
Abstract
A series of 43 papillary renal cell carcinomas (PRCCs) were analyzed to investigate the prognostic value of the morphological subtyping (type 1/type 2) proposed by Delahunt and Eble [6]. Twenty-six cases were type 1 (small cuboid cells arranged in single or double layers), 13 cases were type 2 (voluminous eosinophilic cells with irregular pseudostratification pattern), and four cases with oncocytic cells (large eosinophilic cells with round regular nuclei) were distinct from type 2 and grouped apart. All type-1 and oncocytoid-type PRCCs were staged pT1 or pT2, whereas 8/13 type-2 PRCCs were staged pT3 or pT4. Follow-up information (range, 3–113 months; median, 43 months) showed 12 deaths from disease: 2 in the type-1 group,10 in the type-2 group, 0 in the oncocytoid-type group. The Kaplan-Meier analysis showed that pejorative outcome was associated (P<0.001) with high stage (pT3/pT4), high nuclear grade (3/4), morphological type 2, absence of foam cells, and abundant fibrous stroma. The multivariate analysis showed that stage and morphological type were independently associated with survival (P<0.05). These results support the clinical interest of morphological subtyping of PRCCs in the prognosis evaluation of the patients. The four oncocytoid-type PRCCs had a favorable outcome, but additional data are required to evaluate this type of neoplasm.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the revised classification of renal cell neoplasia [15, 30], papillary renal cell carcinoma (PRCC) is recognized as a distinct tumor type, supported by multiple morphological [1, 4, 8, 18, 20, 22, 24], immunohistochemical [10, 24], and genetic [5, 12, 13, 14, 16] studies. PRCCs represent approximately 15–20% of renal epithelial tumors [3, 18, 20] and are typically characterized by a predominant papillary pattern (more than 50% at least of the tumor) [1, 18, 24], a cytokeratin 7-positive immunophenotype [10], and recurrent cytogenetic alterations consistently showing trisomy or tetrasomy of chromosomes 7 and 17, and with a high-frequency loss of chromosome Y, trisomies 12, 16, and 20 [5, 12, 13, 14, 16]. Most studies suggested that PRCC had a more favorable prognosis than conventional (clear cell) renal cell carcinomas (CRCC) [3, 18, 20, 22]. However, large and recent studies, taking into account the stage and the nuclear grade, have revealed a similar prognosis for PRCC in comparison with CRCC [19, 21]. These discrepancies might be related to the broad range of morphological and cytogenetic variants described in PRCC. Indeed, various architectural variants—including trabecular, tubular, solid, and collecting duct-like patterns—and cytological subtypes—basophilic, eosinophilic, or clear cell—have been described [1, 8, 18, 22, 23, 24, 25, 26, 27, 31], and 20% of PRCCs at least have been reported lacking the typical trisomy 17 [5, 11, 13, 14]. Recently, Delahunt and Eble [6] have proposed the existence of two PRCC subtypes, type 1 with papillae covered by a single or double layer of small cells with scanty cytoplasm, and type 2 with papillae covered by cells with abundant eosinophilic cytoplasm, arranged in a pseudostratified or irregularly stratified manner [6]. Interestingly, the tumor stage at diagnosis was significantly higher in type-2 than in type-1 PRCCs, suggesting these subtypes could be clinicopathologic entities with a different prognosis [6].
In this study, we report a series of 43 consecutively diagnosed cases of PRCC, aiming to: (1) investigate the possibility to classify all the PRCC cases as type 1 or 2, (2) confirm the clinicopathologic features of types 1 and 2, and (3) evaluate the prognostic impact of these morphological subtypes in patients' survival.
Material and methods
Case selection
Cases of PRCC diagnosed between January 1992 and July 1998 were retrieved from the files of the Department of Pathology, Cochin Hospital, Paris, France. We included 43 cases according to the following criteria: (1) the carcinoma had a main diameter of more than 1 cm and showed papillary architecture in at least 50% of the tumor (solid variant with packed papillae were included [25]) according to the revised classification of renal tumor [15, 30]; (2) the differential diagnosis (i.e., metanephric adenoma, oncocytoma, CRCC with papillary pattern, eosinophilic chromophobe cell carcinoma, and collecting duct carcinoma) were excluded; (3) the PRCC was not associated with another renal malignant tumor; (4) the demographic data (age, sex) and follow-up information (disease-free and overall survival) obtained from clinical charts were available. Forty cases were treated by radical nephrectomy; three cases were treated by partial nephrectomy with safe margins (one patient with a 2-cm-diameter tumor, two patients with a congenital single kidney).
Morphological study
Gross examination data (tumor size, extent) were obtained from pathology reports. Hematoxylin–eosin–saffron-stained sections from all cases were reviewed concomitantly by two pathologists (E.B., A.V.), and the following features were evaluated:
-
1.
Type of architecture, classified as papillary, solid-variant, or mixed
-
2.
Cytoplasm characteristics, including size (small or tall) and staining (basophilic or eosinophilic, respectively, if the tumor was predominantly of one cell type, or mixed if both components were conspicuous within the tumor
-
3.
Nuclear size and pleomorphism
-
4.
Tumor necrosis
-
5.
Psammoma bodies
-
6.
Macrophages appearing as foam cells within fibrovascular cores and/or within necrotic area
-
7.
Stroma, described as "scanty" or "abundant and fibrous"
-
8.
Tumor extension through the capsule and/or within (small and large) vessels
The tumors were classified as type-1 PRCC (papillae covered with a single or double layer of small cuboid cells with scanty cytoplasm) or type-2 PRCC (papillae covered by large eosinophilic cells arranged in a pseudostratified or irregularly stratified manner), defined by Delahunt and Eble [6]. Tumor stage was assigned according to the TNM staging system of the International Union Against Cancer (UICC) [29, 30]. Tumor grade was evaluated on the basis of nuclear size and pleomorphism as described by Fuhrman et al. [9].
Statistical analysis
The clinicopathologic features of papillary renal cell carcinoma were tested for their association with the histological types 1 and 2, using for continuous variables the student t-test (age, tumor size, follow-up time), and for qualitative variables Fisher's exact test (ratio of male to female, necrosis, psammoma bodies, foam cells, stroma, deaths), or the Kruskal-Wallis test (stage, grade, cytoplasm). Survival was defined as the time between surgery and patient death. For the analysis, only deaths with PRCC listed as the underlying cause were considered as events. The Kaplan-Meier method was used to analyze the cumulative survival of patients and the effect of the following parameters on survival: age, tumor size, stage, grade, type 1 or type 2, cytoplasm staining, psammoma bodies, foam cells, stroma, and necrosis. Statistical differences between the groups were determined with the log rank test. A multivariate analysis using the Cox proportional hazards regression model was used to test for independent prognostic value. The statistical analysis was performed with Statistica software (version 5.1; Statsoft France, Paris, France).
Results
The results are summarized in Table 1. There were 40 men and 3 women in the series, and the mean age at surgery was 60 years.
Type-1 and type-2 classification
According to the criteria defined by Eble and Delahunt, 28 cases were easily classified as type-1 or -2 PRCCs, but histological subtyping was problematic in 15 cases. Seven of fifteen cases were made of medium-sized cells; we agreed to classify them on the basis of pseudostratification only. Four of fifteen cases displayed significant foci of overlapping features, with papillae covered by small cuboidal cells in the vicinity of large eosinophilic cells; we decided arbitrarily to class them as type-2 PRCCs. Four of fifteen other cases were made of large cells with a striking oncocytic appearance; because of this distinct feature, we decided to consider them separately from the type-2 PRCCs. Eventually, 26 cases were classified as type-1 PRCCs, 13 cases were classified as type-2 PRCCs, and four cases with an oncocytoid pattern were grouped apart.
Comparisons between type-1 and type-2 PRCCs
The mean age of the patients and the male to female ratio was not significantly different between the two groups. The main tumor was associated with distant papillary adenomas (size less than 0.5 cm) in 6 of 26 cases in type-1 PRCCs, and 1 of 13 cases in type-2 PRCCs. The mean size of the main tumor was 5.5 cm in type-1 and 6.7 cm in type-2 PRCCs, without statistical difference. The architecture was predominantly papillary, without difference between the two groups. In type-1 PRCCs, the stalk of papillae was covered by small and cuboid cells with small ovoid nuclei, arranged in a regular and monostratified pattern (Fig. 1). In type-2 PRCCs, the cells lining papillae were large and columnar, predominantly eosinophilic, with large irregular nuclei and an irregular pseudo- or multistratification pattern (Fig. 2). The nuclear grade was significantly higher in type-2 than in type-1 PRCCs (P<0.01). Four type-2 cases had areas with collecting duct-like pattern, with distorted tubules in a desmoplastic stroma. Sarcomatoid cells were noted within two of these latter cases. The diagnosis of collecting-duct carcinoma was excluded by Ulex europaeus lectin negative staining. Foci of necrosis with cholesterol clefts were frequently observed in both type-1 and type-2 PRCCs, without statistical difference. A significant fibrovascular stroma, enlarging papillae or surrounding tumoral tubules, was more often noticed in type-2 (10/13) than in type-1 PRCCs (3/26; P<0.01). Carcinomatous islets filling the lumen of small blood and/or lymphatic vessels surrounding the tumor were frequently noticed in type-2 (8/13), but never in type-1 PRCCs (0/26). The stage was significantly higher in type-2 than in type-1 PRCCs. No type-1 case extended through the renal capsule or into the renal vein. All type-1 cases were staged pT1 or pT2, whereas eight type-2 cases were staged pT3 or pT4 (two cases with extension into the renal vein, and eight cases with extension in the perirenal adipose tissue). Lymph node metastases were noticed at initial surgery in seven cases of type-2, but in none of type-1 PRCCs.

Type-1 papillary renal cell carcinoma with: (A) foam cells within the papillae (asterisk); (B) small, cuboid monostratified cells (arrow); and (C) small, regular nuclei

Type-2 papillary renal cell carcinoma with: (A) abundant fibrous stroma (asterisk); (B) broad pseudostratified papillae; and (C) large and irregular nuclei
Characteristics of oncocytoid-type PRCCs
The mean age and sex ratio of the oncocytoid-type PRCC patients were not significantly different from the patients of type-1 and -2 PRCCs. There was no multifocality, and the mean tumor size was 4.5 cm. Tumoral cells were medium- to large-sized and eosinophilic, with a round regular nucleus, and a low nucleus to cytoplasm ratio. Nuclei had often a conspicuous nucleolus. The nuclear grade was 2 in three cases and 3 in one case. These cytological features gave the cells a distinct oncocytic pattern (Fig. 3), which was lacking in the type-2 PRCCs that were characterized by large, irregular nuclei and abundant eosinophilic cytoplasm. The tumors were made of papillary structures with mono- or pseudostratified pattern. In two of four cases, solid areas were present with packed papillae. Foam cells were focally noticed within the stalks of papillae. The stroma was inconspicuous. All four cases showed extensive necrosis as observed in type-1 and -2 PRCCs. These four cases were staged pT1. We propose to designate them as oncocytoid-type PRCCs.

Oncocytoid-type papillary renal cell carcinoma with: (A) predominant papillary architecture and foam cells within the stalks (arrow); (B) mono- or pseudostratified pattern; and (C) oncocytic feature due to voluminous eosinophilic cytoplasms, regular nuclei, and low nucleus to cytoplasm ratio
Prognosis analysis
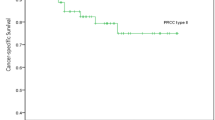
The median follow-up time was 43 months (range 3–113 months). Two of twenty-six patients with type-1 PRCC, and 10 of 13 patients with type-2 PRCC died of disease. No deaths occurred in patients with oncocytoid-type PRCC. The Kaplan-Meier analysis (Fig. 4) showed a worse prognosis for patients with: (1) high stage (pT3 or pT4), (2) tumor size ≥7 cm, (3) high grade (grade 3 or 4), (4) type-2 PRCC, (5) absent or rare foam cells, and (6) abundant fibrous stroma. The prognosis was not related to: (1) age (using median age as cut-off value), (2) psammoma bodies, or (3) necrosis. A multivariate survival analysis using the Cox proportional hazards model showed that only stage and histological type (1 or 2) were independently associated with survival (P<0.05; Table 2).

Cumulative survival of patients, stratified by: (A) the nuclear grade; (B) the tumor stage; (C) the morphological type 1 or 2; and (D) the fibrous stroma
Discussion
Papillary renal cell carcinoma is a well-recognized distinct tumor type among renal tumors, even though this group exhibits a large range of morphological variants [1, 4, 6, 8, 18, 20, 22, 24, 25, 26, 27]. Delahunt and Eble [6] have recently proposed a subtyping of PRCC into two morphological subtypes: type 1 (small cells with scanty cytoplasm arranged in a single or double layer), and type 2 (cells with voluminous and usually eosinophilic cytoplasm, arranged in a pseudo- or irregularly stratified manner), which could be associated with a favorable or a pejorative prognosis, respectively [6]. In the present series, 65% of the cases were easily classified as type 1 or type 2. The other cases were somehow equivocal because of overlapping features, but we proposed to classify them using the following additional criteria: (1) cases with medium-sized cells were classified on the basis of pseudostratification only, (2) cases with papillae covered both by small basophilic cells and large eosinophilic cells were classified as type 2. Despite these difficulties, 39 of 43 cases could be grouped in type 1 or 2, with morphological features mostly similar to those initially described by Delahunt and Eble [6].
In our series, four cases were classified as a distinct group with a oncocytoid pattern due to large eosinophilic cells with round regular nuclei. These tumors were made of a mixture of solid areas with tightly packed papillae and true papillary structures, with scattered foci of foam cells, and cytokeratin 7-positive tumoral cells. Regarding to the oncocytoma's features reviewed by Amin [2], the papillary architecture (either typical or solid variant) and necrosis seemed too extensive to be compatible with the diagnosis of oncocytoma. Such oncocytoid PRCCs have already been reported in adults [22, 31]. Unfortunately, cytogenetic data were lacking, as in the present series, so we cannot demonstrate further whether theses cases belong to the PRCC group. The four patients with oncocytoid PRCC remained free of disease after radical nephrectomy, suggesting a favorable outcome. However, a larger series of similar cases will be necessary to evaluate reliably the prognosis of the oncocytoid PRCC phenotype.
Previous studies on prognosis factors in PRCC have demonstrated that stage, nuclear grade, and DNA aneuploidy are correlated with a poor outcome [1, 3, 8, 19, 21, 22]. The impact of morphological features of PRCC in patients' survival remains controversial [1, 22]. The morphological subtyping proposed by Delahunt and Eble includes cell volume and pseudostratification [6]. Both these authors and Jiang et al. have observed that tumor size and stage are significantly higher in type-2 than in type-1 PRCCs [6, 11]. In our series, eight type-2 cases showed extrarenal extension (seven of which with lymph node metastasis), whereas all type-1 cases were confined to the kidney, confirming that type-1 and type-2 PRCCs might be related to different clinical behavior. Moch et al., reporting a series of 588 renal tumors, including 64 PRCC, found that type 1 behaved less aggressively than type 2 [19]. Recently, Delahunt et al. have studied a series of 66 PRCCs for which morphological type 1 and 2, AgNOR score, and Ki-67 index were independently associated with survival, providing evidence of the clinical relevance of the type-1 and -2 classification [7]. In this study, the univariate analysis showed that stage, tumor size, nuclear grade, morphological subtyping, and fibrous stroma were associated with survival. Furthermore, stage and morphological subtyping were found in multivariate analysis to be independent predicting factors of survival, whereas nuclear grade was not significantly associated with survival. Even though the tumor stage at diagnosis remains the main prognostic factor, our results confirm the prognosis impact of the classification proposed by Delahunt and Eble. Of note, when we compared outcome between type-1 and type-2 PRCCs grouped together with oncocytoid type, the difference was less marked (due to the favorable outcome of the four oncocytoid cases) but remained significant (due to the very poor outcome of the 13 type-2 cases; data not shown).
Given the distinct outcome related to types 1 and 2, future studies will aim to find out the molecular basis. Delahunt et al. have demonstrated that tumor growth kinetics is significantly lower in type-1 than in type-2 PRCCs [7]. The genetic alterations in type-1 and -2 PRCCs were reported to be different, with more frequent chromosome gains of 7p and 17p and allelic imbalance in type 1 than in type 2, and a sporadic c-met mutation restricted to type-1 PRCCs [11, 17, 28]. Delahunt et al. have suggested that these results supported the hypothesis of two different entities, and in particular that type-2 do not evolve from type-1 PRCCs [7]. On the contrary, in our series we observed some cases with mixed features of type 1 and type 2, rather suggestive, at least for these cases, of a type-1 to type-2 tumoral progression. We propose that type 2 might be a heterogeneous group including both cases arising from type 1 and cases arising de novo. Further molecular studies will test this hypothesis.
References
Amin MB, Corless CL, Renshaw AA, Tickoo SK, Kubus J, Schultz DS (1997) Papillary (chromophil) renal cell carcinoma: histomorphological characteristics and evaluation of conventional pathologic prognostic parameters in 62 cases. Am J Surg Pathol 21:621–635
Amin MB, Crotty TB, Tickoo SK, Farrow GM (1997) Renal oncocytoma: a reappraisal of morphological features with clinicopathologic findings in 80 cases. Am J Surg Pathol 21:1–12
Amin MB, Amin MB, Tamboli P, Javidan D, Stricker H, De-Peralta Venturina M, Deshpande A, Menon M (2002) Prognostic impact of histologic subtyping of adult renal epithelial neoplasms. An experience of 405 cases. Am J Surg Pathol 26:281–291
Boczko S, Fromowitz FB, Bard RH (1979) Papillary adenocarcinoma of kidney: a new perspective. Urology 14:491–495
Corless CL, Aburatani H, Fletcher JA, Housman DE, Amin MB Weinberg DS (1996) Papillary renal cell carcinoma: quantitation of chromosomes 7 and 17 by FISH, analysis of chromosome 3p for LOH, and DNA ploidy. Diagn Mol Pathol 5:53–64
Delahunt B, Eble JN (1997) Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 10:537–544
Delahunt B, Eble JN, McCredie MR, Bethwaite PB, Stewart JH, Bilous AM (2001) Morphologic typing of papillary renal cell carcinoma: comparison of growth kinetics and patient survival in 66 cases. Hum Pathol 32:590–595
el-Naggar AK, Ro JY, Ensign LG (1993) Papillary renal cell carcinoma: clinical implication of DNA content analysis. Hum Pathol 24:316–321
Fuhrman SA, Lasky LC, Limas C (1982) Prognostic significance of morphological parameters in renal cell carcinoma. Am J Surg Pathol 6:655–663
Gatalica Z, Kovatich A, Miettinen M (1995) Consistent expression of cytokeratin 7 in papillary renal-cell carcinoma. An immunohistochemical study in formalin-fixed, paraffin-embedded tissues. J Urol Pathol 3:205–211
Jiang F, Richter J, Schraml P, Bubendorf L, Gasser T, Sauter G, Mihatsch MJ, Moch H (1998) Imbalances in papillary renal cell carcinoma: genetic differences between histological subtypes. Am J Pathol 153:1467–1473
Kattar MM, Grignon DJ, Wallis T, Haas GP, Sakr WA, Pontes JE, Visscher DW (1997) Clinicopathologic and interphase cytogenetic analysis of papillary (chromophilic) renal cell carcinoma. Mod Pathol 10:1143–1150
Kovacs G (1989) Papillary renal cell carcinoma. A morphological and cytogenetic study of 11 cases. Am J Pathol 134:27–34
Kovacs G, Fuzesi L, Emanual A, Kung HF (1991). Cytogenetics of papillary renal cell tumors. Genes Chromosom Cancer 3:249–255
Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B, Eble JN, Fleming S, Ljungberg B, Medeiros LJ, Moch H, Reuter VE, Ritz E, Roos G, Schmidt D, Srigley JR, Storkel S, van den Berg E, Zbar B (1997) The Heidelberg classification of renal cell tumours. J Pathol 183:131–133
Lager DJ, Huston BJ, Timmerman TG, Bonsib SM (1995) Papillary renal tumors. Morphologic, cytochemical, and genotypic features. Cancer 76:669–673
Lubensky IA, Schmidt L, Zhuang Z, Weirich G, Pack S, Zambrano N, Walther MM, Choyke P, Linehan WM, Zbar B (1999) Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol 155:517–526
Mancilla-Jimenez R, Stanley RJ, Blath RA (1976) Papillary renal cell carcinoma: a clinical, radiologic, and pathologic study of 34 cases. Cancer 38:2469–2480
Moch H, Gasser T, Amin MB, Torhorst J, Sauter G, Mihatsch MJ (2000) Prognostic utility of the recently recommended histologic classification and revised TNM staging system of renal cell carcinoma: a Swiss experience with 588 tumors. Cancer 89:604–614
Mydlo JH, Bard RH (1987) Analysis of papillary renal adenocarcinoma. Urology 30:529–534
Onishi T, Ohishi Y, Goto H, Suzuki M, Miyazawa Y (1999) Papillary renal cell carcinoma: clinicopathological characteristics and evaluation of prognosis in 42 patients. BJU Int 83:937–943
Orain I, Buzelin F, Ferry N (1987) Tubulopapillary tumors of the kidney. A proposal of 20 new cases and a review of the literature. J Urol 93:1–9
Perot C, Bougaran J, Boccon-Gibod L, Storkel S, Leverger G, Akker J van den, Taillemite JL, Couturier J (1999) Two new cases of papillary renal cell carcinoma with t(X;1)(p11;q21) in females. Cancer Genet Cytogenet 110:54–56
Renshaw AA, Corless CL (1995) Papillary renal cell carcinoma. Histology and immunohistochemistry. Am J Surg Pathol 19:842–849
Renshaw AA, Zhang H, Corless CL, Fletcher JA, Pins MR (1997) Solid variants of papillary (chromophil) renal cell carcinoma: clinicopathologic and genetic features. Am J Surg Pathol 21:1203–1209
Renshaw AA, Morgan TW, Fletcher JA (1998) A sarcomatoid renal cell carcinoma with a "hobnail pattern" and immunohistochemical and cytogenetic features of papillary carcinoma. J Urol Pathol 9:93–102
Renshaw AA, Granter SR, Fletcher JA, Kozakewich HP, Corless CL, Perez-Atayde AR (1999) Renal cell carcinomas in children and young adults: increased incidence of papillary architecture and unique subtypes. Am J Surg Pathol 23:795–802
Sanders ME, Mick R, Tomaszewski JE, Barr FG (2002) Unique patterns of allelic imbalance distinguish type-1 from type-2 sporadic papillary renal cell carcinoma. Am J Pathol 161:997–1005
Sobin LH, Witteking CH (eds) (1997) International Union Against Cancer. TNM classification of malignant tumors (5th edn). Wiley-Liss, New York
Storkel S, Eble JN, Adlakha K, Amin M, Blute ML, Bostwick DG, Darson M, Delahunt B, Iczkowski K (1997) Classification of renal cell carcinoma: workgroup no. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer 80:987–989
Thoenes W, Storkel S, Rumpelt HJ (1986) Histopathology and classification of renal cell tumors (adenomas, oncocytomas and carcinomas). The basic cytological and histopathological elements and their use for diagnostics. Pathol Res Pract 181:125–143
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Allory, Y., Ouazana, D., Boucher, E. et al. Papillary renal cell carcinoma. Virchows Arch 442, 336–342 (2003). https://doi.org/10.1007/s00428-003-0787-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-003-0787-1