent permeability coefficients from basolateral to apical and from apical to basolateral side, respectively.
The permeability–surface area product
Another way to express the BBB permeability is the PS product (i.e. product of permeability coefficient and surface area), quantified as log PS. It is a direct quantitative measure of permeability independent upon plasma and tissue binding, which is the case for log BB (see "The unidirectional influx constant from plasma to brain"). Since brain uptake is also influenced by the capillary surface area, it is a more relevant parameter to quantify the rate of brain penetration behaviour of compound (Jeffrey and Summerfield 2010; Liu et al. 2004; Smith 2003). In contrast to the BB value (see “The unidirectional influx constant from plasma to brain”), which is a function of the total compound concentration, the PS product predicts the level of free compound fraction in the brain (Nicolazzo et al. 2006; Pardridge 2004).
In vivo methods
The parameters obtained with the in vivo methods, as described in “Responses: definitions and calculations”, could be converted into a BBB PS product using the Kety–Renkin–Crone equation (Renkin 1959), slightly adapted to the specific study. Generally this equation takes into account the cerebral blood flow and extraction of test compound:
The regional flow rate F is determined by using the PS value of diazepam, a highly permeable compound reported in the literature as a regional flow rate marker in brain perfusion studies (Liu et al. 2004).
-
1.
Intravenous injection
For both, single and multiple time point analysis (see “The half-life”), the single pass extraction is defined as: \( E = \frac{{K_{\text{in}} }}{F} \), and thus the PS product is expressed as (Cecchelli et al. 2007; Smith 2003):
where F is the cerebral flow rate and K in the unidirectional influx transfer constant.
Deguchi et al. (2004) described the relationship between the apparent brain-to-plasma concentration ratio and the PS product:
Analogously, Dash and Elmquist (2003) formulated the following equation:
where V D [Nm−1] is the brain distribution volume of test compound, V 0 [Nl−3] is the plasma distribution volume of the marker, C p [nl−3] is the terminal plasma concentration and AUC [tnl−3] is the area under the concentration curve.
-
2.
In situ brain perfusion
After calculating the initial uptake clearance (Clup), the PS product is calculated (Liu et al. 2004):
Again Dash and Elmquist (2003; Pardridge 1995) applied Eq. 56 also for perfusion.
-
3.
BUI method
The extraction values (see “The in vivo permeability”) are converted into BBB PS product with the traditionally known Renkin–Crone equation (Bickel 2005; Reichel and Begley 1998).
However, it has been suggested that the conversion into the PS product is not accurate due to the short capillary transit time, making it difficult to calculate PS (Hammarlund-Udenaes 2000; Nicolazzo et al. 2006).
-
4.
Microdialysis
The PS product could also be determined with the microdialysis method (see “Recovery”), but then the apparent brain volume of distribution needs to be measured separately out of the total tissue concentration (Bickel 2005).
-
5.
Indicator diffusion method
Analogous to the BUI method, the unidirectional extraction E is directly used to determine the PS product (Crone 1965; Dash and Elmquist 2003; Knudsen and Paulson 1999) as described in Eq. 55.
-
6.
Mouse brain uptake assay
From Eq. 17, PS product is easily converted to the product of P app and the surface area A (Garberg et al. 2005; Nicolazzo et al. 2006):
-
7.
External imaging detection methods
With these technique, the PS product is determined the same way as in the intravenous injection technique, but measuring brain activity on cryostat sections or real-time with the imaging methods. To calculate the PS product, regional blood flow should be measured with a separated experiment (Bickel 2005; Dash and Elmquist 2003; Taheri and Sood 2006).
In vitro BBB models
The endothelial permeability measured with an in vitro BBB model (see “The in vitro permeability”) can simply be correlated to the PS product when knowing the surface area:
where P e [lt−1m−1] is the measured permeability with the in vitro BBB model and S [l2] is the surface are of the endothelial exchange surface in vitro.
The permeability–surface area product can also be obtained from the slope of the clearance (Eq. 20) versus time curve (Carrara et al. 2007; Feng et al. 2008; Franke et al. 2000; Hansen et al. 2002). However, when using co-cultures, a correction for the filter insert containing the endothelial cell monolayer is required (Cecchelli et al. 1999; Culot et al. 2008; Nakagawa et al. 2009):
where PS e [l3t−1m−1] is the PS product for the endothelial cell monolayer, PS t [l3t−1m−1] is the PS product obtained from the slope by plotting the average clearance volume versus time and PS f [l3t−1m−1] is determined from the slope of the clearance curve with the control filter (Garberg et al. 2005). Similarly, two Michaelis–Menten constants were described as a function of the properties of two individual membranes. Under steady-state conditions, the determined Michaelis–Menten constants describe facilitated diffusion if the two membranes have similar transport properties (Gjedde and Christensen 1984).
The brain uptake rate
From the data obtained with the BUI method (see "The in vivo permeability"), another kinetic parameter, the brain uptake rate (J [nt−1m−1]), could be calculated as (Pardridge 1995; Suzuki et al. 2002):
where F is the cerebral blood flow [l3t−1m−1], C in [nl−3] is the concentration of the test compound in the carotid injection solution.
CSF and plasma elimination
Next to the determination of the CSF concentration obtained with the CSF sampling method (see “Brain-to-plasma partition coefficient”), the elimination half-life could be obtained from the elimination rate constant and the AUC could be calculated. From the plasma samples, the plasma concentration, AUC, the elimination half-life, the distribution volume and total clearance are determined (Okura et al. 2007).
Percentage injected dose
The brain delivery of the compound may be quantified by measuring the percentage of the injected dose (%ID [%]) delivered per gram brain following systemic administration. It is defined as the pharmacokinetic rule (Dash and Elmquist 2003; Pardridge 1995):
where PS [l3t−1m−1] is the BBB permeability–surface area product and AUC [tnl−3] is the plasma area under the concentration curve at a given time after injection.
Theoretical conversion of parameters into each other
Because clearance and permeability are closely related, they could be converted into each other as proposed by de Boer and Breimer (1996). In addition, the PS product is calculated from the slope of the clearance volume versus time by using both, in vitro and in vivo experiments. Moreover, not only the clearance or permeability coefficients are used to determine the PS product, extractions derived from the BUI and indicator diffusion experiments, the brain-to-plasma partition coefficient from microdialysis, and the unidirectional influx rate constant determined with intravenous injection and in situ brain perfusion are also converted into a PS product.
Thus, when comparing PS data, one should be aware of the experimental conditions, which vary between the different methods.
Discussion
It was demonstrated in numerous publications that peptides are able to cross the BBB by transmembrane diffusion as well as active transport mechanisms (Banks 2008; Banks and Kastin 1985, 1990, 1996; Banks et al. 1984, 1989, 2002; Bickel et al. 2001; Bonate 1995; Brasnjevic et al. 2009; Chen et al. 2002; Deguchi et al. 2003, 2004; Dogrukol-Ak et al. 2003; Egleton and Davis 1999; Egleton et al. 2001; Ganapathy and Miyauchi 2005; Gray et al. 1994; Gulya et al. 1988; Hawkins et al. 1989; Kastin and Akerstrom 2003; Kastin et al. 2002; Pan et al. 2006; Wilson et al. 1984). However, not all peptides show the same degree of penetration through the BBB as this is influenced by their structure, experimental setup and the method used to determine their BBB transport. The different BBB methods possess different limits of detection and thus the sensitivity to assign significant influx to the tested compound is different. Owing to these sensitivity differences, different BBB conclusions may be obtained for a peptide depending on the method used. However, at this moment, a full quantitative head-to-head comparison of the different BBB methods is lacking. Moreover, owing to the temporarily and spatially dynamic concept of the BBB, influx of a peptide is influenced by different factors and the BBB cannot be considered as a static system. In order to generalize the obtained data for easier comparison between methods and research groups, we propose to always include a negative as well as a positive control in the evaluation. The experimental value obtained for a peptide can in this way be more generally expressed, i.e. relatively towards the negative and positive control. Although it is widely accepted to include a negative control (e.g. radiolabelled BSA) in some studies, positive controls are lacking, nor any standardization. A possible positive control could be PACAP-27, showing literature K in values of above 2 μl/(g min). Both controls, which should be standardized in the methods, could also serve as system suitability tests, qualifying each time the experimental results.
In addition to differences between methods and experimental setups, up until now, some transport parameters are not clearly defined. For example, a certain parameter obtained by different methods has been given the same symbol, although the meaning is totally different (e.g. the permeability coefficient P). This means that the results are difficult to compare and evaluate. Ewing et al. (2003) defined k i as being the blood-to-tissue transfer constant, whereas Taheri et al. (Taheri and Sood 2007) determined the same parameter, but defining it as permeability coefficient. When looking to the dimension units, it is still confounding, while it has the same units as the influx constant. Moreover, the permeability could also be expressed as the PS product, which has the same units as the influx constant. Therefore, we introduced new symbols by simply adding the method’s name in subscript to the general parameter symbol used. A same parameter could be given another symbol, which would only be evident when looking in detail. This is the case for the blood–brain equilibrium distribution (BB) and the brain-to-plasma partition coefficient (K p). The only difference is that BB could also be calculated from the ratio between the AUC of brain and plasma, next to the ratio of the total concentrations in brain and plasma.
Table 2 gives an overview of the different methods, their mechanistic principles and the obtained responses used in BBB investigations as a summary of “Responses: definitions and calculations”. It should be noted that certain parameters describe facets of permeability as well as of distribution. For example, lipophilicity affects both brain permeability (by its lipid membrane permeability) as well as brain distribution (by its protein and nonspecific binding). Because some parameters could be defined with different methods, we clarified these parameters by adding subscripts such as an abbreviation of the method to the ‘original’ symbol (i.e. as mentioned in the publications) used. Quantitative autoradiography is not mentioned in this table, as it is only indirectly used, in combination with imaging techniques as MRI and (S)PET, to study the permeability.
Links between BBB techniques and responses
As can be seen from the two previous sections, one technique could be used to obtain different responses and on the other hand several techniques could be used to obtain one same response.
In Figure 3, relationships between the BBB methods, responses and equations, used to calculate the responses, are visualized.

Relationship between BBB techniques and responses
Abbreviations
- %ID:
-
Percentage injected dose
- γ :
-
Surface tension
- γ 0 :
-
Surface tension in pure buffer solution
- ε :
-
Dielectric constant
- λ :
-
Terminal rate constant
- π :
-
Surface pressure
- Г:
-
Surface excess concentration
- Г∞ :
-
Limited surface excess concentration
- A :
-
Area of the filter, membrane surface area, capillary surface area
- ABC transporter:
-
ATP-binding cassette transporter
- A D :
-
Surface area of the test compound at the air–water interface
- A m :
-
Amount of test compound in brain
- A s :
-
Cross-sectional area; surface area
- ATP:
-
Adenosine triphosphate
- AUC:
-
Area under the concentration curve
- AUCbrain :
-
Area under the concentration curve in brain
- AUCplasma :
-
Area under the concentration curve in plasma
- BB:
-
Brain-to-plasma concentration ratio at steady-state, blood–brain equilibrium distribution
- BBB:
-
Blood–brain barrier
- BBB-PI:
-
Blood–brain barrier permeability index
- BCRP:
-
Breast cancer resistance protein
- BCSFB:
-
Blood–cerebrospinal fluid barrier
- BEI:
-
Brain efflux index
- BMEC:
-
Brain microvessel endothelial cells
- B/P :
-
Brain-to-plasma concentration ratio at steady-state, blood–brain equilibrium distribution
- Brainpeps:
-
Blood–brain barrier peptide database
- BSA:
-
Bovine serum albumin
- BUI:
-
Brain uptake index
- BW:
-
Body weight
- C :
-
Concentration of test compound
- C 0 :
-
Initial concentration in donor chamber; minimum concentration to induce surface activity
- C a, C A, C acc :
-
Apical (luminal) side concentration, i.e. in the acceptor chamber
- Caco-2:
-
Human colon carcinoma cell line
- Calcein-AM:
-
Calcein acetoxymethyl ester
- CBF:
-
Cerebral blood flow
- C br, C brain :
-
Brain concentration
- C br,tot, C br (total):
-
Total brain concentration
- C buffer :
-
Concentration in buffer
- C CSF :
-
Concentration in the CSF
- CD:
-
Compartmental distribution
- C donor, C d, C D, C DO, C I :
-
Initial concentration of test compound in donor chamber
- C dial :
-
Concentration of the dialysate
- C ECF :
-
Concentration of test compound in brain ECF
- C in :
-
Concentration of the perfusate; concentration in the carotid injection solution
- C iv :
-
Concentration of intravascular marker
- Cl:
-
Clearance
- Cleff :
-
Efflux clearance
- ClH :
-
Hepatic clearance
- Clinf :
-
Uptake clearance
- Clp :
-
Systemic clearance
- ClR :
-
Renal clearance
- ClS :
-
Systemic clearance
- Cltest :
-
Clearance of the test experiment
- Clup :
-
Initial uptake clearance
- CMC:
-
Critical micelle concentration
- CNS:
-
Central nervous system
- CNS− :
-
Low brain penetrator
- CNS+ :
-
High brain penetrator
- Conc(test/ref)brain :
-
Concentration ratio of test and reference compound in brain
- Conc(test/ref)injectate :
-
Concentration ratio of test and reference compound in injection solution
- C org :
-
Concentration test compound in organic phase
- C out, C dial :
-
Concentration of the dialysate
- C p (t):
-
Plasma concentration at time t
- C pa :
-
Arterial plasma concentration
- C perfusion, C perf :
-
Concentration in the perfusion solution
- C pl :
-
Plasma concentration
- C pl,tot :
-
Total plasma concentration
- C ref :
-
Concentration of the reference compound
- C ref,brain :
-
Concentration of the reference compound in the brain
- C ref,inj :
-
Concentration of the reference compound in the injected solutions
- C s :
-
Concentration of the sample
- C sat :
-
Saturated concentration at which the surface pressure collapses
- CSF:
-
Cerebrospinal fluid
- C slice :
-
Free drug concentration in tissue slice
- C test :
-
Concentration of the test compound
- C test,brain :
-
Concentration of the test compound in the brain
- C test,inj :
-
Concentration of the test compound in the injected solutions
- C tissue :
-
Tissue concentration of the indicator
- C tot :
-
Total concentration
- C u :
-
Unbound concentration
- C u,brain :
-
Unbound concentration in brain
- C u,plasma :
-
Unbound concentration in plasma
- C wat :
-
Concentration test compound in aqueous phase
- C wat,ion :
-
Concentration of ionic species of test compound in aqueous phase
- C wat,neut :
-
Concentration of neutral species of test compound in aqueous phase
- D :
-
Distribution coefficient; dilution factor
- dQ/dt :
-
Transport rate of test compound
- E :
-
Extraction of the test compound
- ECF:
-
Extracellular fluid
- ED50 :
-
Dose producing 50% of the maximum response
- EDTA:
-
Ethylenediaminetetraacetic acid
- ER:
-
Efflux ratio; entity-relationship
- E ref :
-
Single pass extraction of the reference compound
- E refp :
-
Extraction of the permeable marker
- E refv :
-
Extraction of the vascular impermeable marker
- E test :
-
Single pass extraction of the test compound
- Exp.t :
-
Exposure time
- F :
-
Regional flow rate
- f :
-
Fraction of plasma exchangeable compound
- FK:
-
Foreign key
- f u :
-
Unbound fraction
- f u,brain :
-
Unbound fraction in brain
- f u,dil :
-
Diluted unbound fraction
- f u,meas :
-
Free fraction measured
- f u,plasma :
-
Unbound fraction in plasma
- GFR:
-
Glomerular filtration rate
- GLUT-1:
-
Glucose transporter 1
- hAAG:
-
Human α1 acid glycoprotein
- HPLC:
-
High-performance liquid chromatography
- IAM:
-
Immobilized artificial membrane
- IUPAC:
-
International Union of Pure and Applied Chemistry
- J :
-
Brain uptake rate
- J max :
-
Maximal flux
- K aw :
-
Air–water partition coefficient
- K d :
-
Dissociation constant
- k eff :
-
Apparent efflux rate constant
- k el :
-
Plasma elimination rate constant
- k el :
-
Efflux rate constant
- k i :
-
Unidirectional influx rate constant determined with imaging techniques
- k IAM :
-
IAM capacity factor
- K in :
-
Unidirectional influx rate constant
- K m :
-
Michaelis–Menten constant
- k out :
-
Brain elimination rate constant
- K p :
-
Brain-to-plasma partition coefficient
- K p,app :
-
Apparent brain-to-plasma partition coefficient
- K p,brain :
-
Brain-to-plasma concentration ratio
- K p,in :
-
Intrinsic plasma-to-brain partition coefficient
- K p,uu :
-
Brain-to-plasma partition coefficient of the free compound concentration
- LAT-1:
-
Large neutral amino acid transporter-1
- Log P cyclohexane/water :
-
Log P value of the cyclohexane/water system
- Log P oct/water :
-
Log P value of the 1-octanol/water system
- MBUA:
-
Mouse brain uptake assay
- MCT:
-
Monocarboxylic transporters
- MDCK:
-
Madin–Darby canine kidney cells
- MDR-1:
-
Multidrug resistance gene
- MID:
-
Method identification
- MRI:
-
Magnetic resonance imaging
- MRP-1:
-
Multidrug resistance associated protein-1
- N A :
-
Avogadro constant
- N serum :
-
Radioactivity measured per volume serum
- N tissue :
-
Radioactivity measured per gram tissue
- OID:
-
Origin identification
- P :
-
Partition coefficient; permeability coefficient
- PAMPA:
-
Parallel artificial membrane permeation assay
- P app :
-
Apparent permeability coefficient, determined with BMEC, co-culture, immortalized cell lines, Caco-2, MDCK and MBUA
- P app (A–B):
-
Apparent permeability coefficient from apical to basolateral side
- P app (B–A):
-
Apparent permeability coefficient from basolateral to apical side
- P app,pampa :
-
Apparent permeability coefficient, determined with PAMPA
- Par-2:
-
Brain parietal cortex area 2
- PBID:
-
Publication identification
- P c :
-
Intrinsic carrier-mediated permeability
- \( P_{\text{c}}^{\prime } \) :
-
Apparent net carrier-mediated permeability
- P e :
-
Endothelial permeability, effective PAMPA permeability
- P eff :
-
Effective PAMPA permeability
- P endoth :
-
Endothelial permeability coefficient, determined with BMEC culture
- PEPT1:
-
Peptide transporter 1
- PET:
-
Positron emission tomography
- P filter :
-
Permeability of the filter
- Pgp:
-
P-glycoprotein
- PI:
-
Permeability index
- PID:
-
Peptide IDentification
- P m :
-
Monolayer permeability determined with BMEC
- P p :
-
Passive diffusive permeability
- PR:
-
Peak response
- PS :
-
Permeability–surface area coefficient
- PS e :
-
PS product for the endothelial cell monolayer
- PS f :
-
PS product of the control filter
- PS t :
-
Total PS product
- P total :
-
Total permeability
- Q :
-
Cerebral blood flow
- \( \dot{Q} \) :
-
Rate of blood flow
- QAR:
-
Quantitative autoradiography
- R :
-
Gas constant; recovery
- Rateeff :
-
In vitro efflux rate
- RBE4:
-
Immortalized primary rat brain endothelial cells
- R dial :
-
Recovery by gain
- RID:
-
Response identification
- RL:
-
Recovery by loss, retrodialysis
- RP-HPLC:
-
Reversed-phase HPLC
- RR:
-
Relative recovery
- S :
-
Capillary surface area
- SMILES:
-
Simplified molecular input line entry specification
- SPECT:
-
Single photon emission tomography
- SV40:
-
Simian vacuolating virus 40
- T :
-
Temperature; perfusion time
- t :
-
Time
- t 0 :
-
Dead time
- t 1/2 :
-
(Terminal) half-life
- t 1/2eq :
-
Equilibration half-life
- t 1/2eq,in :
-
Intrinsic equilibration half-life
- T :
-
Perfusion time
- TEER:
-
Transendothelial electrical resistance
- TM-BBB:
-
Transfected mouse endothelial cells
- t r :
-
Retention time of test compound
- TR-BBB:
-
Transfected rat endothelial cells
- UML:
-
Unified modelling language
- V 0 :
-
Initial distribution volume; plasma distribution volume of marker
- V acc, V A :
-
Volume of the acceptor compartment
- V br, V b :
-
Distribution volume in brain
- V D :
-
Volume of donor compartment; distribution volume in brain
- V e :
-
Volume fraction of brain
- V i :
-
Initial distribution volume; Volume of buffer film remaining around sample slice
- V max :
-
Maximum uptake rate
- V p :
-
Plasma distribution volume
- V R :
-
Volume of acceptor compartment
- V u,br :
-
Unbound brain distribution volume
- V v :
-
Brain vascular volume
- X :
-
Amount of test compound in acceptor compartment
References
Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM (2005) Drug metabolism and pharmacokinetics, the blood–brain barrier, and central nervous system drug discovery. NeuroRx 2:554–571
Albensi BC, Knoblach SM, Chew BGM, O’Reilly MP, Faden AI, Pekar JJ (2000) Diffusion and high resolution MRI of traumatic brain injury in rats: time course and correlation with histology. Exp Neurol 162:61–72
Audus KL, Rose JM, Wang W, Borchardt RT (1999) Brain microvessel endothelial cell culture systems. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 86–93
Banks WA (2008) Delivery of peptides to the brain: emphasis on therapeutic development. Biopolymers 90:589–594
Banks WA, Kastin AJ (1985) Peptides and the blood–brain-barrier—lipophilicity as a predictor of permeability. Brain Res Bull 15:287–292
Banks WA, Kastin AJ (1990) Peptide-transport systems for opiates across the blood–brain-barrier. Am J Physiol 259:E1–E10
Banks WA, Kastin AJ (1996) Passage of peptides across the blood–brain barrier: pathophysiological perspectives. Life Sci 59:1923–1943
Banks WA, Kastin AJ, Coy DH (1984) Evidence that [125I]N-Tyr-delta sleep-inducing peptide crosses the blood–brain barrier by a non-competitive mechanism. Brain Res 301:201–207
Banks WA, Kastin AJ, Conn PM (1989) Quantifying carrier-mediated transport of peptides from the brain to the blood. Method Enzymol 168:652–660
Banks WA, Tschop M, Robinson SM, Heiman ML (2002) Extent and direction of ghrelin transport across the blood–brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther 302:822–827
Becker S, Liu XR (2006) Evaluation of the utility of brain slice methods to study brain penetration. Drug Metab Dispos 34:855–861
Beekman FJ, Vastenhouw B (2004) Design and simulation of a high-resolution stationary SPECT system for small animals. Phys Med Biol 49:4579–4592
Begley DJ (1999) In situ brain perfusion. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 32–40
Berezhkovskiy LM (2008) Some features of the kinetics and equilibrium of drug binding to plasma proteins. Expert Opin Drug Metab Toxicol 4:1479–1498
Bickel U (1999) Intravenous injection/pharmacokinetics. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 41–48
Bickel U (2005) How to measure drug transport across the blood–brain barrier. NeuroRx 2:15–26
Bickel U, Yoshikawa T, Pardridge WM (2001) Delivery of peptides and proteins through the blood–brain barrier. Adv Drug Deliv Rev 46:247–279
Bonate PL (1995) Animal-models for studying transport across the blood–brain-barrier. J Neurosci Methods 56:1–15
Booch G, Rumbaugh J, Jacobson I (1999) The unified modelling language user guide. Addison-Wesley, Reading
Brandsch M, Knutter I, Bosse-Doenecke E (2008) Pharmaceutical and pharmacological importance of peptide transporters. J Pharm Pharmacol 60:543–585
Brasnjevic I, Steinbusch HWM, Schmitz C, Martinez-Martinez P, European NanoBioPharmaceutics R (2009) Delivery of peptide and protein drugs over the blood-brain barrier. Prog Neurobiol 87:212–251
Caboche S, Pupin M, Leclere V, Fontaine A, Jacques P, Kucherov G (2008) NORINE: a database of nonribosomal peptides. Nucleic Acids Res 36:D326–D331
Campbell M, Humphries MM, Kiang AS, Nguyen ATH, Gobbo OL, Tam LCS et al (2011) Systemic low-molecular weight drug delivery to pre-selected neuronal regions. EMBO Mol Med 3:235–245
Carrara S, Reali V, Misiano P, Dondio G, Bigogno C (2007) Evaluation of in vitro brain penetration: optimized PAMPA and MDCKII-MDR1 assay comparison. Int J Pharm 345:125–133
Cecchelli R, Dehouck B, Descamps L, Fenart L, Buée-Scherrer V, Duhem C et al (1999) In vitro model for evaluating drug transport across the blood–brain barrier. Adv Drug Deliv Rev 36:165–178
Cecchelli R, Coisne C, Dehouck L, Miller F, Dehouck M, Buée-Scherrer V, Dehouck B (2006) Modeling the blood–brain barrier. In: Dermietzel R, Spray DC, Nedergaard M (eds) Blood–brain barriers. Wiley, Weinheim, pp 337–355
Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP et al (2007) Modelling of the blood–brain barrier in drug discovery and development. Nat Rev Drug Discov 6:650–661
Chaurasia CS, Muller M, Bashaw ED, Benfeldt E, Bolinder J, Bullock R et al (2007) Microdialysis principles, application, and regulatory perspectives report from the joint AAPS-FDA workshop. AAPS J 9:E48–E59
Chen P (1976) The entity-relationship model—toward a unified view of data. ACM T Database Syst 1:9–36
Chen WQ, Yang JZ, Andersen R, Nielsen LH, Borchardt RT (2002) Evaluation of the permeation characteristics of a model opioid peptide, H-Tyr-D-Ala-Gly-Phe-D-Leu-OH (DADLE), and its cyclic Prodrugs across the blood–brain barrier using an in situ perfused rat brain model. J Pharmacol Exp Ther 303:849–857
Chu TC, Twu KY, Ellington AD, Levy M (2006) Aptamer mediated siRNA delivery. Nucleic Acids Res 34:6
Codd E (1970) A relational model of data for large shared data banks. Commun ACM 13:377–378
Cornford EM (1985) The blood–brain-barrier, a dynamic regulatory interface. Mol Physiol 7:219–260
Cornford EM (1999) The carotid artery single injection technique. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 11–23
Crone C (1965) Facilitated transfer of glucose from blood into brain tissue. J Physiol (Lond) 181:103–113
Cucullot L, Oby E, Hallene K, Aumayr B, Rapp E, Janigro D (2006) Artificial blood–brain barriers. In: Dermietzel R, Spray DC, Nedergaard M (eds) Blood–brain barriers: from ontogeny to artificial interfaces. Wiley, Weinheim, pp 375–401
Culot M, Lundquist S, Vanuxeem D, Nion S, Landry C, Delplace Y et al (2008) An in vitro blood–brain barrier model for high throughput (HTS) toxicological screening. Toxicol In Vitro 22:799–811
Dagenais C, Rousselle C, Pollack GM, Scherrmann JM (2000) Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J Cerebr Blood Flow Metab 20:381–386
Dagenais C, Avdeef A, Tsinman O, Dudley A, Beliveau R (2009) P-glycoprotein deficient mouse in situ blood–brain barrier permeability and its prediction using an in combo PAMPA model. Eur J Pharm Sci 38:121–137
Dai H, Elmquist WF (2003) Drug transport studies using quantitative microdialysis. In: Nag S (ed) The blood–brain barrier: biology and research protocols. Humana, Totowa, pp 249–264
Dash AK, Elmquist WF (2003) Separation methods that are capable of revealing blood–brain barrier permeability. J Chromatogr B 797:241–254
Davies MI, Cooper JD, Desmond SS, Lunte CE, Lunte SM (2000) Analytical considerations for microdialysis sampling. Adv Drug Deliv Rev 45:169–188
de Boer AG, Breimer DD (1996) Reconstitution of the blood–brain barrier in cell culture for studies of drug transport and metabolism. Adv Drug Deliv Rev 22:251–264
de Lange ECM, de Boer AG, Breimer DD (1999) Intracerebral microdialysis. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 94–112
Deguchi Y, Miyakawa Y, Sakurada S, Naito Y, Morimoto K, Ohtsuki S et al (2003) Blood–brain barrier transport of a novel mu(1)-specific opioid peptide, H-Tyr-D-Arg-Phe-beta-Ala-OH (TAPA). J Neurochem 84:1154–1161
Deguchi Y, Naito Y, Ohtsuki S, Miyakawa Y, Morimoto K, Hosoya K et al (2004) Blood–brain barrier permeability of novel [D-Arg2]dermorphin (1-4) analogs: transport property is related to the slow onset of antinociceptive activity in the central nervous system. J Pharmacol Exp Ther 310:177–184
Deli MA, Abraham CS, Kataoka Y, Niwa M (2005) Permeability studies on in vitro blood–brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol 25:59–127
Di L, Kerns EH, Fan K, McConnell OJ, Carter GT (2003) High throughput artificial membrane permeability assay for blood–brain barrier. Eur J Med Chem 38:223–232
Di L, Umland JP, Chang G, Huang Y, Lin Z, Scott DOS, Troutman MD, Liston TE (2011) Species independence in brain tissue binding using brain homogenates. Drug Metab Dispos 39:1270–1277
Dienel GA (2006) Functional brain imaging. In: Dermietzel R, Spray DC, Nedergaard M (eds) Blood–brain barriers: from ontogeny to artificial interfaces. Wiley, Weinheim, pp 551–599
Dogrukol-Ak D, Banks WA, Tuncel N, Tuncel M (2003) Passage of vasoactive intestinal peptide across the blood–brain barrier. Peptides 24:437–444
Ducarme A, Neuwels M, Goldstein S, Massingham R (1998) IAM retention and blood brain barrier penetration. Eur J Med Chem 33:215–223
Ecker GF, Noe CR (2004) In silico prediction models for blood–brain barrier permeation. Curr Med Chem 11:1617–1628
Eddy EP, Maleef BE, Hart TK, Smith PL (1997) In vitro models to predict blood–brain barrier permeability. Adv Drug Deliv Rev 23:185–198
Egleton RD, Davis TP (1999) Transport of the delta-opioid receptor agonist [d-penicillamine(2,5)] enkephalin across the blood–brain barrier involves transcytosis. J Pharm Sci 88:392–397
Egleton RD, Mitchell SA, Huber JD, Palian MM, Polt R, Davis TP (2001) Improved blood–brain barrier penetration and enhanced analgesia of an opioid peptide by glycosylation. J Pharmacol Exp Ther 299:967–972
Ehrlich P (1885) Das sauerstufbudurfnis des organismus. Eine Farbenanalytische Studie. Verlag von August Hirschwald, Berlin
Engelhardt B (2006) Development of the blood–brain interface. In: Dermietzel R, Spray DC, Nedergaard M (eds) Blood–brain barrier: from ontogeny to artificial interfaces. Wiley VCH, Weinheim, pp 11–39
Ermisch A (1992) Peptide receptors of the blood–brain-barrier and substrate transport into the brain. In: Ermisch A, Landgraf R, Ruhle HJ (eds) Circumventricular organs and brain fluid environment: molecular and functional aspects. Elsevier, Amsterdam, pp 155–161
Ermisch A, Ruhle HJ, Landgraf R, Hess J (1985) Blood–brain barrier and peptides. J Cerebr Blood Flow Metab 5:350–357
Ermisch A, Ruhle HJ, Kretzschmar R, Baethmann A (1991) On the blood–brain-barrier to peptides—specific binding of atrial-natriuretic-peptide in vivo and in vitro. Brain Res 554:209–216
Ewing JR, Knight RA, Nagaraja TN, Yee JS, Nagesh V, Whitton PA et al (2003) Patlak plots of Gd-DTPA MRI data yield blood–brain transfer constants concordant with those of C-14-sucrose in areas of blood–brain opening. Magn Reson Med 50:283–292
Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD et al (2008) In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos 36:268–275
Fenstermacher JD, Wei L (1999) Measuring local cerebral capillary permeability-surface area products by quantitative autoradiography. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 122–132
Fiori A, Cardelli P, Negri L, Savi MR, Strom R, Erspamer V (1997) Deltorphin transport across the blood–brain barrier. Proc Natl Acad Sci USA 94:9469–9474
Fischer H, Gottschlich R, Seelig A (1998) Blood–brain barrier permeation: molecular parameters governing passive diffusion. J Membr Biol 165:201–211
Franke H, Galla HJ, Beuckmann CT (2000) Primary cultures of brain microvessel endothelial cells: a valid and flexible model to study drug transport through the blood–brain barrier in vitro. Brain Res Protoc 5:248–256
Friden M, Gupta A, Antonsson M, Bredberg U, Hammarlund-Udenaes M (2007) In vitro methods for estimating unbound drug concentrations in the brain interstitial and intracellular fluids. Drug Metab Dispos 35:1711–1719
Friden M, Ducrozet F, Middleton B, Antonsson M, Bredberg U, Hammarlund-Udenaes M (2009) Development of a high-throughput brain slice method for studying drug distribution in the central nervous system. Drug Metab Dispos 37:1226–1233
Fujikawa M, Ano R, Nakao K, Shimizu R, Akamatsu M (2005) Relationships between structure and high-throughput screening permeability of diverse drugs with artificial membranes: application to prediction of Caco-2 cell permeability. Bioorg Med Chem 13:4721–4732
Gaillard PJ, Voorwinden LH, Nielsen JL, Ivanov A, Atsumi R, Engman H et al (2001) Establishment and functional characterization of an in vitro model of the blood–brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur J Pharm Sci 12:215–222
Ganapathy V, Miyauchi S (2005) Transport systems for opioid peptides in mammalian tissues. AAPS J 7:E852–E856
Garberg P, Ball M, Borg N, Cecchelli R, Fenart L, Hurst RD et al (2005) In vitro models for the blood–brain barrier. Toxicol In Vitro 19:299–334
Gerebtzoff G, Seelig A (2006) In silico prediction of blood–brain barrier permeation using the calculated molecular cross-sectional area as main parameter. J Chem Inf Model 46:2638–2650
Giaginis C, Tsantili-Kakoulidou A (2008) Alternative measures of lipophilicity: from octanol–water partitioning to IAM retention. J Pharm Sci 97:2984–3004
Gjedde A (1981) High-affinity and low-affinity transport of d-glucose from blood to brain. J Neurochem 36:1463–1471
Gjedde A, Christensen O (1984) Estimates of Michaelis–Menten constants for the 2 membranes of the brain endothelium. J Cerebr Blood Flow Metab 4:241–249
Golden PL, Pollack GM (2003) Blood–brain barrier efflux transport. J Pharm Sci 92:1739–1753
Goldmann EE (1913) Vitalfärbung am Zentralnervensystem. Abhandl Konigl preuss Akad Wiss 1:1–60
Grant GA, Abbott NJ, Janigro D (1998) Understanding the physiology of the blood–brain barrier: in vitro models. News Physiol Sci 13:287–293
Gray RA, Vandervelde DG, Burke CJ, Manning MC, Middaugh CR, Borchardt RT (1994) Delta-sleep-inducing peptide—solution conformational studies of a membrane-permeable peptide. Biochemistry 33:1323–1331
Gulya K, Krivan M, Nyolczas N, Sarnyai Z, Kovacs GL (1988) Central effects of the potent and highly selective-mu-opioid antagonist d-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) in mice. Eur J Pharmacol 150:355–360
Gumbleton M, Audus KL (2001) Progress and limitations in the use of in vitro cell cultures to serve as a permeability screen for the blood–brain barrier. J Pharm Sci 90:1681–1698
Gupta A, Chatelain P, Massingham R, Jonsson EN, Hammarlund-Udenaes M (2006) Brain distribution of cetirizine enantiomers: comparison of three different tissue-to-plasma partition coefficients: K-p, K-p, K-u, and K-p, K-uu. Drug Metab Dispos 34:318–323
Hakkarainen JJ, Jalkanen AJ, Kaariainen TM, Keski-Rahkonen P, Venalainen T, Hokkanen J et al (2010) Comparison of in vitro cell models in predicting in vivo brain entry of drugs. Int J Pharm 402:27–36
Hammami R, Ben Hamida J, Vergoten G, Fliss I (2009) PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res 37:D963–D968
Hammarlund-Udenaes M (2000) The use of microdialysis in CNS drug delivery studies—pharmacokinetic perspectives and results with analgesics and antiepileptics. Adv Drug Deliv Rev 45:283–294
Hansen DK, Scott DO, Otis KW, Lunte SM (2002) Comparison of in vitro BBMEC permeability and in vivo CNS uptake by microdialysis sampling. J Pharm Biomed Anal 27:945–957
Hawkins KN, Knapp RJ, Lui GK, Gulya K, Kazmierski W, Wan YP et al (1989) [H-3]-[H-D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2] ([H-3] CTOP), a potent and highly selective peptide for mu-opioid receptors in rat-brain. J Pharmacol Exp Ther 248:73–80
Hoffmann A, Bredno J, Wendland MF, Derugin N, Hom J, Schuster T et al (2011) Validation of in vivo magnetic resonance imaging blood–brain barrier permeability measurements by comparison with gold standard histology. Stroke 42:2054–2060
Hosoya K-i, Ohtsuki S, Terasaki T (2002) Recent advances in the brain-to-blood efflux transport across the blood–brain barrier. Int J Pharm 248:15–29
Hsuchou H, Pan W, Kastin AJ (2007) The fasting polypeptide FGF21 can enter brain from blood. Peptides 28:2382–2386
Innis RB, Cunningham VJ, Delforge J, Fujita M, Giedde A, Gunn RN et al (2007) Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cerebr Blood Flow Metab 27:1533–1539
Isakovic AJ, Abbott NJ, Redzic ZB (2004) Brain to blood efflux transport of adenosine: blood–brain barrier studies in the rat. J Neurochem 90:272–286
Jeffrey P, Summerfield S (2010) Assessment of the blood–brain barrier in CNS drug discovery. Neurobiol Dis 37:33–37
Joo F, Karnushi I (1973) Procedure for isolation of capillaries from rat-brain. Cytobios 8:41–48
Kakee A, Tersaki T, Sugiyama Y (1996) Brain efflux index as a novel method of analyzing efflux transport at the blood–brain barrier. J Pharmacol Exp Ther 277:1550–1559
Kalvass JC, Maurer TS (2002) Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23:327–338
Kansy M, Senner F, Gubernator K (1998) Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J Med Chem 41:1007–1010
Kastin AJ, Akerstrom V (1999) Nonsaturable entry of neuropeptide Y into brain. Am J Physiol Endocrinol Metab 276:E479–E482
Kastin AJ, Akerstrom V (2003) Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes 27:313–318
Kastin AJ, Akerstrom V, Pan WH (2001) Validity of multiple-time regression analysis in measurement of tritiated and iodinated leptin crossing the blood–brain barrier: meaningful controls. Peptides 22:2127–2136
Kastin AJ, Akerstrom V, Pan WH (2002) Interactions of glucagon-like peptide-1 (GLP-1) with the blood–brain barrier. J Mol Neurosci 18:7–14
Kleczkowska P, Kosson P, Ballet S, Van den Eynde I, Tsuda Y, Tourwé D et al (2010) PK20, a new opioid–neurotensin hybrid peptide that exhibits central and peripheral antinociceptive effects. Mol Pain 6:86–92
Knudsen GM, Paulson OB (1999) Measurement of blood–brain barrier in humans using indicator diffusion. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 133–139
Kretzschmar R, Landgraf R, Gjedde A, Ermisch A (1986) Vasopressin binds to microvessels from rat hippocampus. Brain Res 380:325–330
Kumar G, Smith QR, Hokari M, Parepally J, Duncan MW (2007) Brain uptake, pharmacokinetics, and tissue distribution in the rat of neurotoxic N-butylbenzenesulfonamide. Toxicol Sci 97:253–264
Kusuhara H, Terasaki T, Sugiyama Y (2003) Brain efflux index method. In: Nag S (ed) The blood–brain barrier: biology and research protocols. Humana, Totowa, pp 219–231
Lanevskij K, Dapkunas J, Juska L, Japertas P, Didziapetris R (2010) QSAR Analysis of blood–brain distribution: the influence of plasma and brain tissue binding. J Pharm Sci 100:2147–2160
Lazaro E, Rafols CR, Roses M (2005) Characterization of immobilized artificial membrane (IAM) and XTerra columns by means of chromatographic models. J Chromatogr A 1081:163–173
Levin VA (1980) Relationship of octanol–water partition-coefficient and molecular-weight to rat-brain capillary-permeability. J Med Chem 23:682–684
Lin JH (1998) Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab Dispos 26:1202–1212
Liu XR, Tu MH, Kelly RS, Chen CP, Smith BJ (2004) Development of a computational approach to predict blood–brain barrier permeability. Drug Metab Dispos 32:132–139
Liu XR, Smith BJ, Chen CP, Callegari E, Becker SL, Chen X et al (2005) Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood–brain barrier permeability, plasma protein binding, and brain tissue binding. J Pharmacol Exp Ther 313:1254–1262
Liu HM, Liu XF, Yao JL, Wang CL, Yu Y, Wang R (2006a) Utilization of combined chemical modifications to enhance the blood–brain barrier permeability and pharmacological activity of endomorphin-1. J Pharmacol Exp Ther 319:308–316
Liu XR, Smith BJ, Chen C, Callegari E, Becker SL, Chen X et al (2006b) Evaluation of cerebrospinal fluid concentration and plasma free concentration as a surrogate measurement for brain free concentration. Drug Metab Dispos 34:1443–1447
Liu XR, Chen CP, Smith BJ (2008) Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther 325:349–356
Liu Y, Paliwal S, Bankiewicz KS, Bringas JR, Heart G, Mitragotri S et al (2010) Ultrasound-enhanced drug transport and distribution in the brain. AAPS PharmSciTech 11:1005–1017
Logan J (2000) Graphical analysis of PET data applied to reversible and irreversible tracers. Nucl Med Biol 27:661–670
Lundquist S, Renftel M (2002) The use of in vitro cell culture models for mechanistic studies and as permeability screens for the blood-brain barrier in the pharmaceutical industry—background and current status in the drug discovery process. Vascul Pharmacol 38:355–364
Lundquist S, Renftel M, Brillault J, Fenart L, Cecchelli R, Dehouck MP (2002) Prediction of drug transport through the blood–brain barrier in vivo: a comparison between two in vitro cell models. Pharm Res 19:976–981
Madgula VLM, Avula B, Reddy VLN, Khan KA, Khan SI (2007) Transport of decursin and decursinol angelate across Caco-2 and MDR-MDCK cell monolayers: in vitro models for intestinal and blood–brain barrier permeability. Planta Med 73:330–335
Madsen SJ, Hirschberg H (2010) Site-specific opening of the blood–brain barrier. J Biophotonics 3:356–367
Malina KCK, Cooper I, Teichberg VI (2009) Closing the gap between the in vivo and in vitro blood–brain barrier tightness. Brain Res 1284:12–21
Mano Y, Higuchi S, Kamimura H (2002) Investigation of the high partition of YM992, a novel antidepressant, in rat brain—in vitro and in vivo evidence for the high binding in brain and the high permeability at the BBB. Biopharm Drug Dispos 23:351–360
Martin I (2004) Prediction of blood–brain barrier penetration: are we missing the point? Drug Discov Today 9:161–162
Maupetit J, Derreumaux P, Tuffery P (2009) PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res 37:W498–W503
Maurer TS, DeBartolo DB, Tess DA, Scott DO (2005) Relationship between exposure and nonspecific binding of thirty-three central nervous system drugs in mice. Drug Metab Dispos 33:175–181
Mensch J, Oyarzabal J, Mackie C, Augustijns P (2009) In vivo, in vitro and in silico methods for small molecule transfer across the BBB. J Pharm Sci 98:4429–4468
Mensch J, Melis A, Mackie C, Verreck G, Brewster ME, Augustijns P (2010) Evaluation of various PAMPA models to identify the most discriminating method for the prediction of BBB permeability. Eur J Pharm Biopharm 74:495–502
Moriki Y, Suzuki T, Furuishi T, Fukami T, Tomono K, Watanabe J (2005) In vivo evidence for the efflux transport of pentazocine from the brain across the blood–brain barrier using the brain efflux index method. J Drug Target 13:53–59
Mueggler T, Baltes C, Rudin M (2009) Molecular neuroimaging in rodents: assessing receptor expression and function. Eur J Neurosci 30:1860–1869
Murakami H, Takanaga H, Matsuo H, Ohtani H, Sawada Y (2000) Comparison of blood–brain barrier permeability in mice and rats using in situ brain perfusion technique. Am J Physiol Heart Circ Physiol 279:H1022–H1028
Nag S (2003) The blood–brain barrier: biology and research protocols. Humana, Totowa
Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A et al (2009) A new blood–brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int 54:253–263
Nakai D, Kumamoto K, Sakikawa C, Kosaka T, Tokui T (2004) Evaluation of the protein binding ratio of drugs by a micro-scale ultracentrifugation method. J Pharm Sci 93:847–854
Navarro C, Gonzalez-Alvarez I, Gonzalez-Alvarez M, Manku M, Merino V, Casabo VG et al (2011) Influence of polyunsaturated fatty acids on cortisol transport through MDCK and MDCK-MDR1 cells as blood–brain barrier in vitro model. Eur J Pharm Sci 42:290–299
Neuhaus W, Stessl M, Strizsik E, Bennani-Baiti B, Wirth M, Toegel S et al (2010) Blood-brain barrier cell line PBMEC/C1–2 possesses functionally active P-glycoprotein. Neurosci Lett 469:224–228
Nicolazzo JA, Charman SA, Charman WN (2006) Methods to assess drug permeability across the blood–brain barrier. J Pharm Pharmacol 58:281–293
Ohno K, Pettigrew KD, Rapoport SI (1978) Lower limits of cerebrovascular permeability to non-electrolytes in conscious rat. Am J Physiol 235:H299–H307
Okura T, Komiyama N, Morita Y, Kimura M, Deguchi Y, Yamada S (2007) Comparative measurement of spinal CSF microdialysate concentrations and concomitant antinociception of morphine and morphine-6[beta]-glucuronide in rats. Life Sci 80:1319–1326
Oldendorf WH (1970) Measurement of brain uptake of radiolabeled substances using a tritiated water internal standard. Brain Res 24:372–376
Omidi Y, Campbell L, Barar J, Connell D, Akhtar S, Gumbleton M (2003) Evaluation of the immortalised mouse brain capillary endothelial cell line, b.End3, as an in vitro blood-brain barrier model for drug uptake and transport studies. Brain Res 990:95–112
Ottaviani G, Martel S, Escarala C, Nicolle E, Carrupt PA (2008) The PAMPA technique as a HTS tool for partition coefficients determination in different solvent/water systems. Eur J Pharm Sci 35:68–75
Pan WH, Banks WA, Fasold MB, Bluth J, Kastin AJ (1998a) Transport of brain-derived neurotrophic factor across the blood–brain barrier. Neuropharmacology 37:1553–1561
Pan WH, Banks WA, Kastin AJ (1998b) Permeability of the blood–brain barrier to neurotrophins. Brain Res 788:87–94
Pan WH, Yu YM, Cain CM, Nyberg F, Couraud PO, Kastin AJ (2005) Permeation of growth hormone across the blood–brain barrier. Endocrinology 146:4898–4904
Pan WH, Tu H, Kastin AJ (2006) Differential BBB interactions of three ingestive peptides: obestatin, ghrelin, and adiponectin. Peptides 27:911–916
Pardridge WM (1995) Transport of small molecules through the blood–brain-barrier—biology and methodology. Adv Drug Deliv Rev 15:5–36
Pardridge WM (1999a) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge
Pardridge WM (1999b) Isolated brain capillaries: an in vitro model of blood–brain barrier research. In: Pardridge WM (ed) Introduction to the blood–brain barrier. Cambridge University Press, Cambridge, pp 49–61
Pardridge WM (2004) Log(BB), PS products and in silico models of drug brain penetration. Drug Discov Today 9:392–393
Patlak CS, Blasberg RG, Fenstermacher JD (1983) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cerebr Blood Flow Metab 3:1–7
Peremans K, De Spiegeleer B, Buntinx E, Dobbeleir A, Vermeire S, Vandermeulen E et al (2008) Evaluation of serotonin-2A receptor occupancy with I-123–5-I-R91150 and single-photon emission tomography before and after low-dose pipamperone administration in the canine brain. Nucl Med Commun 29:724–729
Petereit AC, Swinney K, Mensch J, Mackie C, Stokbroekx S, Brewster M et al (2010) Prediction of blood–brain barrier penetration of poorly soluble drug candidates using surface activity profiling. Eur J Pharm Biopharm 75:405–410
Pidgeon C, Venkataram UV (1989) Immobilized artificial membrane chromatography—supports composed of membrane-lipids. Anal Biochem 176:36–47
Plattner VE, Germann B, Neuhaus W, Noe CR, Gabor F, Wirth M (2010) Characterization of two blood–brain barrier mimicking cell lines: distribution of lectin-binding sites and perspectives for drug delivery. Int J Pharm 387:34–41
Price TO, Samson WK, Niehoff ML, Banks WA (2007) Permeability of the blood–brain barrier to a novel satiety molecule nesfatin-1. Peptides 28:2372–2381
Prieto P, Blaauboer BJ, de Boer AG, Boveri M, Cecchelli R, Clemedson C et al (2004) Blood–brain barrier in vitro models and their application in toxicology—the report and recommendations of ECVAM workshop 49. Altern Lab Anim 32:37–50
Raub TJ, Lutzke BS, Andrus PK, Sawada GA, Staton BA (2006) Early preclinical evaluation of brain exposure in support of hit identification and lead optimization. In: Borchardt RT, Kerns EH, Hageman MJ, Thakker DR, Stevens JL (eds) Optimizing the “Drug-Like” properties of leads in drug discovery. Springer, New York, pp 355–410
Raybon JJ, Boje KMK (2001) A critical evaluation of the brain efflux index method as applied to the nitric oxide synthase inhibitor, aminoguanidine. Biopharm Drug Dispos 22:391–401
Read KD, Braggio S (2010) Assessing brain free fraction in early drug discovery. Expert Opin Drug Metab Toxicol 6:337–344
Reichel A (2009) Addressing central nervous system (CNS) penetration in drug discovery: basics and implications of the evolving new concept. Chem Biodivers 6:2030–2049
Reichel A, Begley DJ (1998) Potential of immobilized artificial membranes for predicting drug penetration across the blood–brain barrier. Pharm Res 15:1270–1274
Reichel A, Begley DJ, Abbott NJ (2003) An overview of in vitro techniques for blood–brain barrier studies. In: Nag S (ed) The blood–brain barrier: biology and research protocols. Humana, Totowa, pp 307–324
Renkin EM (1959) Transport of potassium-42 from blood to tissue in isolated mammalian skeletal muscles. Am J Physiol 197:1205–1210
Rossi JJ (2011) Inducible and reversible breaching of the blood brain barrier by RNAi. EMBO Mol Med 3:186–188
Roux F, Couraud PO (2005) Rat brain endothelial cell lines for the study of blood–brain barrier permeability and transport functions. Cell Mol Neurobiol 25:41–58
Saunders NR, Dziegielewska KM (1999) Transport in the developing brain. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 277–289
Scheller D, Kolb J (1991) The internal reference technique in microdialysis—a practical approach to monitoring dialysis efficiency and to calculating tissue concentration from dialysate samples. J Neurosci Methods 40:31–38
Seelig A, Gottschlich R, Devant RM (1994) A method to determine the ability of drugs to diffuse through the blood–brain-barrier. Proc Natl Acad Sci USA 91:68–72
Shayan G, Choi YS, Shusta EV, Shuler ML, Lee KH (2011) Murine in vitro model of the blood–brain barrier for evaluating drug transport. Eur J Pharm Sci 42:148–155
Shen DD, Artru AA, Adkison KK (2004) Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev 56:1825–1857
Siissalo S, Hannukainen J, Kolehmainen J, Hirvonen J, Kaukonen AM (2009) A Caco-2 cell based screening method for compounds interacting with MRP2 efflux protein. Eur J Pharm Biopharm 71:332–338
Smith QR (2003) A review of blood–brain barrier transport techniques. In: Nag S (ed) The blood–brain barrier: biology and research protocols. Humana, Totowa, pp 193–208
Smith M, Omidi Y, Gumbleton M (2007) Primary porcine brain microvascular endothelial cells: biochemical and functional characterisation as a model for drug transport and targeting. J Drug Target 15:253–268
Stenken JA (1999) Methods and issues in microdialysis calibration. Anal Chim Acta 379:337–358
Stewart BH, Chan OH (1998) Use of immobilized artificial membrane chromatography for drug transport applications. J Pharm Sci 87:1471–1478
Su Y, Sinko PJ (2006) Drug delivery across the blood–brain barrier: why is it difficult? How to measure and improve it? Expert Opin Drug Del 3:419–435
Subramanian G, Kitchen DB (2003) Computational models to predict blood–brain barrier permeation and CNS activity. J Comput Aided Mol Des 17:643–664
Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S et al (2007) Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J Pharmacol Exp Ther 322:205–213
Suzuki T, Moriki Y, Goto H, Tomono K, Hanano M, Watanabe J (2002) Investigation on the influx transport mechanism of pentazocine at the blood–brain barrier in rats using the carotid injection technique. Biol Pharm Bull 25:1351–1355
Taheri S, Sood R (2006) Kalman filtering for reliable estimation of BBB permeability. Magn Reson Imaging 24:1039–1049
Taheri S, Sood R (2007) Partial volume effect compensation for improved reliability of quantitative blood–brain barrier permeability. Magn Reson Imaging 25:613–625
Takasato Y, Rapoport SI, Smith QR (1984) An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol 247:H484–H493
Terasaki T (1999) Development of brain efflux index (BEI) method and its application to the blood–brain barrier efflux transport study. In: Pardridge WM (ed) Introduction to the blood–brain barrier: methodology, biology and pathology. Cambridge University Press, Cambridge, pp 24–31
Terasaki T, Ohtsuki S, Hori S, Takanaga H, Nakashima E, Hosoya K-i (2003) New approaches to in vitro models of blood–brain barrier drug transport. Drug Discov Today 8:944–954
Triguero D, Buciak J, Pardridge WM (1990) Capillary depletion method for quantification of blood–brain-barrier transport of circulating peptides and plasma-proteins. J Neurochem 54:1882–1888
Tsai TH (2003) Assaying protein unbound drugs using microdialysis techniques. J Chromatogr B Technol Biomed Life Sci 797:161–173
Tunblad K, Jonsson EN, Hammarlund-Udenaes M (2003) Morphine blood–brain barrier transport is influenced by probenecid co-administration. Pharm Res 20:618–623
Uchida M, Fukazawa T, Yamazaki Y, Hashimoto H, Miyamoto Y (2009) A modified fast (4 day) 96-well plate Caco-2 permeability assay. J Pharmacol Toxicol Methods 59:39–43
Urquhart BL, Kim RB (2009) Blood–brain barrier transporters and response to CNS-active drugs. Eur J Clin Pharmacol 65:1063–1070
Usansky HH, Sinko PJ (2003) Computation of log BB values for compounds transported through carrier-mediated mechanisms using in vitro permeability data from brain microvessel endothelial cell (BMEC) monolayers. Pharm Res 20:390–396
Van Belle K, Dzeka T, Sarre S, Ebinger G, Michotte Y (1993) In vitro and in vivo microdialysis calibration for the measurement of carbamazepine and its metabolites in rat brain tissue using the internal reference technique. J Neurosci Methods 49:167–173
Van Dorpe S, Adriaens A, Polis I, Peremans K, Van Bocxlaer J, De Spiegeleer B (2010) Analytical characterization and comparison of the blood–brain barrier permeability of eight opioid peptides. Peptides 31:1390–1399
Van Dorpe S, Adriaens A, Vermeire S, Polis I, Peremans K, De Spiegeleer B (2011) Desirability function combining metabolic stability and functionality of peptides. J Pept Sci 17:398–404
Vanhee P, Reumers J, Stricher F, Baeten L, Serrano L, Schymkowitz J et al (2010) PepX: a structural database of non-redundant protein-peptide complexes. Nucleic Acids Res 38:D545–D551
Verma RP, Hansch C, Selassie CD (2007) Comparative QSAR studies on PAMPA/modified PAMPA for high throughput profiling of drug absorption potential with respect to Caco-2 cells and human intestinal absorption. J Comput Aided Mol Des 21:3–22
Walker I, Coleman MD (1995) The blood–brain barrier: in vitro methods and toxicological applications. Toxicol In Vitro 9:191–204
Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li JB et al (2005) Evaluation of the MDR-MDCK cell line as a permeability screen for the blood–brain barrier. Int J Pharm 288:349–359
Wang GS, Li X, Wang Z (2009) APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res 37:D933–D937
Webb S, Ott RJ, Cherry SR (1989) Quantitation of blood–brain barrier permeability by positron emission tomography. Phys Med Biol 34:1767–1771
Wilson JF, Anderson S, Snook G, Llewellyn KD (1984) Quantification of the permeability of the blood–CSF barrier to [alpha]-MSH in the rat. Peptides 5:681–685
Yazdanian M, Glynn SL, Wright JL, Hawi A (1998) Correlating partitioning and Caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm Res 15:1490–1494
Yoon CH, Kim SJ, Soo BS, Lee SC, Yoo SD (2006) Rapid screening of blood–brain barrier penetration of drugs using the immobilized artificial membrane phosphatidylcholine column chromatography. J Biomol Screen 11:13–20
Zlokovic BV, Hyman S, McComb JG, Lipovac MN, Tang G, Davson H (1990) Kinetics of arginine-vasopressin uptake at the blood–brain-barrier. Biochim Biophys Acta 1025:191–198
Zlokovic BV, Mackic JB, McComb JG, Weiss MH, Kaplowitz N, Kannan R (1994) Evidence for transcapillary transport of reduced glutathione in vascular perfused guinea-pig brain. Biochem Biophys Res Commun 201:402–408
Acknowledgments
This work was supported by a PhD grant of ‘Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen)’ (73402 to S.V.D.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Van Dorpe, S., Bronselaer, A., Nielandt, J. et al. Brainpeps: the blood–brain barrier peptide database. Brain Struct Funct 217, 687–718 (2012). https://doi.org/10.1007/s00429-011-0375-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-011-0375-0