
Abstract
Giant cells tumors of bone (GCTB) are benign in nature but cause osteolytic destruction with a number of particular characteristics. These tumors can have uncertain biological behavior often contain a significant proportion of highly multinucleated cells, and may show aggressive behavior. We have studied differential gene expression in GCTB that may give a better understanding of their physiopathology, and might be helpful in prognosis and treatment. Rapid subtractive hybridization (RaSH) was used to identify and measure novel genes that appear to be differentially expressed, including KTN1, NEB, ROCK1, and ZAK using quantitative real-time polymerase chain reaction (qRT-PCR) and immunohistochemistry in the samples of GCTBs compared to normal bone tissue. Normal bone was used in the methodology RaSH for comparison with the GCTB in identification of differentially expressed genes. Functional annotation indicated that these genes are involved in cellular processes related to their tumor phenotype. The differential expression of KTN1, ROCK1, and ZAK was independently confirmed by qRT-PCR and immunohistochemistry. The expression of the KTN1 and ROCK1 genes were increased in samples by qRT-PCR and immunohistochemistry, and ZAK had reduced expression. Since ZAK have CpG islands in their promoter region and low expression in tumor tissue, their methylation pattern was analyzed by MSP-PCR. The genes identified KTN1, ROCK1, and ZAK may be responsible for loss of cellular homeostasis in GCTB since they are responsible for various functions related to tumorigenesis such as cell migration, cytoskeletal organization, apoptosis, and cell cycle control and thus may contribute at some stage in the process of formation and development of GCTB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Giant cell tumor of bone (GCTB) is a rare benign lesion that comprises ∼5% of primary bone tumors [1, 2]. They can be locally aggressive and destructive with rapid growth leading to thinning and rupture of the cortical bone with invasion of adjacent soft tissues, but without invading and ulcerating the skin and subcutaneous tissue [3, 4]. Generally, GCTBs most frequently occur in young adults between 20 and 40 years of age, with a slight predominance in females. They affect mainly the ends of long bones, and frequently occur in the distal end of the femur and the proximal end of the tibia [5–8].
GCTB is characterized by the presence of numerous osteoclast-like giant cells, and a mononuclear component that includes proliferating mononuclear stromal cells and infiltrating macrophages [9–11]. The mononuclear stromal cell represents the neoplastic component of the tumor, whereas monocytes represent a minor component of the mononuclear cell population [10, 12]. There is consensus among most authors regarding the true histogenesis of these lesions, with the evidence favoring a mesenchymal and mononuclear cells as stem cells of GCTB [13].
Despite its benign histological appearance, GCTB has an unpredictable clinical course [14]. The rate of local recurrence following surgical curettage is relatively high at 18–60%, and some occasionally undergo malignant transformation [15]. Metastasis occurs most commonly to the lung, variously reported in 1–9% of cases [16]. The outcome cannot be predicted on the basis of histological or radiographical criteria. In the absence of a clear histogenetic origin, GCTB is currently classified among lesions with an uncertain derivation and named after its peculiar morphologic appearance [14].
A better understanding of the mechanisms underlying the complex physiopathology of GCTBs, including a characterization of the gene expression profile and the functions of the genes involved in this phenomenon, and the molecular environment that might typify the tumor. We have investigated the molecular biology characteristics of GCTB to determine whether a more directed targeted clinical intervention can be adopted in the future.
The rapid subtraction hybridization approach (RaSH) [17] has been used to identify genes involved in tumorigenesis and the development of GCTBs. Subtraction hybridization methods are designed to identify the expression of complementary DNAs (cDNAs) in one of the two analyzed groups, thereby detecting differentially expressed messenger RNAs (mRNAs).
Tumorigenesis is a process of accumulation of genetic and epigenetic abnormalities that lead to cellular dysfunction and malignant transformation. Epigenetic mechanisms involving DNA methylation, histone modifications, and non-coding RNAs regulate and maintain gene expression states. Similar to genetic mutations, alterations in epigenetic regulation can lead to uncontrolled cell division, tumor initiation and growth, and invasiveness and metastasis [18].
The role of DNA methylation in cancer has received much attention; it is accepted that the methylation of the promoter region of many genes is associated with gene silencing [19]. The DNA methylation is the addition of a methyl group for the carbon atom 5′cytosine, present in the dinucleotide CpG, resulting in the formation of a 5-methylcytosine [20]. The methyl groups found mainly on the islands of CpGs promoter regions of genes can influence the changes between the molecule of DNA and proteins, reducing the binding affinity between the promoter regions and transcription factors through mechanisms involving changes in chromatin structure or levels of histone acetylation [21].
Thus, our aim has been to identify and characterize differentially expressed genes in GCTBs compared to normal bone tissue that may be involved in the molecular biology and development of the disease, generating insight for the new prognosis, and treatment and understanding of this tumor.
Materials and methods
Patients and tissues
GCTB tissue samples were obtained from the tumor bank at The Pio XII Foundation/IBILCE-UNESP, São Paulo, Brazil. The use of all patient-derived material was approved by institution’s research ethics board, The Pio XII Foundation—Cancer Hospital Barretos, and informed consent was obtained individually from the patients. The diagnosis of GCTB was established by biopsy prior to surgical excision by pathologists. Tissues were obtained at surgery from patients undergoing tumor resection. Microdissection was performed to collect samples.
Fifty-six samples were collected from primary GCTB, 23 fresh frozen samples of GCBT, 24 paraffin-embedded samples of GCTB, and 9 fresh histologically normal medullar bone tissue evaluated by a pathologist for confirmation of any morphological alteration. (Table 1 gives the clinicopathologic features of these tumors).
The study population included 43 patients. Twenty-one (48%) were male and 22 (52%) female, and the minimum and maximum ages were 13 and 74 years, respectively, mean 35.6 years ± 14.9. Main localizations of the GCTBs were femur (26%), tibia (21%), and radius (14%). Eight patients (17%) had recurrence between 11 and 42 months after surgery and six patients (12, 7%) had metastases between 1 and 65 months after surgery.
RNA extraction and RT-PCR
Total RNA was isolated from GCTB tissue and normal tissue using the TRIzol reagent (solution for extraction of RNA, Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. RNA integrity post-purification was checked using the Agilent 2100-Bioanalyser, giving a minimal RIN value of 5.5.
For quantitative real-time PCR (qRT-PCR), ∼5 μg of total RNA from each sample were used to synthesize cDNA with a high-capacity cDNA Archive Kit (Applied Biosystems), according to the manufacturer’s instructions. The quality of the cDNA was checked by PCR of the housekeeping gene, β-ACTIN. The primer sets were 5′ to 3′ GGCATCGTGATGGACTCCG and GCTGGAAGGTGGACAGCG. The PCR products were analyzed by electrophoresis on 1% agarose gel and stained with ethidium bromide.
Rapid subtractive hybridization
For this methodology, paired samples of the same patient (2F) were used, a sample of GCTB and a sample of histologically normal bone tissue. RaSH cDNA libraries were performed as described previously [17] with modifications. From the 25 μg total RNA pool, cDNAs were synthesized and digested with MboI (Invitrogen Life Technologies) at 37°C for 1 h and extracted with phenol–chloroform followed by ethanol precipitation. The digested cDNAs were mixed with 20 mmol/L of primers XDPN-14 (50CTGATCACTCGAGA30) and XDPN-12 (50GATCTCTCGAGT30) in 30 μL of 1X T4 DNA Ligase Buffer (Invitrogen Life Technologies), heated at 55°C for 1 min, and cooled to 14°C within 1 h. Ligation was carried out overnight at 14°C After adding 9 units of T4 DNA ligase to the mixtures individually.
The mixtures were diluted to 100 μL and >40 mL of the mixtures were used for PCR amplification with primer XDPN-18 (5′CTGATCACTCGAGAGATC′3). Portions (10 μg) of the tester PCR products (CGT or normal tissue) were digested with 20 units of XhoI and purified with phenol–chloroform extraction and ethanol precipitation. The fragments were inserted into XhoI-digested pZERO plasmid (1 μg/μl) at 16°C for 3 h. The constructs were introduced into the TOP10 competent cells. Two RaSH cDNA libraries were prepared, one using cDNA from the CGT as tester with the normal bone as driver, and the other using cDNA from the normal bone as tester with cDNA from the CGT as driver.
All bacterial colonies were analyzed by PCR with use of the M13 forward and M13 reverse primers to verify those with an insert. The sequences of these clones were determined with DNA sequencer (ABI PRISM 377, Applied Biosystems) and DYEnamic ET dye terminator sequencing kit (Amersham Biosciences). The sequences were analyzed using an annotation pipeline that had 4 steps: (1) quality checking, phred base-calling, cutoff 0.05; (2) vector trimming and removal of undesirable sequences, such as bacterial, mitochondrial, and rRNA sequences; (3) masking of repetitive elements and screening of low-complexity regions by Repeat Masker, using the default settings; and (4) annotation against existing databases, using BLASTN with default parameters. Significant hits were determined by using an E value threshold of 10–15 for searches against nucleotide sequence databases [22].
Validation by quantitative real time
The qRT-PCR was used to assess the expression of genes found by the RaSH method in all fresh samples of CGTB individually. For analysis, we used 23 fresh samples of CGTBs and a pool of total RNA from a subset of nine fresh tissue of normal bone, defined as normal reference. Gene-specific primers for qRT-PCR were designed for optimal hybridization kinetics, using the Primer 3.0 program (provided by the Whitehead/MIT Center for Genome Research, Cambridge, MA).
Quantitative real-time PCR was performed using an ABI prism 7300 sequencer detector system and Sybr Green PCR Core Reagent (Applied Biosystems), following the manufacturer’s protocol. In brief, the reaction mixture (20 μl total volume) contained 25 ng of cDNA, gene-specific forward and reverse primers for each gene, and 10 μL of 2× quantitative Sybr Green PCR Master Mix. Relative quantification is given by the CT values, determined for triplicate reactions for GCTB samples and reference sample from each gene, and for the endogenous control (glyceraldehyde 3-phosphate dehydrogenase; GAPDH). The primer sequences are given in Table 2.
Thus, the relative expression of each specific gene was calculated by using the formula: \( R = {\left( {E{\text{ target}}} \right)^{{\Delta {\text{Ct target }}\left( {{\text{control}} - {\text{sample}}} \right)}}}/{\left( {E{\text{ endogenous}}} \right)^{{\Delta {\text{Ct endogenous }}\left( {{\text{control}} - {\text{sample}}} \right)}}} \), as previously described [23].
DNA extraction
DNA samples of fresh tissue were isolated using TRIzol Reagent (Life Technologies). For the extraction of DNA from paraffin, samples were first deparaffinized with xylene, and the tissue samples digested in a buffer (100 mmol/L NaCl, 10 mmol/L Tris-HCl pH 8.0, 25 mmol/L ethylenediamine tetraacetic acid [EDTA], and 1% sodium dodecyl sulfate) containing 20 mg/mL proteinase K at 50°C for 3 days. Total DNA was isolated by phenol–chloroform extraction and ethanol precipitation. The DNA pellets were suspended with 20 mL TE buffer (10 mmol/L Tris-HCl, 1 mmol/L EDTA, pH 8.0) and stored at −20°C until PCR amplification.
Methylation-specific PCR
Genomic DNA from both tumoral and normal tissues was treated with sodium bisulfite to modify unmethylated cytosine to uracil [24]. After the DNA conversion, hypermethylation in CGTs was determined by the MSP method [25], to analyze the methylation pattern of the ZAK gene. The primers used for PCR reaction were specific for methylated and unmethylated DNA (Table 3). PCR was individually performed in 25-μL reaction volumes, containing 1 × Platinum Taq buffer, 1.5 mmol/L MgCl2, 0.2 mmol/L of each dNTP, 0.4 μmol/L of each primer set, 1 U of Platinum Taq DNA Polymerase (Invitrogen), and 1 μL of treated DNA. In vitro methylated DNA (IVD) was used as a positive control, and DNA from lymphocytes of healthy donors were used as negative controls. PCR products were separated on silver-staining 8% non-denaturing polyacrylamide gels. PCR amplification conditions are available on request.
Statistical analysis of quantitative real-time PCR data
Statistical analysis was performed using the Minitab Student 14 software, and the significance level was set at p ≤ 0.05. Relative expression levels detected by qRT-PCR for the four genes in CGTs samples were transformed into natural logarithms. The Wilcoxon Signed Ranks Test was applied to compare the gene expression levels in tumor tissue and normal bone tissue.
Immunohistochemical staining
Immunohistochemistry was used to assess the expression of proteins found by the RaSH method in paraffin-embedded samples of CGTB individually. For analysis, we used 22 paraffin-embedded samples of CGTBs.
Immunohistochemical staining was performed on 4-μm sections obtained from formalin-fixed, paraffin-embedded blocks. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 30 min. Antigen retrieval was carried out in citrate buffer (10 mM, pH 6) for 30 min at 95°C in a Pan Steam. Polyclonal antibodies used were rabbit anti-human KTN1, rabbit anti-human ROCK1, and polyclonal rabbit anti-human ZAK (Sigma, St. Louis, MO, USA) antibodies at 1:100 were applied incubated at 4°C overnight. Afterward, sections were incubated with a biotinylated secondary antibody and then exposed to a streptavidin complex (HRP ready-to-use, DakoCytomation, Carpinteria, CA). Positive reactions were visualized with 3,3′ diaminobenzidine tetrahydrochloride (DAB, Signet® Laboratories, Dedham, MA, USA), followed by counterstaining with hematoxylin.
Normal testicle was used as a positive control for KTN1 and normal liver were used as the positive control to ROCK1 and ZAK. Sections treated without primary antibodies were used as negative controls. Immunoreactivity was evaluated blindly by two observers, who independently assessed the immunostainings with a semi-quantitative grading system (− no staining; + weak staining in 10–30% of cells; ++ moderate staining in 31–65% of cells; +++ strong staining of more than 66% of cells) [15].
Results
The RaSH approach was adopted to identify differentially expressed genes in GCTBs compared with the normal bone tissue. A total of 619 cDNA clones were sequenced, 169 clones were obtained from the reverse library (downregulated genes) and 450 clones were obtained from the upregulated genes library. To analyze the sequences, phred and phrap software were used to select the best quality sequences according to the following pattern: phred cutoff of 0.09, minmatch 10, and minscore 20; sequences with >100 base pairs of quality were accepted. The best quality sequences were submitted to the BLAST alignment program [26]. After alignment with the RefSeq database, the sequences that presented >90% of the target sequence length at alignment were selected. These included KTN1, NEB, ROCK1, and ZAK.
We compared the relative expression levels of four genes by qRT-PCR, using triple determination and normalization based on the GAPDH level. In the validation of the target genes, GCTBs samples were used and normal bone tissue as reference (group control).
KTN1 and ROCK1 were significantly upregulated in 100% tumor tissues compared to normal bone tissue (p < 0.000). ZAK was significantly downregulated in 100% tumor tissues compared to normal bone tissue (p < 0.001). NEB gene was not differentially expressed in GCTBs compared with normal bone tissue (p = 0.964). Relative gene expression data are presented in Fig. 1.

Relative expression values of the selected genes for validation by qRT-PCR. Expression of gene ZAK was significantly lower in GCTB compared to normal reference (normal tissue bone). ROCK1 and KTN1 are overexpressed in GCTB. The samples used correspond to the samples 1F–23F Table 1
The methylation pattern of the promoter region of ZAK by MSP-PCR was investigated after gene expression analysis. Two pairs of primers for the gene ZAK were designed. ZAK hypermethylation was detected in 18.2% (4/22) (p = 0.82) of the samples with the P1 primers and none (p = 1.000) of the samples when the P2 primer was used (Fig. 2). The sets of primers for the gene ZAK did not indicate hypermethylation in the lymphocytes analyzed. The methylation pattern data are presented in Table 4.

Representative examples of MSP reaction for gene ZAK P1 and ZAK P2. Tumor (3F) of a GCTB patient. Lanes U and M correspond to unmethylated and methylated reactions, respectively. In each case, C− indicates DNA from lymphocyte, C+ indicates IVD, H2O indicates negative PCR control. On the left: molecular weight marker; and below: the size of methylated PCR product
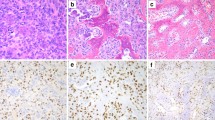
We used immunohistochemistry to evaluate protein expression of genes found by RaSH in different cell types due to the heterogeneity of histological CGTBs. Immunohistochemical staining of GCTB samples confirmed the presence of KTN1 and ROCK1 in the GCTB microenvironment. KTN1 immunostaining revealed cellular strong staining in 90% (20/22) of GCTB tissue samples, localized mainly in the multinucleated giant cells and stromal cells and occasionally monocytic cell (Fig. 3a). ROCK1 staining was strongly positive in 59% (13/22) of the samples in the cytoplasm of multinucleated giant cells, stromal cells, and monocytic cells (Fig. 3b). In contrast, the expression of ZAK was weakly localized mainly in 82% (18/22) of the samples in the cytoplasm of the multinucleated giant cells and stromal cells, and occasionally in some of the monocytic cells (Fig. 3b). In normal bone tissue, there was no immunohistochemical staining in different cell types for the three genes analyzed.

Immunolocalization of KTN1, ROCK1, and ZAK in human specimens of primary GCTB samples. a KTN1; b ROCK1; c ZAK; d negative control (original magnification 400×). These are representative figures from 22 GCTB patient tissue samples. The staining patterns for KTN1, ROCK1, and ZAK were generally consistent among all the patient samples. The presence of multinucleated giant cells is marked with stars, mesenchymal stromal cells with caret, and monocytes cell with arrow. Positive stainings of the corresponding antigens show brownish color in various degrees, in contrast to the bluish color in negative stainings
The results showed that genes ROCK1 and KTN1 were upregulated in GCTB, confirmed by qRT-PCR and immunohistochemistry, ZAK was significantly lower, by qRT-PCR and immunohistochemistry, in the GCTB samples, and that the NEB gene was not differentially expressed by qRT-PCR.
Discussion
GCTB does not fit well into a strictly defined category. Therefore, it has been approached in the context of its morphological definition of reactive rounded mononuclear cells, reactive osteoclastic giant cells, and neoplastic mononuclear spindle-shaped cells. It is clinically defined as a benign bone tumor, but it has a tendency to recur and can behave aggressively in a local manner, occasionally metastasizing to the lungs. Although both morphologic and radiologic examination can lead to the correct diagnosis in most cases, diagnosis of GCTB can often be difficult, with no clear diagnostic marker currently being available.
RaSH in allowing us to analyze genes that are expressed differentially in two samples, including genes that have not previously been characterized and rare transcripts, led to the identification of specific genes and signaling pathways involved in the regulation of the disease process. This method has simpler hybridization and subtraction steps than other subtractive hybridization methods.
Three potential biomarkers of giant cell tumor of bone were identified, validated by qRT-PCR and immunohistochemistry, and proved to have distinct differences in expression between tumor and non-neoplastic samples. The markers were genes KTN1, ROCK1, and ZAK, the first two being significantly upregulated and the last downregulated. The expression of genes ROCK1 and KTN1 were significantly high in GCTBs than in normal bone tissue and ZAK was significantly lower in tumor tissues than in the non-tumor adjacent tissue.
ROCK1 located in 18q11.1, encodes a protein associated with Rho kinase and belongs to a family of serine/threonine kinases activated via interaction with Rho GTPases. ROCK is involved in a wide range of fundamental cellular functions such as regulation the reorganization of the actin cytoskeleton, formation of the focal adhesion, smooth muscle contraction, cell migration, gene expression, and apoptosis [27, 28]. Rho-associated serine-threonine protein kinase, ROCK, one of the best characterized downstream effectors of Rho, is activated when it selectively binds to the active GTP-bound form of Rho. Activated ROCK interacts with the actin cytoskeleton to promote stress fiber formation and assembly of focal contacts [29]. There is considerable evidence to suggest that Rho–ROCK signalling is deregulated in cancer, and thereby contributes to invasive and metastatic behavior [30].
Upregulation of Rho–ROCK signaling in tumors is well-known to be linked to increased invasion and metastatic potential, which may occur as a result of increased RhoA, RhoC, or ROCK1 expression [30]. Cell migration is pivotal in metastasis, and rearrangements in the actin cytoskeleton are certainly involved. Overexpression of Rho has been linked to progression of human cancers, and therefore the Rho/ROCK pathway is a major player in cancer progression through its regulation of actin cytoskeleton reorganization; indeed, a specific ROCK inhibitor can suppress the tumor growth and metastasis [29], and therefore could be a prime molecular target for the prevention of cancer invasion and metastasis [30].
Rho kinase (ROCK) through its regulating of cytoskeletal events increases with the degree of malignancy, and is known to contribute to the invasion of hepatocellular carcinoma [31], bladder cancer [29], melanoma [30], and to be upregulated in anaplastic astrocytomas and glioblastomas [32]. Since ROCK1 was upregulated in multinucleated giant cells, stromal cells, and monocytic cells of GCTBs, we believe that overexpression in these tumors may contribute to their occasional invasion, metastasis, and increased malignancy.
KTN1 gene is located in the region 14q22.1 and encodes the full kinectin, membrane receptor for kinesin that is found in the endoplasmic reticulum and is responsible for the transport of vesicles along microtubules. It is expressed in the brain, liver, ovarian, and hematopoietic cells [33, 34]. The kinectin has an important role in vesicle transport as a receptor for kinesin and dynein, thereby aiding the movement of motor proteins within the cell. However, the exact mechanism by which cells control the transport of vesicles remains unclear [35]. According to Santama et al. [36], kinectin was demonstrated in vivo by overexpression and RNA interference assay, and is selectively involved in the transport of specific types of organelles.
Besides interacting with the kinesin, the kinectin also interacts with other proteins, such as Rho protein, which forms the complex Rho-kinectin responsible for transport of microtubules between the actin filaments and the elongation factor of translation-1δ [37, 38]. The latter plays an important role in regulating the synthesis of proteins that participate in the elongation phase of mRNA translation.
The exact function of KTN1 and its role in tumor progression remain unclear. There are no reports in the literature about the relationship of KTN1 gene in tumors, but this gene was highly expressed in all GCTBs.
ZAK gene is located in the region 2q24.2 and belongs to the family of mixed lineage kinase, which comprises a group of serine/threonine kinases that works with MAP3K [39, 40]. It is expressed in most tissues and is regulated by environmental stresses [39]. The expression of ZAK arrests cells in G2, due to the effect that its protein somehow decreases the expression of cyclin E [41].
In addition to this important function for example, overexpression can induce apoptosis in hepatocyte cell line [39. The gene NF-κB belongs to a gene family of transcription factors regulating the transcription of several genes related to immune response, cell proliferation, apoptosis, and the progression and maintenance of the status factor [42, 43]. NF-κB super can regulate the expression of proteins that interfere with the receptor via apoptosis, and induce the expression of apoptosis inhibitors and some family members of anti-apoptotic Bcl-2 [44]. Another way of induction of apoptosis by ZAK gene is via the JNK/SAPK, which belongs to the MAPK family and is activated by many types of cellular stresses or extracellular signals. The activation of this pathway is associated with regulation of cell proliferation, survival, apoptosis, and immune response by regulating the gene expression of cytokines. Furthermore, this protein is associated with the regulation of mRNA stabilization, cell migration, and integrity of the cytoskeleton [44, 45].
The low expression of the ZAK gene can therefore lead to poorer inhibition of cell proliferation by cycle arrest in tumor cells and more sustained growth of the cells of CGTBs.
Regarding the methylation pattern of gene ZAK presenting island CpGs in their promoter region, we found that this gene was downregulated in GCTBs, although no methylation occurred in the promoter region. Other mechanisms of gene silencing might be eliminating their activities, such as microRNAs and RNA interference.
In conclusion, differentially expressed genes in the CGTBs include those whose deregulation is potentially associated with disease progression. Methylation of the promoter region is not responsible for the downregulation of gene ZAK. These findings will contribute to the understanding of the molecular basis of GCTB, thus helping to improve diagnosis, treatment, and patient outcome. Further studies are required to evaluate whether the identified genes are specifically altered in GCTB or might also be deregulated in other tumors.
References
Mendenhall WM, Zlotecki RA, Scarborough MT, Gibbs CP, Mendenhall NP (2006) Giant cell tumor of bone. Am J Clin Oncol 29(1):96–99. doi:10.1097/01.coc.0000195089.11620.b7
Luther N, Bilsky MH, Härtl R (2008) Giant cell tumor of the spine. Neurosurg Clin N Am 19(1):49–55. doi:10.1016/j.nec.2007.09.009
De Camargo O, Croci AT, De Oliveira CRGC AM, Caiero MT, Giannotti CM (2001) Giant cells tumor—diagnosis and treatment history evolution in Instituto de Ortopedia eTraumatologia from FMUSP. Acta Ortop Bras 9(4):46–52. doi:10.1590/S1413-78522001000400006
Lee CH, Espinosa I, Jensen KC, Subramanian S, Zhu SX, Varma S, Montgomery KD, Nielsen TO, van de Rijn M, West RB (2008) Gene expression profiling identifies p63 as a diagnostic marker for giant cell tumor of the bone. Mod Pathol 21(5):531–539. doi:10.1038/modpathol.3801023
Khalil ESA, Younis A, AziZ SA, Shahawy ME (2004) Surgical management for giant cell tumor of bones. J Egypt Natl Canc Inst 16(3):145–152. doi:10.1038/modpathol.3801023
Turcotte RE (2006) Giant cell tumor of bone. Orthop Clin North Am 35(1):35–51. doi:10.1016/j.ocl.2005.08.005
Haque AU, Moatasim A (2008) Giant cell tumor of bone: a neoplasm or a reactive condition? Int J Clin Exp Pathol 1:489–501
Bassiony AA, Abdelrahman M, Abdelhady A, Assal MK (2009) Resection arthrodesis for the management of aggressive giant cell tumor of the distal femur. Indian J Orthop 43(1):67–71. doi:10.4103/0019-5413.44432
Wülling M, Engels C, Jesse N, Werner M, Delling G, Kaiser E (2001) The nature of giant cell tumor of bone. J Cancer Res Clin Oncol 127(8):467–474. doi:10.1007/s004320100234
Morgan T, Atkins GJ, Trivett MK, Johnson SA, Kansara M, Schlicht SL, Slavin JL, Simmons P, Dickinson I, Powell G, Choong PF, Holloway AJ, Thomas DM (2005) Molecular profiling of giant cell tumor of bone and the osteoclastic localization of ligand for receptor activator of nuclear factor kappaB. Am J Pathol 167(1):117–128
Alberghini M, Kliskey K, Krenacs T, Picci P, Kindblom L, Forsyth R, Athanasou NA (2010) Morphological and immunophenotypic features of primary and metastatic giant cell tumour of bone. Virchows Arch 456(1):97–103. doi:10.1007/s00428-009-0863-2
Goldring SR, Roelke MS, Petrison KK, Bhan AK (1987) Human giant cell tumors of bone identification and characterization of cell types. J Clin Invest 79(2):483–491. doi:10.1172/JCI112838
Salerno M, Avnet S, Alberghini M, Giunti A, Baldini N (2008) Histogenetic characterization of giant cell tumor of bone. Clin Orthop Relat Res 466(9):2081–2091. doi:10.1007/s11999-008-0327-z
Mak IW, Cowan RW, Popovic S, Colterjohn N, Singh G, Ghert M (2009) Upregulation of MMP-13 via Runx2 in the stromal cell of giant cell tumor of bone. Bone 45(2):377–386. doi:10.1016/j.bone.2009.04.253
Viswanathan S, Jambhekar NA (2010) Metastatic giant cell tumor of bone: are there associated factors and best treatment modalities? Clin Orthop Relat Res 468(3):827–833. doi:10.1007/s11999-009-0966-8
Jiang H, Kang DC, Alexandre D, Fisher PB (2000) RaSH, a rapid subtraction hybridization approach for identifying and cloning differentially expressed genes. Proc Natl Acad Sci USA 97(23):12684–12689
Fouse SD, Costello JF (2009) Epigenetics of neurological cancers. Future Oncol 5(10):1615–1629. doi:10.2217/fon.09.132
Fourkala EO, Hauser-Kronberger C, Apostolidou S, Burnell M, Jones A, Grall J, Reitsamer R, Fiegl H, Jacobs I, Menon U, Widschwendter M (2009) DNA methylation of polycomb group target genes in cores taken from breast cancer centre and periphery. Breast Cancer Res Treat 120(2):345–355. doi:10.1007/s10549-009-0384-3
Van der Auwera I, Elst HJ, Van Laere SJ, Maes H, Huget P, van Dam P, Van Marck EA, Vermeulen PB, Dirix LY (2009) The presence of circulating total DNA and methylated genes is associated with circulating tumour cells in blood from breast cancer patients. Br J Cancer 100(8):1277–1286. doi:10.1038/sj.bjc.6605013
Lopez-Serra L, Esteller M (2008) Proteins that bind methylated DNA and human cancer: reading the wrong words. Br J Cancer 98(12):1881–1885. doi:10.1038/sj.bjc.6604374
Calmon MF, Rodrigues RV, Kaneto CM, Moura RP, Silva SD, Mota LD, Pinheiro DG, Torres C, de Carvalho AF, Cury PM, Nunes FD, Nishimoto IN, Soares FA, da Silva AM, Kowalski LP, Brentani H, Zanelli CF, Silva WA Jr, Rahal P, Tajara EH, Carraro DM, Camargo AA, Valentini SR (2009) Epigenetic silencing of CRABP2 and MX1 in head and neck tumors. Neoplasia 11(12):1329–1339. doi:10.1593/neo.91110
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):2002–2007
Calmon MF, Colombo J, Carvalho F, Souza FP, Filho JF, Fukuyama EE, Camargo AA, Caballero OL, Tajara EH, Cordeiro JA, Rahal P (2007) Methylation profile of genes CDKN2A (p14 and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer Genet Cytogenet 173(1):31–37. doi:10.1016/j.cancergencyto.2006.09.008
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93(18):9821–9826
Meola J, Rosa E, Silva JC, Dentillo DB, da Silva WA, Jr Veiga-Castelli LC, de Souza Bernardes LA, Ferriani RA, Paro de Paz CC, Giuliatti S, Martelli L (2009) Differentially expressed genes in eutopic and ectopic endometrium of women with endometriosis. Fertil Steril 93(6):1750–1773. doi:10.1016/j.fertnstert.2008.12.058
Shi J, Wei L (2007) Rho kinase in the regulation of cell death and survival. Arch Immunol Ther Exp (Warsz) 55(2):61–75. doi:10.1007/s00005-007-0009-7
Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, Hiroi Y, Shimizu K, Luscinskas FW, Sun J, Liao JK (2008) ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest 118(5):1632–1644. doi:10.1172/JCI29226
Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, Oshima H (2003) Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res 9(7):2632–2641
Pinner S, Sahai E (2008) PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol 10(2):127–137. doi:10.1038/ncb1675
Man K, Ng KT, Xu A, Cheng Q, Lo CM, Xiao JW, Sun BS, Lim ZX, Cheung JS, Wu EX, Sun CK, Poon RT, Fan ST (2010) Suppression of liver tumor growth and metastasis by adiponectin in nude mice through inhibition of tumor angiogenesis and downregulation of Rho kinase/IFN-inducible protein 10/matrix metalloproteinase 9 signaling. Clin Cancer Res 16(3):967–977. doi:10.1158/1078-0432
Oellers P, Schröer U, Senner V, Paulus W, Thanos S (2009) ROCKs are expressed in brain tumors and are required for glioma-cell migration on myelinated axons. Glia 57(5):499–509. doi:10.1002/glia.20777
Hirano N, Butler MO, Guinan EC, Nadler LM, Kojima S (2005) Presence of anti-kinectin and anti-PMS1 antibodies in Japanese aplastic anaemia patients. Br J Haematol 128(2):221–223. doi:10.1111/j.1365-2141.2004.05317.x
Bai JZ, Mon Y, Krissansen GW (2006) Kinectin participates in microtubule-dependent hormone secretion in pancreatic islet beta-cells. Cell Biol Int 30(11):885–894
Tran H, Pankov R, Tran SD, Hampton B, Burgess WH, Yamada KM (2002) Integrin clustering induces kinectin accumulation. J Cell Sci 115:2031–2040
Santama N, Connie PNE, Lee-Lee O, Hanry Y (2004) Distribution and functions of kinectin isoforms. J Cell Sci 117(19):4537–4549
Hotta K, Tanaka K, Mino A, Kohno H, Takai Y (1996) Interaction of the Rho family small G proteins with kinectin, an anchoring protein of kinesin motor. Biochem Biophys Res Commun 225(1):69–74. doi:10.1006/bbrc.1996.1132
Ong LL, Lin PC, Zhang X, Chia SM, Yu H (2006) Kinectin-dependent assembly of translation elongation factor-1 complex on endoplasmic reticulum regulates protein synthesis. J Biol Chem 281(44):33621–33634. doi:10.1074/jbc.M607555200
Liu TC, Huang CJ, Chu YC, Wei CC, Chou CC, Chou MY, Chou CK, Yang JJ (2000) Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-alpha motif. Biochem Biophys Res Commun 274(3):811–816. doi:10.1006/bbrc.2000.3236
Huang CY, Kuo WW, Chueh PJ, Tseng CT, Chou MY, Yang JJ (2004) Transforming growth factor-beta induces the expression of ANF and hypertrophic growth in cultured cardiomyoblast cells through ZAK. Biochem Biophys Res Commun 324(1):424–431. doi:10.1016/j.bbrc.2004.09.067
Yang JJ (2003) A novel zinc finger protein, ZZaPK, interacts with ZAK and stimulates the ZAK-expressing cells re-entering the cell cycle. Biochem Biophys Res Commun 301(1):71–77. doi:10.1016/S0006-291X(02)02980-7
Tergaonkar V (2006) NFKB pathway: a good signaling paradigm and therapeutic target. Int J Biochem Cell Biol 38:1647–1653. doi:10.1016/j.biocel.2006.03.023
Dolcet X, Llobet D, Pallares J, Matias-Guiu X (2005) NF-kB in development and progression of human cancer. Virchows Arch 446(5):475–482. doi:10.1007/s00428-005-1264-9
Wang TH, Wang HS, Soong YK (2000) Regulation and functions of c-Jun N-terminal kinase/stress-activated protein kinase. Chang Gung Med J 23(2):57–72
Nishina H, Nakagawa K, Azuma N, Katada T (2003) Activation mechanism and physiological roles of stress-activated protein kinase/c-Jun NH2-terminal kinase in mammalian cells. J Biol Regul Homeost Agents 17(4):295–302
Acknowledgments
The authors thank the State of São Paulo Research (FAPESP), The National Council for Scientific and Technological Development (CNPq), and Coordination for the Improvement of Higher Education Personnel (CAPES) for their financial support.
We are also grateful to Sandra Morini MD, Valter Penna MD, and André Stellet MD for their collaboration in this work.
Conflict of interest
The authors declare that they have no conflict of interest with the organization that sponsored the research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Babeto, E., Conceição, A.L.G., Valsechi, M.C. et al. Differentially expressed genes in giant cell tumor of bone. Virchows Arch 458, 467–476 (2011). https://doi.org/10.1007/s00428-011-1047-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-011-1047-4