
Abstract
Mature T-cell and natural killer (NK)-cell neoplasms account for a small proportion of non-Hodgkin lymphomas (NHL). Despite the fact that they are relatively uncommon, T-cell lymphoproliferations often are submitted to the clinical molecular laboratory for testing. The diagnostic need for molecular testing is because T cells do not have a definitive immunophenotypic marker of clonality, like kappa and lambda antigen receptor immunophenotyping in B cells, so molecular methods can be used. A variety of molecular assays are available to evaluate T-cell receptor (TR) gene rearrangements, structural and numeric chromosomal abnormalities, and viral sequences associated with mature T- and NK-cell neoplasms. However, many of these molecular tests, especially PCR-based TR gene rearrangement testing, have important caveats that must be considered when interpreting the assays. Therefore, molecular results must be considered in the context of available clinical information, histology, immunophenotype, and other laboratory data for proper interpretation and clinical use.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Molecular Basis of Disease
T-cell and natural killer (NK)-cell neoplasms are relatively rare entities, collectively accounting for approximately 12 % of non-Hodgkin lymphomas (NHL) [1]. Subtypes of mature T- and NK-cell neoplasms are defined according to the World Health Organization (WHO) classification system [2], listed in Table 43.1. Despite the fact that they are relatively uncommon, most T-cell lymphoproliferations are assessed in the clinical molecular laboratory. The diagnostic need for molecular testing is because T cells do not have a definitive immunophenotypic marker of clonality, like kappa and lambda antigen receptor immunophenotyping in B cells, so molecular methods can be used. More specifically, two broad categories of molecular changes are used for clinical testing of T-cell lymphomas (TCL): T-cell receptor (TR) gene rearrangements and chromosomal alterations such as translocations, insertions, or deletions.
TR Gene Rearrangements
Pluripotent bone marrow (BM) stem cells give rise to progenitor T cells, which migrate to the thymus for primary ontogeny. There, early in T lymphocyte development TR genes undergo somatic rearrangement of germline gene sequences, similar to the process that occurs with immunoglobulin heavy chain (IGH) and kappa and lambda light chain (IGK and IGL) genes. The four TR genes are: TR delta (TRD) at chromosome 14q11, TR gamma (TRG) at 7p14, TR beta (TRB) at 7q34, and TR alpha (TRA) at 14q11. In the germline configuration, TRD and TRB contain multiple variable (V), diversity (D), and joining (J) region segments. The TRG and TRA loci do not contain D segments. Depending on the locus, the number of segments is 8–67 V segments, 2–3 D segments, and 4–61 J segments [3]. For TRD and TRB, the somatic rearrangement initially involves the joining of one D to one J segment, followed by the joining of one V segment to the DJ segment (Fig. 43.1). For TRG and TRA, the V segment is joined to the J segment. The rearrangement process results in the deletion of the intervening coding and noncoding DNA sequences between the linked V, D, and J segments. Thus, the TRD and TRB gene rearrangements result in V-D-J juxtaposition similar to IGH, and TRG and TRA rearrangements result in V-J rearrangements similar to IGK and IGL. In all cases, the V-(D)-J segment is joined to the downstream constant (C) region by mRNA splicing. The TR genes are ultimately translated into two types of receptors, which exist as heterodimers (αβ or γδ). Of note, the TR genes do not undergo somatic hypermutation as occurs with antigen stimulation for the immunoglobulin (Ig) genes.

The TRB locus is used as an example to demonstrate the rearrangement of the variable (V), diversity (D), joining (J), and constant (C) region gene segments. The hashed white boxes between the rearranged V-D and D-J gene segments represent variable regions of nucleotide deletion and addition that occur during TR gene rearrangement
The TR genes generally rearrange in the following order: TRD, TRG, TRB, and finally TRA [4]. The TRD genes are located within the TRA locus, so rearrangement of TRA will result in TRD deletion on that allele. Also of note for clonality assessment, this hierarchical order of rearrangements results in both αβ- and γδ-T cells containing TRG rearrangements. For this and other reasons, TRG is commonly used for PCR-based clonality assessment.
The numerous different V, (D), and J segments present within each TR locus enable a large number of different V-(D)-J segments to be generated. The TR repertoire is further expanded by deletion and addition of nucleotides between the D-J and V-D junctions by terminal deoxynucleotidyl transferase (TdT), and by the combinations of different α–β or γ–δ chains. Collectively, these mechanisms generate extensive diversity of TRs with varying antigen specificity.
More than 95 % of mature, circulating T cells express the αβ receptor. In contrast, γδ T cells are mainly found in the skin, spleen, gastrointestinal tract, and other extranodal sites, which are often sites of origin for γδ T-cell lymphomas. T-cell neoplasms ensue after maturation arrest at one of the stages of T-cell development, such as from immature T cells in T lymphoblastic leukemia/lymphoma, or from more mature T cells in peripheral TCL and mycosis fungoides. Due to a common progenitor as well as some shared functional and immunophenotypic features, NK-cell neoplasms often are classified with mature T-cell neoplasms. Since NK cells do not rearrange the TR genes, neoplasms derived from these cells do not demonstrate TR gene rearrangements. The mature T- and NK-cell neoplasms of the WHO classification are listed in Table 43.1 [2].
Somatic Chromosomal Abnormalities
A variety of somatic structural and numeric chromosomal abnormalities have been observed in mature TCL, and a subset of these is listed in Table 43.2. ALK-positive anaplastic large cell lymphoma (ALCL), is associated with translocations involving the anaplastic lymphoma receptor tyrosine kinase (ALK) gene on chromosome 2. The most common ALK translocation is t(2;5)(p23;q35), which occurs in approximately 75 % of ALK-positive ALCL cases and results in the fusion of nucleophosmin (NPM1) on chromosome 5 with ALK [5, 6]. The t(1;2)(q21;p23) occurs in approximately 15 % of ALK-positive ALCL cases and results in the fusion of the tropomyosin 3 (TPM3) gene on chromosome 1 with ALK [5, 7]. The numerous other less common ALK fusion partners include ATIC at 2q35, TFG at 3q12, CLTC at 17q23, MSN at Xq12, TPM4 at 19p13, MYH9 at 22q12, and RNF213 (ALO17) at 17q25 [5, 8]. The various translocations result in the activation of ALK, which is not normally expressed in lymphocytes, with subsequent oncogenic actions [5, 6].
Other somatic structural chromosomal abnormalities commonly associated with mature TCL include an inversion of chromosome 14 [inv(14)(q11q32)] associated with T-cell prolymphocytic leukemia [9], and an isochromosome involving the long arm of chromosome 7 [i(7)(q10)] often present with trisomy 8 and associated with hepatosplenic TCL [10]. In addition, amplification of oncogenes and loss of tumor suppressor genes can contribute to the development of mature TCL. As an example, gains involving the long arm of chromosome 9 are observed in more than half of enteropathy-associated TCL [11]. Likewise, loss of function of the CDKN2B (P15) and CDKN2A (P16) genes on the short arm of chromosome 9 due to allelic loss and aberrant promoter methylation occurs in mycosis fungoides and Sézary syndrome [12].
Indications for Testing
The diagnosis of a mature T- or NK-cell neoplasm is primarily based on histology, immunophenotype, and clinical information. For a subset of cases, distinguishing between a reactive and neoplastic process is difficult. Furthermore, in contrast to restricted Ig light-chain expression in mature B-cell lymphomas, T cells do not have a definitive immunophenotypic marker of clonality. Therefore, detection of a clonal TR gene rearrangement can assist in classifying the suspected lymphoproliferation as reactive or neoplastic. Due to specimen and test availability, clonality assessment most commonly involves PCR-based analysis of the TRG locus, or less frequently the TRB locus. If sufficient fresh or frozen neoplastic specimen is available, Southern blot analysis of the TRB locus may be considered.
Another potential application of TR gene rearrangement assays includes determination of clonal relatedness of multiple lesions derived from the same patient. T-cell neoplasms that share the same clonal origin will generally demonstrate identical TR gene rearrangement; however, ongoing and secondary rearrangements of the TR loci may result in alteration or loss of this initial TR rearrangement. Evolution of the TR rearrangement has primarily been observed with precursor lymphoid neoplasms [13]. Other reported applications of TR gene rearrangement assays include staging, minimal residual disease (MRD) monitoring, and lineage assignment, but most clinical assays do not have sufficient sensitivity for the first two applications and cross-lineage rearrangements are observed at a high enough rate to limit the utility of use for lineage assignment.
Although it is less commonly applied in routine clinical practice, demonstration of somatic chromosomal structural or numeric abnormalities by molecular or cytogenetic methods can assist in classifying the suspected lymphoproliferation as reactive or neoplastic. Only a few T-cell neoplasms are associated with specific chromosomal structural abnormalities, but these may assist with subclassification. As an example, the t(2;5)(p23;q35) involving NPM1 and ALK, as well as other ALK rearrangements, are found in ALK-positive ALCL. More recently, data from multiple groups collectively indicate that IRF4 rearrangements (Fig. 43.2) are found in 25–30 % of primary cutaneous anaplastic large cell lymphomas, but are uncommon in T-cell neoplasms considered in the differential diagnosis, including cutaneous involvement by systemic ALK-negative ALCL, lymphomatoid papulosis, and transformed mycosis fungoides [14–16]. Consequently, testing for IRF4 rearrangements by fluorescence in situ hybridization (FISH) may assist with the classification of cutaneous CD30-positive T-cell lymphoproliferative disorders in the context of histology, immunophenotype, and clinical information.

Interphase fluorescence in situ hybridization (FISH) image demonstrating rearrangement of the IRF4 locus with an IRF4 break-apart probe. The yellow arrow points to a cell with an IRF4 rearrangement with one overlapping green and red fusion signal (indicating a normal unrearranged IRF4 locus) and separate red and green signals (indicating an abnormal rearranged IRF4 locus) (Image provided by Andrew Feldman, Mayo Clinic.)
Detection of certain viruses can assist with the subclassification of mature T- and NK-cell neoplasms. Epstein-Barr virus (EBV) is strongly associated with aggressive NK-cell leukemia, EBV-positive T-cell lymphoproliferative disorders of childhood, nasal-type extranodal NK/TCL, and angioimmunoblastic TCL [2]. In situ hybridization (ISH) for EBV-encoded RNA (EBER) is the preferred method of testing paraffin-embedded tissue sections. In addition, the retrovirus human T-cell lymphotropic virus type 1 (HTLV-1) is involved in the pathogenesis of adult T-cell leukemia/lymphoma (ATLL) [17], and demonstration of HTLV-1 infection in ATLL cases is performed by HTLV-1 serology and PCR [18].
Available Assays
Clonality assessment is broadly available to assist in the classification of suspected lymphoproliferative disorders. Likewise, other molecular methods are available for the detection of chromosomal abnormalities and viral sequences associated with mature TCL and NK-cell lymphomas.
PCR-Based Clonality Assessment
Clonality assessment is much more frequently performed by PCR methods in clinical practice than by Southern blot analysis. Advantages of PCR-based assays include good sensitivity, short turnaround time, minimal quantity of required DNA, and the ability to utilize partially degraded DNA such as that derived from formalin-fixed, paraffin-embedded (FFPE) tissues. In addition, genomic DNA from fresh or frozen tissues, blood, BM, or body fluids can be used for assessment of T-cell clonality.
PCR-Based Clonality Assessment Using TRG
The majority of laboratories that perform PCR-based assessment of T-cell clonality examine the TRG locus. The TRG contains nine commonly rearranged V segments, which are further grouped into four V gene families based on sequence homology, and 5 J segments [19, 20]. The TRG locus is less complex than the TRB locus, making primer design to detect all rearrangements simpler. Because the TRG locus is rearranged prior to TRB, clonal rearrangements involving TRG can be detected in both αβ and γδ T cells.
Multiple approaches are used for PCR primer design for the detection of TRG rearrangements. Consensus V and J segment primers can amplify the majority of TRG rearrangements observed in lymphoid cells [21, 22]. Alternatively, primers directed against the four Vγ families coupled with a group of J-region primers can be multiplexed [3, 23, 24], which is currently the most common primer design for PCR-based detection of TRG rearrangements. Even with this common approach, significant differences in the assays used by different laboratories affect assay interpretation and performance. After PCR amplification, the products can be visualized by methods including capillary electrophoresis, heteroduplex analysis, or denaturing gradient gel electrophoresis. Examples of two different PCR-based TRG assays using capillary electrophoresis are shown in Fig. 43.3.

Examples of capillary electropherograms for PCR-based analysis of the TRG locus with two different primer sets. The assays show polyclonal results for a normal lymph node specimen (a, b) and clonal results for a T-cell neoplasm (c, d), using two different methods. The assay depicted in panels (a) and (c) is based on the BIOMED-2 Concerted Action, and the resulting PCR products are distributed among multiple size ranges. This assay uses two multiplex reactions, and the results from one multiplex reaction are shown. In contrast, the assay depicted in (b) and (d) uses a single multiplex reaction and results in PCR products distributed within a single size range. The T-cell neoplasm specimen was derived from a patient with mycosis fungoides, and multiple lesions from this patient demonstrated a dominant peak at the size shown. Note that for illustrative purposes, the vertical axes have been rescaled; the dominant clonal peaks in (c) and (d) are actually tenfold higher than the peak of the corresponding Gaussian distribution in (a) and (b), respectively
PCR-Based Clonality Assessment Using TRB
Although it is less commonly examined for PCR-based assessment of T-cell clonality than TRG, the TRB locus may be assessed as an adjunct or alternative to TRG. The TRB locus is significantly more complex than the TRG locus, containing approximately 65 V segments, two D segments, and 13 J segments [25]. The most common primer set used for analysis of TRB was developed by the BIOMED-2 study [3] and contains 23 Vβ, two Dβ, and 13 Jβ primers divided among three reaction tubes. The resulting PCR products are examined by capillary electrophoresis or heteroduplex analysis to detect the majority of TRB rearrangements.
Southern Blot Analysis Clonality Assessment
Although it is much less frequently performed than the PCR-based assays described above, Southern blot analysis can be a valuable tool for diagnostically challenging lymphoproliferative cases when sufficient fresh or frozen tissue is available. In general, Southern blot analysis of TR loci has better inter-observer agreement and fewer issues with false-positive results than PCR-based analysis. However, most biopsies for suspected lymphoproliferative disorders are small and preserved as FFPE blocks, and thus yield neither sufficient quantity nor quality of DNA for Southern blot analysis. In addition, Southern blot analysis is both time consuming and technically demanding.
Southern blot analysis for T-cell clonality most commonly examines the TRB locus. Although Southern blot analysis detects a clonal rearrangement in > 90 % of T-cell neoplasms, it is generally unable to detect a clonal rearrangement in NK-cell or γδ T-cell neoplasms. Southern blot analysis of the TRB locus uses a probe directed against the TRB constant regions (Cβ) or probes directed against one or both of the TRB J segments (Jβ1 and Jβ2) [26]. An example of a TRB Southern blot is shown in Fig. 43.4. Even though numerous rearrangements derived from T lymphocytes are present in a lane, only the clonal rearrangement reaches the detection level.

Example of a TRB Southern blot. Three restriction enzymes (B, BamHI; E, EcoRII; H, HindIII) were used to digest genomic DNA from germline (left panel) and TCL (right panel) specimens. The lymphoma specimen demonstrates one or two additional rearrangements (arrows) compared to the germline specimen with all three restriction enzymes
Guidelines suggest using at least three different restriction enzymes for digestion of the genomic DNA [27]. The presence of up to two non-germline bands in at least two of the three digests is considered definitive evidence of a clonal rearrangement. One or two non-germline bands in only one of the three digests may represent a clonally rearranged allele or a benign germline polymorphism, and performing digestion with an additional restriction endonuclease may assist in distinguishing between these possibilities. Only two of the three digests are required to demonstrate non-germline bands because one of the digests may have a rearrangement that is a similar size to and obscured by the germline band. The allowance for up to two rearranged bands per digest accounts for rearrangement of both TRB alleles or for the creation of a new restriction site in the rearranged allele within the probe region. The presence of more than two non-germline bands may indicate oligoclonality or a chromosomal abnormality, but incomplete digestion of genomic DNA or germline polymorphisms also may lead to multiple rearranged bands and must be excluded.
Detection of Chromosomal Abnormalities
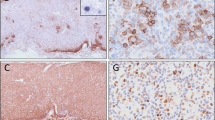
Reverse transcription PCR (RT-PCR) and real-time quantitative PCR (RT-qPCR) can be used for the detection or detection and quantitation, respectively, of fusion transcripts such as NPM1–ALK found in the majority of cases of ALK-positive ALCL. NPM1–ALK and other fusion transcripts involving ALK result in the expression of the ALK protein, which is not normally expressed in lymphocytes. ALK expression is a favorable prognostic marker, and patients with ALK-positive ALCL have improved overall survival compared to patients with ALK-negative ALCL [28, 29]. This association appears to be independent of the translocation partner [30]. Furthermore, up to 25 % of ALK-positive ALCL cases involve translocation partners other than NPM1, and some NPM1–ALK translocations will not be detected by RT-PCR. For these and other reasons, ALK immunohistochemistry is the most widely used test for examining ALK expression in ALCL (Fig. 43.5). RT-PCR and RT-qPCR for NPM1–ALK may provide prognostic information or allow MRD monitoring in select patients with ALK-positive ALCL and the NPM1–ALK fusion [31]. These sensitive methods should be used with caution in diagnostic situations because low levels of NPM1–ALK fusion transcripts have been reported in ALK-negative ALCL, Hodgkin lymphoma, reactive tissue, and even peripheral blood from healthy individuals [32, 33].

Immunohistochemistry demonstrating nuclear and cytoplasmic ALK staining in an ALK-positive anaplastic large cell lymphoma secondarily involving the skin
FISH on fresh or FFPE tissues also can be used to detect NPM1–ALK fusions and other ALK rearrangements, and FISH for ALK rearrangements correlates well with ALK immunohistochemistry [34]. Likewise, FISH can be performed for other structural and numeric chromosomal abnormalities, but FISH testing for mature T- and NK-cell lymphomas is offered in only a few clinical laboratories.
Detection of Viral Sequences Associated with T-Cell Lymphomas
ISH is commonly used for the detection and localization of EBV in tumor cells of the various EBV-positive mature T- and NK-cell neoplasms, especially nasal-type extranodal NK/T-cell lymphoma. ISH can be performed on FFPE tissue sections and identifies EBV RNA within the nuclei of virtually all tumor cells. EBER RNA is most commonly targeted due to the very high copy number in EBV-infected tumor cells. PCR detection of EBV in lymphoid tissue specimens is less useful because the vast majority of adults have been infected with EBV and the assay may detect latently infected background B cells that are not associated with the tumor.
The diagnosis of ATLL involves the demonstration of HTLV-1 infection. This may be performed by HTLV-1 serology or PCR.
Interpretation of PCR-Based T-Cell Clonality Assays
Interpretation of PCR-based T-cell clonality assays is a challenging and commonly debated area of molecular pathology. Interpretation is dependent on assay design and the method used to evaluate the PCR products. Different interpretive criteria are required depending on assay design [35]. Key features of assay design that influence choice of interpretive criteria include primer selection, multiplexing strategy, use of one or multiple different fluorescently labeled primers, and whether the resulting PCR products result in a single or multiple Gaussian regions. In gel-based assays, interpretation as a clonal rearrangement is based on the finding of a discrete band of an appropriate size in contrast to a smear or ladder of bands derived from polyclonal T cells. With capillary electrophoresis-based assays, a wide variety of interpretive criteria is used, and each laboratory must establish and validate its own criteria for a positive result while considering the many issues described in this section. Commonly applied interpretive criteria for the detection of a clonal T-cell rearrangement include observing a reproducible peak above baseline or establishing a cutoff ratio based on the height of the peak in question relative to that of the polyclonal background [3, 36–38]. Computer-aided algorithms also are used to identify peaks that deviate from the normal distribution curve [39]. Less-stringent criteria can be applied with capillary electrophoresis in follow-up specimens when the size of the expected clonal PCR product is known from prior testing.
Oligoclonal proliferations should be differentiated from monoclonal proliferations. The number of bands on a gel or prominent peaks on a capillary electrophoresis tracing necessary to define an oligoclonal vs monoclonal proliferation depends on the assay design and locus examined. In general, the presence of more than two bands or prominent peaks is considered compatible with an oligoclonal proliferation when examining the TRG locus.
Clonality Assay Limit of Detection
The limit of detection for TR gene rearrangements by Southern blot analysis has been generally reported to vary from 1–10 % clonal cells [40, 41], with 5 % representing a reasonable estimate in standard clinical practice [26]. The limit of detection for PCR-based TR gene rearrangement assays depends on the assay design, extent of polyclonal background, and detection method. As an example, commonly used primer sets developed by the BIOMED-2 study are reported to have limits of detection of 1–10 % when heteroduplex analysis is used and 0.5–5 % when capillary electrophoresis is used [3]. Lower limits of detection can be achieved with patient-specific primers, in the range of 0.01–0.001 %. However, this approach is labor and time intensive, and is not generally utilized for mature TCL outside of the research or clinical trial setting.
Clinical Sensitivity
In principle, virtually all TR gene rearrangements can be detected by Southern blot analysis if a sufficiently large clonal T-cell population is present in the specimen. In practice, low tumor burden is a major cause of false-negative TR Southern blot analysis of T-cell neoplasms, and consequently PCR-based assays have higher clinical sensitivity [42]. A clonal TRG rearrangement is detected in >90 % of mature T-cell neoplasms when family-specific V-region primers are used, and a clonal TRB rearrangement is detected in >75 % of mature T-cell neoplasms [43–45]. Rates of detection >95 % in fresh/frozen and FFPE tissues have been reported when both TRG and TRB loci are examined. False-negative results may occur when the family or consensus primers fail to efficiently anneal to the rearranged V, D, or J segments due to limited homology or alteration of the targeted gene region caused by the rearrangement. False-negative results also may be attributed to primer design. As an example, the BIOMED-2 study did not include a Jγ1.2 (JγP) primer to avoid false-positive TRG results from canonical rearrangements [3]. This J segment is used in approximately 3 % of T-cell neoplasms [46]. Consequently, a false-negative TRG result may rarely occur if the other TRG allele has not rearranged. Finally, specimen sampling issues may result in a false-negative TR gene rearrangement if an insufficient number of clonal T cells are present within the tested specimen. An appropriately trained pathologist must review and select the tissue for testing to ensure sufficient abnormal cells are present.
Clinical Specificity
Lack of clinical specificity is a major limitation of PCR-based clonality studies, especially involving the TR loci. Clonal rearrangement of TR loci has been reported in a variable percentage of myeloid and B-cell neoplasms as well as in a variety of nonneoplastic conditions. In some cases, the detected clonal rearrangement represents a true cross-lineage rearrangement within the neoplastic cell population (e.g., detection of a TR rearrangement in B lymphoblastic leukemia/lymphoma [B-ALL]). In many other cases, the detected clonal TR gene rearrangement represents either a limited number or a restricted repertoire of T cells in the specimen.
TR gene rearrangements generally should not be used for lineage determination due to lineage infidelity, which is the rearrangement of TR genes in non-T cells (or the rearrangement of Ig genes in non-B cells). Clonal rearrangements involving TRD or TRG occur in approximately 10 % of acute myeloid leukemia cases [47, 48]. Cross-lineage TR gene rearrangements are very common in B-ALL, with over half of cases demonstrating TRG rearrangements [49]. In contrast, cross-lineage TR gene rearrangements are relatively rare in mature B-cell neoplasms when examined by Southern blot analysis, generally observed in <10 % of cases [50–53]. However, a large study by the BIOMED-2 group using their PCR-based assays examined 369 mature B-cell neoplasms and identified a clonal TR gene rearrangement in approximately 25 % of cases [54]. They noted that these cases could be divided into two groups based on the pattern of TR gene rearrangements. In approximately 10 % of the cases, the rearrangement involved a single locus and exhibited a strong clonal pattern, probably representing true cross-lineage rearrangements present within the malignant B-cell population. In approximately 15 % of cases, TR gene rearrangements were observed at two or more loci and exhibited a weak clonal pattern, probably representing a coexisting restricted or small T-cell population. This underscores that such weak bands or small peaks observed in clonality assays should be interpreted with caution to avoid false-positive results. Subsequent work further illustrates that application of more stringent interpretive criteria to this primer set may improve the clinical specificity of the assay [35].
Detection of Ig gene rearrangements in mature T-cell neoplasms has been observed, but generally at a lower rate than TR rearrangements in B-cell neoplasms [51, 52]. In a study of 188 mature T-cell neoplasms by the BIOMED-2 study, 10 % demonstrated clonal Ig gene rearrangements by PCR, but most of these cases exhibited a weak clonal pattern probably representing a coexisting restricted or small B-cell population [44]. Of the different mature T-cell neoplasm classifications examined, the highest level of Ig gene rearrangements was found in angioimmunoblastic T-cell lymphoma (AITL). In this and other studies, up to one-third of AITL cases have clonal Ig gene rearrangements [44, 55]. Likewise, a high percentage of clonal Ig rearrangements have been reported in peripheral TCL, NOS with rates of 10–35 % [44, 55, 56].
TR gene rearrangement studies are typically performed to distinguish monoclonal from polyclonal lymphoproliferations, but the detection of a monoclonal TR gene rearrangement is not always indicative of malignancy. TR gene rearrangements have been reported in nonneoplastic conditions. Clonal T-cell populations are observed in peripheral blood specimens, especially from older individuals [57–59]. Likewise, oligoclonal and clonal TR gene rearrangements occur in individuals with autoimmune disorders, viral infections, reactive lymphoproliferations, and benign cutaneous lesions [60, 61]. As an example, lymphomatoid papulosis (LyP), a benign primary cutaneous CD30-positive T-cell lymphoproliferative disorder, is associated with the development of a malignant lymphoma in approximately 20 % of cases [62]. Over 40 % of LyP cases are associated with clonally rearranged TR genes, but this is not indicative of malignancy or predictive of progression to lymphoma [63, 64]. Consequently, in LyP cases, TR gene rearrangement studies are of little value.
Pseudoclonality
Pseudoclonality refers to the artifactual detection of an apparently clonal or oligoclonal lymphoid population and is a common problem in PCR-based clonality assessment of certain specimen types. PCR can result in the selective amplification of one or a limited number of TR gene rearrangements when there are a limited number of T cells. Small numbers of T cells and consequently pseudoclonal results are commonly encountered in small skin biopsy specimens, needle biopsies, and B-cell malignancies with very high tumor burdens. Duplicate or triplicate testing of these specimen types often will demonstrate different size gene rearrangements, indicating pseudoclonality. If available, testing of other lesions from the patient can assist in distinguishing clonality from pseudoclonality. This approach appears to be particularly useful in distinguishing inflammatory dermatoses from cutaneous TCL [65, 66].
Oligoclonality
Oligoclonal proliferations have a restricted TR gene repertoire and occur in processes such as antigen-stimulated expansion of distinct subclones, immune reconstitution, or immunosenescence. The number of predominant peaks or bands required to distinguish monoclonality from oligoclonality depends on the assay and locus examined. For TRG, the presence of more than two predominant peaks or bands generally is interpreted as an oligoclonal proliferation. As noted previously, oligoclonal proliferations are commonly observed in individuals with autoimmune disorders, viral infections, reactive lymphoproliferations, and benign cutaneous lesions [60]. Likewise, reduced diversity of the TR repertoire and TR gene oligoclonality is observed in older adults and during immune reconstitution after chemotherapy or stem cell transplantation [67, 68]. If selective amplification of one of the clones occurs in these oligoclonal proliferations, the result may appear clonal (i.e., pseudoclonality). Replicate testing or testing of multiple neoplastic specimens from the same patient will frequently reveal different sized gene rearrangements, indicating pseudoclonality in these cases.
PCR Products Outside of Size Range
In T-cell clonality assessment by PCR, bands outside the defined size range may represent true TR gene rearrangements, but these must be distinguished from nonspecific PCR products. For size ranges established between the 5th and 95th percentiles, bands or peaks just outside of this size range likely represent true rearrangements. Likewise, significantly oversized or undersized bands or peaks also may represent true rearrangements due to amplification from downstream J segments or partial V segment deletions, respectively [69]. In cases in which the peaks or bands are outside of the size range, sequencing the potential rearrangement product allows definitive characterization.
Weak Clonal Results
Weak bands or small peaks in PCR-based TR gene rearrangement assays must be interpreted with caution to avoid false-positive results. Such results should be repeated to ensure that the results are reproducible. In addition, examination of other loci and other involved specimens from the same patient, as well as using an orthogonal detection method, can assist in determining the significance of these weak clonal results. At this time, specific criteria for interpretation of PCR-based clonality testing have not been universally embraced, although (as is discussed above) multiple approaches have been proposed. Each laboratory is required to design, validate, and establish interpretive criteria for their specific assay given the issues and limitations discussed in this section.
Laboratory Issues
Appropriate controls for PCR-based TR gene rearrangement assays include positive, negative, sensitivity, and no DNA template reactions. Positive controls can be derived from previously positive patient specimens or characterized cell lines [70, 71]. A polyclonal control is typically derived from tonsillar tissue processed in the same way as clinical specimens. Sensitivity controls can be made by dilution of DNA from a positive cell line in DNA from a specimen with a polyclonal T-cell population, such as tonsil, to determine and then monitor the limit of detection of the assay. The limit of detection for a clonal population in a non-lymphoid background will be lower than in the presence of a polyclonal lymphoid background and may lead to inaccurate assessment of sensitivity. Finally, no DNA template control reactions, which include all PCR primers and reagents but no DNA, should be included to monitor for exogenous DNA or PCR product contamination. For Southern blot analysis, control germline DNA may be obtained from a variety of non-lymphoid cells, such as placenta [26].
In many laboratories, the majority of specimens will be FFPE tissues. Although some DNA degradation is inherent to formalin fixation, the extracted DNA is usually of sufficient quality for PCR-based TR gene rearrangement testing. However, some cases, approximately 20 % of cases in one study [45], will not have sufficient DNA quality for successful analysis. Consequently, a non-TR or -IG control gene must be amplified for each specimen to prevent a false-negative interpretation due to inadequate DNA quality (or the presence of a PCR inhibitor such as heparin). Furthermore, the control gene should be the same size or larger than the largest amplicon in the assay.
Conclusions and Future Directions
A variety of molecular pathology assays are available to evaluate TR gene rearrangements, structural and numeric chromosomal abnormalities, and viral sequences associated with mature T- and NK-cell neoplasms. In a subset of TCL, these molecular studies are important for disease classification or determining prognosis. However, many of these molecular studies, especially PCR-based TR gene rearrangement studies, have important caveats that must be considered during validation and subsequent clinical use and interpretation of the assays. Therefore, molecular test results must be considered in the context of available clinical information, histology, immunophenotype results, and other laboratory data to ensure proper clinical use of the results.
With the widespread application of genomic and other high-throughput methodologies such as massively parallel sequencing in the research setting, the amount of data regarding the genetics underlying hematolymphoid neoplasms is rapidly increasing. Recurrent somatic mutations as well as structural chromosomal alterations have been identified in lymphoid malignancies. Most of this work has focused on B-cell neoplasms, but projects involving mature T-cell neoplasms are underway. Recently, massively parallel sequencing has led to identification of a recurrent translocation in ALK-negative ALCL [72]. In addition, massively parallel sequencing strategies are being applied to analysis of TR loci [73], and clinical assays based on these methods are now in use. These data and new methods have the potential to improve the diagnosis, determination of prognosis, and ability to monitor response to therapy in mature T- and NK-cell neoplasms.
References
A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997; 89 (11):3909–18
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press; 2008.
van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, Garcia-Sanz R, van Krieken JH, Droese J, Gonzalez D, Bastard C, White HE, Spaargaren M, Gonzalez M, Parreira A, Smith JL, Morgan GJ, Kneba M, Macintyre EA. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–317.
Dik WA, Pike-Overzet K, Weerkamp F, de Ridder D, de Haas EFE, Baert MRM, van der Spek P, Koster EEL, Reinders MJT, van Dongen JJM, Langerak AW, Staal FJT. New insights on human T cell development by quantitative T cell receptor gene rearrangement studies and gene expression profiling. J Exp Med. 2005;201(11):1715–23.
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8(1):11–23.
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4.
Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood. 1999;93(9):3088–95.
Delsol G, Falini B, Muller-Hermelink HK, Campo E, Jaffe ES, Gascoyne RD, Stein H, Kinney MC. Anaplastic large cell lymphoma (ALCL), ALK-positive. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press; 2008. p. 312–6.
Brito-Babapulle V, Catovsky D. Inversions and tandem translocations involving chromosome 14q11 and 14q32 in T-prolymphocytic leukemia and T-cell leukemias in patients with ataxia telangiectasia. Cancer Genet Cytogenet. 1991;55(1):1–9.
Alonsozana EL, Stamberg J, Kumar D, Jaffe ES, Medeiros LJ, Frantz C, Schiffer CA, O’Connell BA, Kerman S, Stass SA, Abruzzo LV. Isochromosome 7q: the primary cytogenetic abnormality in hepatosplenic gammadelta T cell lymphoma. Leukemia. 1997;11(8):1367–72.
Zettl A, Ott G, Makulik A, Katzenberger T, Starostik P, Eichler T, Puppe B, Bentz M, Muller-Hermelink HK, Chott A. Chromosomal gains at 9q characterize enteropathy-type T-cell lymphoma. Am J Pathol. 2002;161(5):1635–45.
Scarisbrick JJ, Woolford AJ, Calonje E, Photiou A, Ferreira S, Orchard G, Russell-Jones R, Whittaker SJ. Frequent abnormalities of the p15 and p16 genes in mycosis fungoides and sezary syndrome. J Invest Dermatol. 2002;118(3):493–9.
Pongers-Willemse MJ, Seriu T, Stolz F, d’Aniello E, Gameiro P, Pisa P, Gonzalez M, Bartram CR, Panzer-Grumayer ER, Biondi A, San Miguel JF, van Dongen JJ. Primers and protocols for standardized detection of minimal residual disease in acute lymphoblastic leukemia using immunoglobulin and T cell receptor gene rearrangements and TAL1 deletions as PCR targets: report of the BIOMED-1 CONCERTED ACTION: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13(1):110–8.
Wada DA, Law ME, Hsi ED, Dicaudo DJ, Ma L, Lim MS, Souza A, Comfere NI, Weenig RH, Macon WR, Erickson LA, Ozsan N, Ansell SM, Dogan A, Feldman AL. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24(4):596–605.
Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, Vergier B, Jouary T, Beylot-Barry M, Merlio JP. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130(3):816–25.
Feldman AL, Law M, Remstein ED, Macon WR, Erickson LA, Grogg KL, Kurtin PJ, Dogan A. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009;23(3):574–80.
Mortreux F, Gabet AS, Wattel E. Molecular and cellular aspects of HTLV-1 associated leukemogenesis in vivo. Leukemia. 2003;17(1):26–38.
Tsukasaki K, Hermine O, Bazarbachi A, Ratner L, Ramos JC, Harrington Jr W, O’Mahony D, Janik JE, Bittencourt AL, Taylor GP, Yamaguchi K, Utsunomiya A, Tobinai K, Watanabe T. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27(3):453–9.
Chen Z, Font MP, Loiseau P, Bories JC, Degos L, Lefranc MP, Sigaux F. The human T-cell V gamma gene locus: cloning of new segments and study of V gamma rearrangements in neoplastic T and B cells. Blood. 1988;72(2):776–83.
Quertermous T, Strauss WM, Van Dongen JJ, Seidman JG. Human T cell gamma chain joining regions and T cell development. J Immunol. 1987;138(8):2687–90.
Benhattar J, Delacretaz F, Martin P, Chaubert P, Costa J. Improved polymerase chain reaction detection of clonal T-cell lymphoid neoplasms. Diagn Mol Pathol. 1995;4(2):108–12.
Bourguin A, Tung R, Galili N, Sklar J. Rapid, nonradioactive detection of clonal T-cell receptor gene rearrangements in lymphoid neoplasms. Proc Natl Acad Sci U S A. 1990;87(21):8536–40.
Greiner TC, Raffeld M, Lutz C, Dick F, Jaffe ES. Analysis of T cell receptor-gamma gene rearrangements by denaturing gradient gel electrophoresis of GC-clamped polymerase chain reaction products. Correlation with tumor-specific sequences. Am J Pathol. 1995;146(1):46–55.
Theodorou I, Bigorgne C, Delfau MH, Lahet C, Cochet G, Vidaud M, Raphael M, Gaulard P, Farcet JP. VJ rearrangements of the TCR gamma locus in peripheral T-cell lymphomas: analysis by polymerase chain reaction and denaturing gradient gel electrophoresis. J Pathol. 1996;178(3):303–10.
Rowen L, Koop BF, Hood L. The complete 685-kilobase DNA sequence of the human beta T cell receptor locus. Science. 1996;272(5269):1755–62.
van Dongen JJ, Wolvers-Tettero IL. Analysis of immunoglobulin and T cell receptor genes. Part I: basic and technical aspects. Clin Chim Acta. 1991;198(1-2):1–91.
Enns RK, Altmiller DH, Hinkel CS, Madej RM, O’Leary TJ. Immunoglobulin and T-cell receptor gene rearrangement assays; approved guideline, second edition, NCCLS document MM2-A2, 2002
Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, Verhoef G, Menestrina F, Todeschini G, Paulli M, Lazzarino M, Giardini R, Aiello A, Foss HD, Araujo I, Fizzotti M, Pelicci PG, Flenghi L, Martelli MF, Santucci A. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999;93(8):2697–706.
Gascoyne RD, Aoun P, Wu D, Chhanabhai M, Skinnider BF, Greiner TC, Morris SW, Connors JM, Vose JM, Viswanatha DS, Coldman A, Weisenburger DD. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999;93(11):3913–21.
Falini B, Pulford K, Pucciarini A, Carbone A, De Wolf-Peeters C, Cordell J, Fizzotti M, Santucci A, Pelicci PG, Pileri S, Campo E, Ott G, Delsol G, Mason DY. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood. 1999;94(10):3509–15.
Damm-Welk C, Busch K, Burkhardt B, Schieferstein J, Viehmann S, Oschlies I, Klapper W, Zimmermann M, Harbott J, Reiter A, Woessmann W. Prognostic significance of circulating tumor cells in bone marrow or peripheral blood as detected by qualitative and quantitative PCR in pediatric NPM-ALK-positive anaplastic large-cell lymphoma. Blood. 2007;110(2):670–7.
Maes B, Vanhentenrijk V, Wlodarska I, Cools J, Peeters B, Marynen P, de Wolf-Peeters C. The NPM-ALK and the ATIC-ALK fusion genes can be detected in non-neoplastic cells. Am J Pathol. 2001;158(6):2185–93.
Trumper L, Pfreundschuh M, Bonin FV, Daus H. Detection of the t(2;5)-associated NPM/ALK fusion cDNA in peripheral blood cells of healthy individuals. Br J Haematol. 1998;103(4):1138–44.
Cataldo KA, Jalal SM, Law ME, Ansell SM, Inwards DJ, Fine M, Arber DA, Pulford KA, Strickler JG. Detection of t(2;5) in anaplastic large cell lymphoma: comparison of immunohistochemical studies, FISH, and RT-PCR in paraffin-embedded tissue. Am J Surg Pathol. 1999;23(11):1386–92.
Cushman-Vokoun AM, Connealy S, Greiner TC. Assay design affects the interpretation of t-cell receptor gamma gene rearrangements: comparison of the performance of a one-tube assay with the BIOMED-2-based TCRG gene clonality assay. J Mol Diagn. 2010;12(6):787–96.
Greiner TC, Rubocki RJ. Effectiveness of capillary electrophoresis using fluorescent-labeled primers in detecting T-cell receptor gamma gene rearrangements. J Mol Diagn. 2002;4(3):137–43.
Sprouse JT, Werling R, Hanke D, Lakey C, McDonnel L, Wood BL, Sabath DE. T-cell clonality determination using polymerase chain reaction (PCR) amplification of the T-cell receptor gamma-chain gene and capillary electrophoresis of fluorescently labeled PCR products. Am J Clin Pathol. 2000;113(6):838–50.
Lee SC, Berg KD, Racke FK, Griffin CA, Eshleman JR. Pseudo-spikes are common in histologically benign lymphoid tissues. J Mol Diagn. 2000;2(3):145–52.
Kuo FC, Hall D, Longtine JA. A novel method for interpretation of T-cell receptor gamma gene rearrangement assay by capillary gel electrophoresis based on normal distribution. J Mol Diagn. 2007;9(1):12–9.
Minden MD, Toyonaga B, Ha K, Yanagi Y, Chin B, Gelfand E, Mak T. Somatic rearrangement of T-cell antigen receptor gene in human T-cell malignancies. Proc Natl Acad Sci U S A. 1985;82(4):1224–7.
Aisenberg AC, Wilkes BM, Jacobson JO, Harris NL. Immuno-globulin gene rearrangements in adult non-Hodgkin’s lymphoma. Am J Med. 1987;82(4):738–44.
Sandberg Y, van Gastel-Mol EJ, Verhaaf B, Lam KH, van Dongen JJ, Langerak AW. BIOMED-2 multiplex immunoglobulin/T-cell receptor polymerase chain reaction protocols can reliably replace Southern blot analysis in routine clonality diagnostics. J Mol Diagn. 2005;7(4):495–503.
van Krieken JH, Langerak AW, Macintyre EA, Kneba M, Hodges E, Sanz RG, Morgan GJ, Parreira A, Molina TJ, Cabecadas J, Gaulard P, Jasani B, Garcia JF, Ott M, Hannsmann ML, Berger F, Hummel M, Davi F, Bruggemann M, Lavender FL, Schuuring E, Evans PA, White H, Salles G, Groenen PJ, Gameiro P, Pott C, Dongen JJ. Improved reliability of lymphoma diagnostics via PCR-based clonality testing: report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia. 2007;21(2):201–6.
Bruggemann M, White H, Gaulard P, Garcia-Sanz R, Gameiro P, Oeschger S, Jasani B, Ott M, Delsol G, Orfao A, Tiemann M, Herbst H, Langerak AW, Spaargaren M, Moreau E, Groenen PJ, Sambade C, Foroni L, Carter GI, Hummel M, Bastard C, Davi F, Delfau-Larue MH, Kneba M, van Dongen JJ, Beldjord K, Molina TJ. Powerful strategy for polymerase chain reaction-based clonality assessment in T-cell malignancies Report of the BIOMED-2 Concerted Action BHM4 CT98-3936. Leukemia. 2007;21(2):215–21.
Liu H, Bench AJ, Bacon CM, Payne K, Huang Y, Scott MA, Erber WN, Grant JW, Du MQ. A practical strategy for the routine use of BIOMED-2 PCR assays for detection of B- and T-cell clonality in diagnostic haematopathology. Br J Haematol. 2007;138(1):31–43.
Lawnicki LC, Rubocki RJ, Chan WC, Lytle DM, Greiner TC. The distribution of gene segments in T-cell receptor gamma gene rearrangements demonstrates the need for multiple primer sets. J Mol Diagn. 2003;5(2):82–7.
Boeckx N, Willemse MJ, Szczepanski T, van der Velden VH, Langerak AW, Vandekerckhove P, van Dongen JJ. Fusion gene transcripts and Ig/TCR gene rearrangements are complementary but infrequent targets for PCR-based detection of minimal residual disease in acute myeloid leukemia. Leukemia. 2002;16(3):368–75.
Przybylski G, Oettle H, Ludwig WD, Siegert W, Schmidt CA. Molecular characterization of illegitimate TCR delta gene rearrangements in acute myeloid leukaemia. Br J Haematol. 1994;87(2):301–7.
Szczepanski T, Flohr T, van der Velden VH, Bartram CR, van Dongen JJ. Molecular monitoring of residual disease using antigen receptor genes in childhood acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. 2002;15(1):37–57.
Kneba M, Bergholz M, Bolz I, Hulpke M, Batge R, Schauer A, Krieger G. Heterogeneity of immunoglobulin gene rearrangements in B-cell lymphomas. Int J Cancer. 1990;45(4):609–13.
Pelicci PG, Knowles 2nd DM, Dalla Favera R. Lymphoid tumors displaying rearrangements of both immunoglobulin and T cell receptor genes. J Exp Med. 1985;162(3):1015–24.
Griesser H, Feller A, Lennert K, Minden M, Mak TW. Rearrangement of the beta chain of the T cell antigen receptor and immunoglobulin genes in lymphoproliferative disorders. J Clin Invest. 1986;78(5):1179–84.
Chen Z, Le Paslier D, Dausset J, Degos L, Flandrin G, Cohen D, Sigaux F. Human T cell gamma genes are frequently rearranged in B-lineage acute lymphoblastic leukemias but not in chronic B cell proliferations. J Exp Med. 1987;165(4):1000–15.
Evans PA, Pott C, Groenen PJ, Salles G, Davi F, Berger F, Garcia JF, van Krieken JH, Pals S, Kluin P, Schuuring E, Spaargaren M, Boone E, Gonzalez D, Martinez B, Villuendas R, Gameiro P, Diss TC, Mills K, Morgan GJ, Carter GI, Milner BJ, Pearson D, Hummel M, Jung W, Ott M, Canioni D, Beldjord K, Bastard C, Delfau-Larue MH, van Dongen JJ, Molina TJ, Cabecadas J. Significantly improved PCR-based clonality testing in B-cell malignancies by use of multiple immunoglobulin gene targets. Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia. 2007;21(2):207–14.
Tan BT, Warnke RA, Arber DA. The frequency of B- and T-cell gene rearrangements and epstein-barr virus in T-cell lymphomas: a comparison between angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified with and without associated B-cell proliferations. J Mol Diagn. 2006;8(4):466–75.
Tan BT, Seo K, Warnke RA, Arber DA. The frequency of immunoglobulin heavy chain gene and T-cell receptor gamma-chain gene rearrangements and Epstein-Barr virus in ALK+ and ALK- anaplastic large cell lymphoma and other peripheral T-cell lymphomas. J Mol Diagn. 2008;10(6):502–12.
Posnett DN, Sinha R, Kabak S, Russo C. Clonal populations of T cells in normal elderly humans: the T cell equivalent to “benign monoclonal gammapathy”. J Exp Med. 1994;179(2):609–18.
Delfau-Larue MH, Laroche L, Wechsler J, Lepage E, Lahet C, Asso-Bonnet M, Bagot M, Farcet JP. Diagnostic value of dominant T-cell clones in peripheral blood in 363 patients presenting consecutively with a clinical suspicion of cutaneous lymphoma. Blood. 2000;96(9):2987–92.
Fitzgerald JE, Ricalton NS, Meyer AC, West SG, Kaplan H, Behrendt C, Kotzin BL. Analysis of clonal CD8+ T cell expansions in normal individuals and patients with rheumatoid arthritis. J Immunol. 1995;154(7):3538–47.
Hodges E, Krishna MT, Pickard C, Smith JL. Diagnostic role of tests for T cell receptor (TCR) genes. J Clin Pathol. 2003;56(1):1–11.
Langerak AW, Molina TJ, Lavender FL, Pearson D, Flohr T, Sambade C, Schuuring E, Al Saati T, van Dongen JJ, van Krieken JH. Polymerase chain reaction-based clonality testing in tissue samples with reactive lymphoproliferations: usefulness and pitfalls. A report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2007;21(2):222–9.
Bekkenk MW, Geelen FA, van Voorst Vader PC, Heule F, Geerts ML, van Vloten WA, Meijer CJ, Willemze R. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95(12):3653–61.
Kadin ME. Pathobiology of CD30+ cutaneous T-cell lymphomas. J Cutan Pathol. 2006;33 Suppl 1:10–7.
el-Azhary RA, Gibson LE, Kurtin PJ, Pittelkow MR, Muller SA. Lymphomatoid papulosis: a clinical and histopathologic review of 53 cases with leukocyte immunophenotyping, DNA flow cytometry, and T-cell receptor gene rearrangement studies. J Am Acad Dermatol. 1994;30(2 Pt 1):210–8.
Thurber SE, Zhang B, Kim YH, Schrijver I, Zehnder J, Kohler S. T-cell clonality analysis in biopsy specimens from two different skin sites shows high specificity in the diagnosis of patients with suggested mycosis fungoides. J Am Acad Dermatol. 2007;57(5):782–90.
Ponti R, Fierro MT, Quaglino P, Lisa B, Paola F, Michela O, Paolo F, Comessatti A, Novelli M, Bernengo MG. TCR[gamma]-chain gene rearrangement by PCR-based GeneScan: diagnostic accuracy improvement and clonal heterogeneity analysis in multiple cutaneous T-cell lymphoma samples. J Invest Dermatol. 2007;128(4):1030–8.
Sarzotti M, Patel DD, Li X, Ozaki DA, Cao S, Langdon S, Parrott RE, Coyne K, Buckley RH. T cell repertoire development in humans with SCID after nonablative allogeneic marrow transplantation. J Immunol. 2003;170(5):2711–8.
Mariani S, Coscia M, Even J, Peola S, Foglietta M, Boccadoro M, Sbaiz L, Restagno G, Pileri A, Massaia M. Severe and long-lasting disruption of T-cell receptor diversity in human myeloma after high-dose chemotherapy and autologous peripheral blood progenitor cell infusion. Br J Haematol. 2001;113(4):1051–9.
Langerak AW. Undersized, oversized? It is not one-size-fits-all in lymphoid clonality detection. Leuk Res. 2008;32(2):203–4.
Yao R, Rich SA, Schneider E. Validation of sixteen leukemia and lymphoma cell lines as controls for molecular gene rearrangement assays. Clin Chem. 2002;48(8):1344–51.
Sandberg Y, Verhaaf B, van Gastel-Mol EJ, Wolvers-Tettero IL, de Vos J, Macleod RA, Noordzij JG, Dik WA, van Dongen JJ, Langerak AW, Lam KH. Human T-cell lines with well-defined T-cell receptor gene rearrangements as controls for the BIOMED-2 multiplex polymerase chain reaction tubes. Leukemia. 2007;21(2):230–7.
Feldman AL, Dogan A, Smith DI, Law ME, Ansell SM, Johnson SH, Porcher JC, Ozsan N, Wieben ED, Eckloff BW, Vasmatzis G. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117(3):915–9.
Benichou J, Ben-Hamo R, Louzoun Y, Efroni S. Rep-Seq: uncovering the immunological repertoire through next-generation sequencing. Immunology. 2012;135(3):183–91.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Merker, J.D., Arber, D.A. (2016). T-Cell Lymphomas. In: Leonard, D. (eds) Molecular Pathology in Clinical Practice. Springer, Cham. https://doi.org/10.1007/978-3-319-19674-9_43
Download citation
DOI: https://doi.org/10.1007/978-3-319-19674-9_43
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19673-2
Online ISBN: 978-3-319-19674-9
eBook Packages: MedicineMedicine (R0)