
Abstract
Main conclusion
Comparative morphological, transcriptomic and phytohormone analyses reveal a defence network leading to PCD involved in cabbage hybrid lethality.
Abstract
Hybrid lethality (HL) plays an essential role in the stability of a population by blocking gene exchange between species, but the molecular mechanism remains largely undetermined. In this study, we performed phenotype, transcriptome and plant hormone analyses of HL in cabbage. Phenotype analysis confirmed that HL is characterised by a typical programmed cell death (PCD) process. A time-resolved RNA-Seq identified 2724 differentially expressed genes (DEGs), and functional annotations analyses revealed that HL was closely associated with the defence response. A defence regulation network was constructed based on the plant–pathogen interaction pathway and MAPK signalling pathway, which comprised DEGs related to Ca2+ and hydrogen peroxide (H2O2) leading to PCD. Moreover, important DEGs involved in hormone signal transduction pathways including salicylic acid (SA) and jasmonic acid (JA) were identified, which were further confirmed by endogenous and exogenous SA and JA measurements. Our results identified key genes and pathways in the regulating network of HL in cabbage, and might open the gate for revealing the molecular mechanism of HL in plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reproductive segregation is a common phenomenon in which offspring or fertile offspring are not produced after mating under natural conditions in a population with close kinship; this process maintains stability within a population by blocking gene exchange between species (Orr and Presgraves 2000; Yamada and Marubashi 2003). Reproductive segregation can be divided into pre-mating and post-mating reproductive isolation based on the time of occurrence; the latter is mainly caused by the incompatibility of internal genetic factors, such as hybrid sterility, hybrid weakness, hybrid breakdown and hybrid lethality (HL) (Maheshwari and Barbash 2010). HL manifests as the death of F1 hybrids before flowering, and offspring are not obtained, with phenotypic manifestations including wilting, chlorosis, dwarf-type growth, and even lethality (Hollingshead 1929). The Dobzhansky–Muller (DM) genetic model appropriately explains the complex molecular mechanism of HL, including a dual-locus and two-locus mode (Muller 1942; Dobzhansky 1959). Most HL cases result from the two-locus model, and the interaction of the two sites causes genetic incompatibility, which leads to reproductive isolation. Many factors can cause hybrid weakness from a genetic point of view, such as chromosomal rearrangement, gene transposition, sequence variation, repeated noncoding sequences and unbalanced doses (Pickersgill 1971; Maheshwari and Barbash 2010; Tezuka and Marubashi 2006). Thus, studies on the complex genetic mechanism underlying HL can help us understand species evolution mechanisms and promote inter-species communication.
HL is an extreme phenotype that occurs during the process of growth and development. At present, HL cases have been reported involving many species, such as Arabidopsis, lettuce, rice, ginseng, bean, mimulus, cotton, potato, and tomato (Hollingshead 1929; Wiebe 1934; Shii et al. 1980; Chen et al. 2013; Bomblies et al. 2007; Jeuken et al. 2009; Chae et al. 2014; Zuellig et al. 2018). Some genes that cause HL, such as Hwi1 and Hwi2, have been isolated and cloned; these genes encode a leucine-rich repeat receptor-like kinase (LRR-RLK) and a secreted putative subtilisin-like protease, respectively, resulting in hybrid weakness in rice (Chen et al. 2013). In species of the cotton genus, there is an incompatible dominant locus called Le4. Here, they report that a coiled coil nucleotide-binding site leucine-rich repeat (CC-NBS-LRR) gene is responsible (Deng et al. 2019). In Mimulus hybridus, the incompatible dominant locus hl13 encodes a critical photosynthetic gene pTAC14 (Zuellig et al. 2018). In lettuce, one of the two interacting genes involved in HL is RPM1-interacting protein 4 (Rin4), which is known to interact with multiple resistance (R) gene products (Jeuken et al. 2009). In terms of Arabidopsis HL, DM1 and DM2 encode two nucleotide-binding domain and leucine-rich repeat (NB-LRR) disease resistance genes (Bomblies et al. 2007; Chae et al. 2014). These findings show that most candidate genes triggering HL are associated with R genes. Furthermore, some studies have shown that the interactions between R genes activate the autoimmune response and defence responses in the absence of biotic/abiotic stress, leading to HL. By performing a transcriptome analysis, Deng et al. (2019) identified 106 pathogen-related genes reportedly related to the immune response. In Arabidopsis, classes of genes associated with the immune response were overrepresented in the larger list of transcriptome analyses, and the hypersensitive response (HR), which is the common consequence of plant immune responses, was determined to correlate with HL (Bomblies et al. 2007). Additionally, Chen et al. (2013) reported that PCD-related and biotic stress-responsive genes were highly enriched in interspecific hybrid weakness of rice. However, why and how these two genes interact and trigger the immune response is unclear, and additional work is needed to reveal this genetic mechanism.
When HL occurs, there are many complex physiological changes in plants. Some evidence shows that plant hormones are involved in the regulation of hybrid mortality. Alcazar et al. (2009) reported that salicylic acid (SA) pathway activation is necessary for HL and that overexpression of salicylate hydroxylase gene (NahG), which can inhibit the accumulation of SA in incompatible interaction lines, did not result in cell death (Rubén et al. 2009). Hannah et al. (2007) reported that the accumulation of SA leads to the death of roots, which is the main result of HL common bean. The results indicated that the plant defence response mediated by SA is an important factor in the formation of HL. Auxin (AUX) and ethylene (ET) also play roles in the regulation of HL. N. glutinosa × N. repanda exhibits temperature-sensitive HL with a higher IAA content than do its parents, and exogenous IAA treatment prevents death (Zhou et al. 1991). Additionally, exogenous ET treatment achieved the same effect. In addition, temperature is the main environmental factor that regulates plant growth, and most of the reported hybrids are sensitive to temperature (Jeuken et al. 2009).
Cabbage (Brassica oleracea var. capitata) is an important Brassica vegetable crop cultivated worldwide. Hu et al. (2016) reported a HL case in cabbage, and they concluded that the incompatible interactions of two dominant complementary HL genes BoHL1 and BoHL2 caused death. Then, 12 and 6 candidate genes, including differentially expressed genes (DEGs) and resistance-related genes were predicted for BoHL1 and BoHL2, respectively (Xiao et al. 2017). However, the complex molecular mechanism of HL in cabbage was largely unclarified. In the current study, we performed comparative morphological, transcriptomic and phytohormone analyses of HL divided into four stages, with one control. Endogenous substances such as hydrogen peroxide (H2O2), SA and jasmonic acid (JA) were measured, and exogenous hormone treatments were also performed. Our results identified key genes and pathways in the regulating network of HL in cabbage, and might open the gate for revealing the molecular mechanism of HL in plants.
Materials and methods
Plant materials
Cabbage (Brassica oleracea var. capitata) inbred lines 09–211 and 09–222 were used, and F1 of 09–211 and 09–222 appeared normal at the initial growth stage but exhibited 100% seedling mortality. The plants were incubated in a greenhouse at 20–25 °C with a 16-h dark photoperiod.
Phenotyping
We observed the growth of the seedlings every day to clearly describe the HL phenotype. To further investigate the characteristics of the lethal hybrids, the seedling development characteristics (height, stem diameter, length and width of the maximum outer leaf) of F1 and the parents were measured. Haematoxylin–Eosin (TE) staining, transmission electron microscopy (TEM) observations and TdT-mediated dUTP Nick-End Labeling (TUNEL) assays were performed to determine the changes in leaves or stems of HL at the cell, organelle and DNA levels, respectively. TE staining was performed according to the manufacturer’s protocol of a Haematoxylin–Eosin/HE Staining Kit (Solarbio, Beijing, China). TUNEL assays were performed according to the manufacturer’s protocol of a TUNEL Apoptosis Assay Kit (Solarbio). The leaves were fixed with 4% formaldehyde fixative solution for 0.5 h. After washing the cells 2–3 times with phosphate buffer saline (PBS), the fluorescence intensity of the stained cells was monitored with a fluorescence microplate. TEM observations were performed using the method of Xing et al. (2019), and sections were photographed using a HT7700 transmission electron microscope (Hitachi, Tokyo, Japan).
Sampling and RNA-seq
For sampling, we chose young leaves for transcriptome analysis because the HL genes were thought to be more active. The young leaves from the five stages of the hybrid F1 plants were harvested and stored at − 80 °C for further analyses. Three biological replicates were set, with each including three randomly selected plants.
Total RNA was extracted using an EasyPure Plant RNA Kit (Vazyme, Nanjing, China) according to the manufacturer’s instructions. The RNA purity was assessed utilising an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). A total of fifteen cDNA libraries were constructed using a TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA, USA) following the manufacturer’s protocol. All libraries were sequenced on an Illumina HiSeq 4000 platform (Illumina). The raw data were processed by removing the low-quality sequences (> 50% bases with quality scores ≤ 5), adaptor-contaminated sequences, and sequences with ambiguous base reads accounting for more than 5%. The GC content of the total data was calculated using the GC-Profile tool (http://tubic.tju.edu.cn/GC-Profile/). Clean reads were acquired after raw sequence data processing and were aligned to the ‘TO1000′ B. oleracea reference genome (http://plants.ensembl.org/Brassica_oleracea) using HISAT2 (http://ccb.jhu.edu/software/hisat2/index.shtml).
Functional annotation and differential expression analysis of genes
To analyse the gene expression levels of different genes and different samples, the fragments per kilobase per transcript per million mapped reads (FPKM) values were calculated and used to estimate the sequencing depth and gene length on the mapped read counts. The differential expression analysis was performed using the DESeq R package (1.10.1), with a false discovery rate (FDR)-adjusted P value (q value) ≤ 0.05 and |fold change (FC)|> 1 used in this study as thresholds. Heatmaps were generated and hierarchical clustering was performed to analyse the different expression profiles. To further and systematically predict the complex biological functions and pathways associated with HL, Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were enriched by differentially expressed genes (DEGs) if the P values were ≤ 0.05. Transcription factors (TFs) and family members in this study were compared against the Plant Transcription Factor Database families of transcription factors or regulatory motifs (http://plntfdb.bio.uni-potsdam.de/v3.0/).
Validation of the RNA-Seq data and expression analysis by qRT-PCR
Quantitative real-time PCR was performed to validate the expression patterns obtained from the RNA-Seq data. Gene-specific primers were designed according to the gene sequences from the ‘TO1000′ reference genome by Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA), and primers used in qRT-PCR are shown in Table S1. First-strand cDNA was synthesised using an EasyPure Plant RNA Kit (Vazyme). The programme and reaction conditions for a PrimeScript RT Reagent Kit (Tiangen, Beijing, China) in conjunction with QuantStudio 6 Flex (Life Technologies Corporation, Carlsbad, CA, USA) were the same as those described in the manufacturers’ protocols. Three technical replicates were performed for each gene, and three biological replicates were included for each sample. Relative expression levels were calculated by the comparative 2−ΔΔCT method (Livak and Schmittgen 2001) using the expression of cabbage Actin (GenBank accession No. XM_013731369.1) as an internal control (Xing et al. 2019).
H 2 O 2 and endogenous JA and SA measurements
Leaf samples of lethal individuals and normal individuals from the F2 separated population at five stages were harvested. The H2O2 content was measured with reagents provided in a Micro Hydrogen Peroxide (H2O2) Assay Kit (Solarbio) and measured by a UV spectrophotometer (Unico Instrument Co. Ltd., Shanghai, China) at wavelength 415 nm. Endogenous SA and JA were measured via liquid chromatography–tandem mass spectrometry (LC–MS/MS) using an AB SCIEX 5500 QTrap mass spectrometer (Shimadzu, Tokyo, Japan), with three biological replications. The samples were achieved on an ACE UltraCore 2.5 SuperC18 (100 × 2.1 mm) column. The LC conditions were optimised as follows: solvent A was 0.1% formic acid in water, and solvent B was 100% acetonitrile. The gradient programme for pump B was as follows: 0–1 min, 10–20%; 1–6 min, 20–100%; 6–6.1 min, 100–10%; and 6.1–9.5 min, 10%. The flow rate was set to 0.5 mL/min and the column temperature was set at 40 °C. The mass spectra were acquired in both positive and negative modes using electrospray ionisation and quantification of all analytes was carried out in multiple reaction monitoring (MRM). LabSolutions software (Shimadzu) was used to control the instruments and process the data. Three biological replicates were set, with each group including three plants. The error bars represent the ± SEs from three independent experiments. The data were analysed by ANOVA using SAS software (SAS Institute, Cary, NC, US).
Exogenous hormone treatment
To further analyse the relationship between plant hormones and HL, exogenous hormone treatment was conducted by spraying the hybrid F1 seedlings with SA (100 mg/l), JA (100 mg/l), and H2O (as the mock treatment), with three replications for each treatment. Based on the lethality phenotyping, the seedlings were treated at 5 days before HLW1 stage.
Results
HL is a typical PCD process
To clearly describe the HL phenotype, we divided this period into five stages, based on seedling characteristics of F1 hybrids and their parents (Fig. 1, Fig. S1). At stage one, HLN (15 days after sowing; DAS) F1 hybrids were normal at the two-leaf stage (Fig. 1a). HLW1 (21 DAS) F1 hybrids showed slight growth stunting at three-leaf stage (Fig. 1b). HLW2 (24 DAS) F1 hybrids exhibited apparent growth stunting and slight yellowing (Fig. 1c). HLW3 (27 DAS) F1 hybrids showed serious growth arrest and apparent chlorosis (Fig. 1d). The leaves of HLW4 (30 DAS) appeared completely chlorotic and the plants become wilt and dead (Fig. 1e). The parent lines 09–211 and 09–222 grow normally with two to five leaves, respectively, corresponding to the five HL stages (Fig. 1f, g, Fig. S1).

Phenotypes of the cabbage F1 hybrids and the 09–211 control. a The F1 hybrids was normal at two-leaf stage (HLN). b The F1 hybrids showed slight growth stunting at three-leaf stage (HLW1). c The F1 hybrids showed apparent growth stunting and slight yellowing (HLW2). d The F1 hybrids showed seriously growth arrest and apparent chlorosis (HLW3). e The leaves appeared completely chlorotic and the plants become wilt and dead (HLW4). f, g The parent line 09–211 grows normally with two to five leaves, respectively, corresponding to the five HL stages. Scale bars = 5 cm
HE staining and light microscopy showed shrinkage of leaf and stem cells after HL (Fig. 2d–f), while normal plant cells showed plump, round cells (Fig. 2a–c). TEM observations revealed that the cells of plants undergoing HL exhibited obvious separation of the cytoplasm as well as marginalisation (Fig. 2h, i) and agglutination of chromatin and disintegration and disappearance of organelles (Fig. 2k, l). In addition, staining results of the TUNEL assay showed that there were a lot of TUNEL-positive photoreceptors in hybrid lethal plants (Fig. 2q) in comparison with normal ones (Fig. 2n), showing that DNA fragmentation of the cells increased in the lethal plants (Fig. 2p, r) compared with the normal ones (Fig. 2m–o). These results confirmed that HL is characterised by the typical PCD process, which lays a theoretical foundation for future studies of HL.

Haematoxylin–Eosin (TE) staining, transmission electron microscopy (TEM) observations and TUNEL assays of cabbage F1 hybrids and the 09–211 control. a−c TE staining of leaf longitudinal sections and stem cross-sections of the 09–211 control. e−g TE staining of leaf longitudinal section and stem cross-sections of the control and F1 hybrids. The TE staining shows that the cells shrank during hybrid lethality. g, j TEM observations of the cell and organelle morphology of the 09–211 control. h, i, k, l TEM observations of cell and organelle morphology of the F1 hybrids. The cells of the plants undergoing hybrid lethality displayed obvious separation of cytoplasm and marginalisation (h, i), as well as agglutination of chromatin and the disintegration and disappearance of organelles (k, l). m −o DAPI staining. TUNEL assay results and the merged images of the 09–211 control. p, q, r DAPI staining. TUNEL assay results and the merged images of the F1 hybrids. Scale bars = 200 μm (a,b, d, e and m–r), 100 μm (c, f), 80 μm (g–i), 20 μm (j–l)
Transcriptome sequencing and gene expression profiles
HL in cabbage is a phenotypic change of the whole plant, and the young leaves showed similar characteristics with the old ones. However, we found that the timepoint of the young leaves showing HL phenotype slightly lagged the old ones, which was more suitable for transcriptome analysis because the HL genes were more active.
To understand the general overview of differences in the gene expression associated with HL in cabbage, we prepared young leaves samples of F1 hybrids at the five different stages for transcriptome sequencing. In total, 15 samples were sequenced on a HiSeq 2000 sequencing platform. A total of 687,767,424 raw read pairs were generated, and 92.68% of clean reads were obtained after removing the adaptors and low-quality reads. In addition, Q20 values (reads with average quality scores > 20) were obtained for more than 96% of the reads, and Q30 values (reads with average quality scores > 30) were obtained for more than 92% of the reads. These results indicate that the accuracy and quality of the sequencing data were sufficient for further analysis. The average percentage of total mapped reads ranged from 75 to 84%. The percentage of reads mapped to the genes and exons was highest, which indicated that our reference genome was relatively complete. A summary of the sequencing data is shown in Table S2. The sequencing data have been deposited into NCBI sequence read archive (SRA) under BioProject accession PRJNA655525 (alias: SUB7867970).
DEGs were screened based on set criteria (q value ≤ 0.05 and |log2FC|> 1), and totals of 7144 (HLW1vsHLN), 8220 (HLW2vsHLN), 17,304 (HLW3vsHLN) and 15,240 (HLW4vsHLN) DEGs were detected, which indicated the complex molecular mechanism of HL associated with a large number of DEGs (Fig. 3a). The number of DEGs showed that they gradually increased when lethality symptoms appeared, and the largest number of differences in expression levels in the plants occurred during the HLW3 stage. The numbers of DEGs in HLW1 and HLW2 are essentially the same, suggesting that genes associated with HL have begun to exhibit different expression levels before the HLW1 stage. To assess the diversity of DEGs at the different stages, Venn diagrams were constructed (Fig. 3c). A total of 23,088 DEGs were shared, and 2724 DEGs overlapped during four stages. Furthermore, a general overview of the expression pattern was visualised by a heat map (Fig. 3b), which provided an overall understanding of the changes in gene expression. We observed that there were more upregulated DEGs than downregulated DEGs, and most of the DEGs exhibited the same expression pattern, suggesting that more genes contribute to positive regulation of HL in cabbage. The coexpression of 2724 DEGs may play a key role in HL in cabbage, and our subsequent analysis will also focus on these genes.

Analysis of DEGs in the four comparison groups. a Histogram showing the number of DEGs in the four comparison groups. b Venn diagram showing the mutual overlaps of DEGs in the four comparison groups. c Heat map showing a general overview of the expression patterns
HL was closely associated with the defence response
GO and KEGG analyses were performed between each HL stage (HLW1, HLW2, HLW3, and HLW4) and control stage (HLN) to understand the mechanisms underlying HL. GO and KEGG enrichments show difference at four comparisons groups, the top 10 GO-enriched categories and KEGG-enriched pathways for each groups are shown in Table S3. For GO enrichment terms, the early stage (HLW1vsHLN, HLW2vsHLN) mainly involved in ‘kinase activity’ and ‘binding’, and the end stage (HLW3vsHLN, HLW4vsHLN) mainly involved in ‘ribosome’ and ‘intracellular’. For KEGG pathways, we noticed that ‘plant–pathogen interaction’, ‘plant hormone signal transduction’, and ‘MAPK signalling pathway-plant’ were the most enriched at early stages (HLW1vsHLN), which indicated that these KEGGs were well correlated with HL in cabbage. Then, other stage mainly involved in ‘Ribosome’, ‘Plant–pathogen interaction’, ‘Glucosinolate biosynthesis’ (HLW2vsHLN), ‘Ribosome Porphyrin’ and ‘chlorophyll metabolism ‘Photosynthesis-antenna proteins’ (HLW3vsHLN) and ‘Ribosome’, ‘Photosynthesis-antenna proteins’ and ‘Porphyrin and chlorophyll metabolism’ (HLW4vsHLN). And ‘Photosynthesis-antenna proteins’ and ‘Porphyrin and chlorophyll metabolism’ are corresponding to the lethal phenomenon of chlorosis and wilting. This result suggested that ‘plant–pathogen interaction’, ‘plant hormone signal transduction’, and ‘MAPK signalling pathway-plant’ have worked at early stage and this net and related genes may play an important role in reveal the molecular mechanism of HL.
To characterise the complex functions of the core DEGs, the DEGs were evaluated via GO and KEGG enrichment pathway analyses. The coexpression of 2724 DEGs during four stages were assigned to 2915 GO enrichment terms, which were then grouped into three main categories: biological processes, molecular functions and cellular components. The DEGs in GO-enriched categories of biological process were mainly involved in multiorganism processes (GO:0051704) and protein phosphorylation (GO:0006468), and there were multiple GO terms related to the defence response (response to external biotic stimuli, response to other organisms, response to biotic stimuli, etc.). For cellular components, the most represented terms were ribosome (GO:0005840), MCM complex (GO:0042555) and DNA packaging complex (GO:0044815), and for molecular functions, the most represented terms were adenyl ribonucleotide binding (GO:0032559), adenyl nucleotide binding (GO:0030554) and protein kinase activity (GO:0004672) (Fig. 4a).

Functional annotation and classification and qRT-PCR verification of DEGs. a GO classifications. The GO terms are summarised on the basis of the three main categories (cellular components (CCs), molecular functions (MFs) and biological processes (BPs). The degree of significance of the enrichment of DEGs in a pathway is represented by −log10 (P value). b Top 20 pathways of KEGG functional enrichment among the DEGs. c qRT-PCR verification of the expression patterns of PR genes. The error bars represent the ± SE of three independent experiments (P ≤ 0.05)
Furthermore, 2724 DEGs were assigned to 122 KEGG pathways. The pathway ‘plant–pathogen interaction’ was the most common term, containing 45 DEGs, followed by ‘plant hormone signal transduction’ (43), ‘ribosome’ (40), ‘MAPK signalling pathway-plant’ (23) and ‘phenylpropanoid biosynthesis’ (23). The top 20 KEGG pathways with the highest representation of DEGs are shown in Fig. 4b. From the independent analysis results of each comparison group, we noticed that ‘plant–pathogen interaction’, ‘plant hormone signal transduction’, and ‘MAPK signalling pathway-plant’ were the most enriched across the four stages, which indicated that these KEGGs were well correlated with HL in cabbage. These three KEGG pathways are the typical and critical pathways associated with response reactions, which implies that HL has a close relationship with defence responses. Thus, the expression levels of most representative pathogenesis-related (PR) genes (pathogenesis-related proteins 1 (PR1), PR2, PR5, nonexpressor of-related genes 1 (NPR1), peptidylarginine deiminase 4 (PAD4), beta-glucanase (BGL2), and enhanced disease susceptibility 1 (EDS1)) were analysed by qRT-PCR (Fig. 4c). The results showed that all PR genes were upregulated during HL, which was consistent with the RNA-Seq data at the different time points. These results also demonstrated the accuracy of the transcriptome analysis in the present study.
Analysis of DEGs involved in the defence response network
To further investigate whether the DEGs activated by HL are involved in the defence network, the most enriched KEGG pathways in this study (‘plant–pathogen interaction’, ‘plant hormone signal transduction’ and ‘MAPK signalling pathway-plant’) were subjected to a combined analysis. According to comparisons of plants at the different stages, the percentage of DEGs of the three pathways was determined (Table S4). The results indicated that approximately 25% of the DEGs at the HLW1 and HLW2 stages and approximately 50% of the DEGs at the HLW3 and HLW4 stages were identified in the three pathways. The similar percentages of DEGs in each stage also suggested that these pathways may have the same expression profile. Integrated correlation networks of the three pathways and their genes were constructed (Fig. 5). Among them, Bo3g088360 (PR1), encoding pathogenesis-related gene 1, Bo6g042530 (MKK4), encoding mitogen-activated protein kinase kinase 4, and Bo3g170710 (BAK1), encoding BRI1-associated receptor kinase, contributed to all three pathways, and twelve DEGs contributed to two pathways, suggesting that the DEGs involved in the three response pathways were interconnected to play roles in the defence response for HL. Furthermore, 26 GO terms related to the defence response were enriched, and 12 were significantly enriched (P value ≤ 0.05); 12 GO terms related to the immune response were enriched, and 6 were significantly enriched (P value ≤ 0.05) (Table 1). Three core defence response pathways and GO terms related to the immune response and defence response were significantly enriched and interconnected, which indicated that HL was closely related to the defence response and immune response.

Association graph showing the relationships between three important pathways (‘plant–pathogen interaction’, ‘plant hormone signal transduction’, ‘MAPK signalling pathway-plant’) and the related core DEGs
The ‘plant–pathogen interaction’ pathway is a complex process that is controlled by the genotype and determined by the interaction of resistance genes and the corresponding virulence (Fig. S2). The downstream processes triggered by resistance genes included the hypersensitive response (HR), PCD, and the immune response, which is consistent with a previous study about HL triggered by R gene interactions (Bomblies et al. 2007; Chen et al. 2013). A total of 123 DEGs were identified during the whole stage, and 45 of them showed different expression levels at each stage. Most DEGs in the pathway were mainly receptor kinases (BAK1, CPK, MKK4), pto-interacting proteins (PTI1, RIN4), transcription factors (WRKY25), PR1, disease resistance proteins (RPS2), and EDS1. Interestingly, 44 of 45 DEGs were upregulated according to the heatmap (Fig. S3), suggesting that the ‘plant–pathogen interaction’ pathway has a significant positive response to HL and may activate downstream HR and PCD processes in cabbage. Only CNGCs (Bo5g122750) were downregulated, while another six variants (Bo1g119310, Bo4g158880, Bo5g122720, Bo4g009240, Bo3g054400, and Bo1g119320) were upregulated. More than half of the DEGs were located in the Ca2+ pathway, including cyclic nucleotide-gated channels (CNGCs)(7), calcium-dependent protein kinases (CDPKs)(6), calmodulin and calmodulin-like proteins (CALM/CMLs)(17), respiratory burst oxidase homologs (Rbohs) (1), WRKY25s (5), which cause the HR, cell wall reinforcement, defence-related gene induction and stomatal closure (Fig. 6). Many studies have reported that calcium can regulate stomatal closure, playing an important role in cell death and PCD (Pauwels et al. 1991; Aleo 2002; Verbert et al. 2007; Wang et al. 2018). On the other hand, the HR can also be triggered by interactions between two genes (RIN4 and RPS2), which then activate the upregulation of required for Mla12 resistance (RAR1) and SGT1 and the downregulation of heat-shock protein 90 (HSP90). PCD and defence amplification may also result from the upregulation of EDS1, which regulates both the intracellular oxygen burst and SA accumulation and inhibits the JA/ET pathway (Beckers and Spoel 2006). Coexpression of Rboh with CDPK5 or CDPK13 can induce ROS production, leading to HR and cell wall reinforcement (Yamauchi et al. 2017). In PAMP-trigger immunity (PTI), CDPK and Rboh were significantly upregulated, suggesting that ROS may increase during HL (Fig. 6). H2O2, one of the important representatives of ROS, was enriched in ‘MAPK signalling pathway-plant’, with eight upregulated DEGs involved with oxidative signal-Inducible1 (OXI1), mitogen-activated protein kinase kinase 4 (MKK4), MPK3, WRKY25, MAP kinase (MPK1) and PR1. The upregulated expression profile suggests that pathogen defence, cell death and H2O2 production may play a positive role in HL. A possible functional defence network of HL is shown in Fig. 6. The H2O2 content of the F1 hybrids gradually became higher than that of normal plants during the lethality stage, which is consistent with the results obtained from RNA-Seq (Fig. 6b). The HLW1 of the F1 hybrids showed the most significant upregulation, suggesting that the increase in H2O2 played an important role in the early stage of HL in cabbage. H2O2 can trigger cell death and contribute to pathogen defence, and the increased H2O2 content further indicated that HL is closely linked to the defence network at the physiological level. The expression levels of some ‘defence network’ genes in Fig. 6a were analysed by RT-PCR which also demonstrated the accuracy of the transcriptome analysis in the present study (Fig. S4).

Possible functional defence network of cabbage hybrid lethality and a heat map of related DEGs. a Possible functional defence network of hybrid lethality. The upregulated DEGs in this category are red, the downregulated DEGs are green, and DEGs that are both, upregulated and downregulated, are orange. b H2O2 content. The error bars represent the ± SE of three independent experiments (P ≤ 0.05). c Heatmap of DEGs in the defence network
Analysis of DEGs involved in the PCD process
On the basis of HL phenotyping of cabbage and the results of previous studies, we assumed that HL in cabbage was associated with the PCD process. And the defence response network further confirms that the immune and defence response lead to cell death and PCD. The GO terms involved in PCD and the cell death process were screened to further elucidate the PCD process involved in HL. Thus, nine GO terms were identified, including regulation of cell death (GO: 0043067), programmed cell death (GO: 0012501), and cell death (GO: 0008219), that were associated with six core DEGs (Table S5). A network between these nine GO terms and related DEGs was subsequently constructed (Fig. 7). Six of these DEGs were mechanosensitive channel candidate (MCA2) (Bo1g039710), Rboh (Bo9g033770), ATL55 (Bo9g172200), lectin receptor kinase (LecRK) (Bo3g097940), RPS2 (Bo1g041880), and BI-1 (Bo9g069310). In addition, the molecular functions are associated with calcium cations (MCA2, RbohF), the endoplasmic reticulum (ATL55, BI-1) and disease resistance (LecRK, RPS2), with the same upregulated expression pattern. Within this network, MCA2, RbohF, BI-1 and ATL55 contribute to more than six GO terms related to PCD; however, cell death is associated with all six, and regulation of cell death and programmed cell death is associated with five DEGs. The results showed that they play a positive role in PCD and the cell death process response to HL in cabbage. Our results may provide evidence for an analysis of key genes and molecular mechanisms of HL in cabbage.

Nine GO terms related to PCD and cell death and their expression patterns
Plant hormones, especially SA and JA, were closely related to HL
Within the ‘plant hormone signal transduction’ pathway, 43 of the significantly enriched DEGs were involved in the Auxin (AUX, 16), cytokine (2), abscisic acid (ABA) (1), ethylene (ET, 5), brassinosteroid (BRs, 10), JA (6) and SA (5) signalling pathways. The results revealed DEGs associated with multiple endogenous hormone signalling responses to HL, especially those involving AUX, ET, BRs, JA and SA (Fig. 8). AUX is a well-known phytohormone that contributes to plant growth and cell enlargement (Leopold 1955). However, the DEGs related to AUX showed complex differences in their expression; AUX (Bo8g102050, Bo3g042430) and SAUR (Bo2g014230, Bo7g119560, Bo7g117030, Bo3g167560, Bo5g027860, Bo4g197270, Bo2g085040, Bo9g111470) were downregulated, whereas AUX1/IAA, encoding auxin influx carriers (Bo5g004310, Bo1g103500), and GH3 (Bo9g167830, Bo2g011190) were upregulated. These results suggest that the complex differences in AUXs, SAURs and GH3s response to HL. The same expression profiles occurred for BR, which is involved in cell division and elongation (Ting et al. 2018). CYCD3 (Bo1g083500, Bo7g116660, Bo3g169030, Bo3g169030, Bo1g007590, Bo7g063880) was downregulated, while others (BAK1, BSK, BZR1/2, TCH4) were upregulated. In addition, the DEGs related to the ET, JA and SA signalling pathways were enriched are associated with senescence, the stress response and disease resistance (Tang 2005; Dhirendra 2014; Häffner et al. 2014; Yang et al. 2016). For the ET signalling pathway, SIMKK (Bo6g042530) and ERF1 (Bo9g069400, Bo2g132270)/ERF2 (Bo4g176080, Bo4g052480) were downregulated, indicating that the ET signalling pathway responds negatively to HL in cabbage. JAZ is the key regulator of the signalling pathway that can transduce the JA signal from the receptor to the nucleus and affects gene transcription (Chini et al. 2007), and five upregulated DEGs (Bo8g104890, Bo8g068340, Bo8g102890, Bo6g086720, Bo6g112300) were detected. JAR1 (Bo4g009300), which encodes a JA-amino synthetase that is required to activate JA for optimal signalling, was upregulated (Koo et al. 2009). Specifically, in the SA signalling pathway, the DEGs were associated with all three genes (NPR1, TGA, PR-1), including Bo7g070020, Bo9g018570, Bo00615s190, Bo8g071300 and Bo9g035510, whose expression was markedly upregulated. The significantly upregulated expression of DEGs in the SA signalling pathway indicated that SA may significantly be involved in the response of HL in cabbage. In addition, many DEGs involved in hormone signalling were also enriched in the ‘MAPK signalling pathway-plant’ pathway, such as those involved with ET, BRs and ABA, providing more evidence for their close relationships with HL.

Plant hormone signal transduction pathway components and corresponding heat map expression profiles
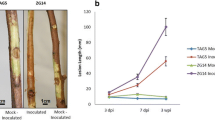
To further analyse the relationships between plant hormones and HL, the contents of key endogenous hormones were measured, and exogenous hormone treatments were performed. In our study, we chose respective hormones that may be associated with HL (SA/JA) based on RNA-Seq. The results showed that SA markedly increased, while JA presented no change (Fig. 9a, b). Thus, these results suggest that HL is accompanied by the accumulation and outbreak of SA, which is in agreement with the transcriptome analysis of the SA response. Furthermore, hormone treatment was conducted by spraying the F1 hybrid seedlings. The results of the exogenous application showed that exogenous SA slows the onset of HL and increases the growth duration. However, SA cannot prevent HL from occurring. In contrast, JA accelerated HL, shortened the lethality time and aggravated the symptoms (Fig. 9c). The results revel their possible antagonistic roles in HL. The marked increase in SA content and delay or advance of HL treated with exogenous SA and JA showed that SA and JA can contribute to or respond to HL in cabbage.

Fluctuations in endogenous SA and JA between the lethal individuals and normal individuals (control) from the F2 separated population and F1 hybrids in response to exogenous SA and JA treatment. a Endogenous SA content between the lethal individuals and normal individuals. b Endogenous JA content between the lethal individuals and normal individuals, the error bars represent the ± SE of three independent experiments (P ≤ 0.05). c F1 hybrids in response to exogenous SA and JA treatment. F1 hybrids treated with H2O exhibited initial hybrid lethal phenotyping (HLW1), F1 hybrids treated with SA grew normally, and F1 hybrids treated with JA exhibited moderate hybrid lethal phenotyping (HLW2). Scale bars = 5 cm
Transcription factors responded globally to HL
Increasing evidence has indicated that transcription factors (TFs) play an important role in plant morphogenesis regulation, metabolic regulation and growth and development. In the present study, 58 TF families involving 33,381 genes were identified. Among these TF families, eleven included more than one thousand numbers (the bHLH, MYB-related, NAC, B3, ERF, WRKY, G2-like, C2H2, MYB, GRAS, and C3H families). 1776 of 2724 DEGs (65%) encoding TFs were identified and belonged to 56 TF families, which suggested that a large number of TFs displayed different expression responded globally to HL. Among them, the top five families were the NAC (9.01%), bHLH (8.33%), ERF (8.22%), WRKY (6.93%), and MYB-related (6.48%) families (Fig. 10). These TFs play an important role in different biological processes. For example, the upregulation of WRKY25 and downregulation of WRKY29 may contribute to defence-related gene induction; Bo7g070020 (NPR1) belongs to the MYB TF family and induces defence gene expression (Mou et al. 2003). Kjaersgaard et al. (2011) reported that members of the NAC (CUC, ATAF1, 2, NAM) TF family are upregulated during senescence in barley and that they interact with barley radical-induced cell death 1 (RCD1) to regulate cell death. These identified differentially expressed TFs provide additional information for revealing the regulatory mechanism for HL in cabbage.

Numbers and classification of differentially expressed TFs involved in hybrid lethality. Categories of transcription factors that constitute less than 1% of the total are not marked on the pie chart
Discussion
HL genes and defence response
Previous studies have shown that most cloned genes triggering HL are associated with defence response (Hollingshead 1929; Muller 1942; Dobzhansky 1959; Tezuka and Marubashi 2006). To better understand the molecule mechanism of HL in plants, the sequences of these reported HL genes were blasted on the Brassica genome (http://plants.ensembl.org/Brassica_oleracea/Info/Index?db=core), including DM1 and DM2 from Arabidopsis, Hiw1 and Hiw2 from rice, Rin4 from lettuce and pTAC14 from Mimulus (Bomblies et al. 2007; Jeuken et al. 2009; Chen et al. 2013; Chae et al. 2014; Zuellig et al. 2018). The six homologous genes were identified with 55–82% sequence similarity. However, they were not located in the mapping region of HL genes in cabbage (Xiao et al. 2017). Moreover, expression levels and functional annotation also were analysed (Table S6). Among them, Bo4g142440 and Bo3g006960 were upregulated at three HL stages; Bo3g083790 was upregulated at two HL stages. And Bo3g162060, encoding chloroplast protein, was downregulated at two end stages, consistent with the phenotype of HL. These genes are associated with response to stimulus (Bo4g142440, Bo3g006960), immune system and hypersensitive response (Bo3g083790), which showed a close relationship between HL and defence responses. These results also provide some clues to reveal the molecular mechanism of HL in cabbage. Similarly, our transcript results showed that many DEGs were enriched in GO terms and pathways related to immune response and defence response, and the defence response network based on the ‘plant–pathogen interaction’ pathway and ‘MAPK signalling’ pathway was constructed. ‘Plant–pathogen interactions’ pathway revealed a complex process involving the interaction of resistance genes and the corresponding virulence, causing HR, immune response, cell death, and PCD (Flor 2003; Mitsuhara et al. 2008; Bari and Jones 2009). This pathway involves some gene interaction pairs, such as RIN4 and RPS2, both of which were upregulated according to the transcript results. In lettuce, one of the two interacting genes involved in HL is Rin4, which is known to interact with multiple R gene products (Jeuken et al. 2009). Chen et al. (2013) also indicated that the interactions between R genes activate the autoimmune response and defence responses in the absence of biotic/abiotic stress, leading to HL. Although HL is activated according to the gene-to-gene model, unlike the plant–pathogen interaction model, we deduced that ‘plant–pathogen interactions’ pathway may be an important reference to study HL. Based on a previous study and our transcript results, we infer that R genes interactions might activate the defence response and immune response, ultimately leading to PCD.
Plant hormones and HL
Plant hormones are activated by incompatible interactions between plants and pathogens to enhance plant resistance (Bari and Jones 2009). The AUX, ABA, ET, BR, JA, SA and GA signalling pathways respond to the plant immune response, forming complex and interconnected plant hormone signalling networks to coordinate different stress responses and growth (Pre et al. 2008; Wu et al. 2008; Bari and Jones 2009; Clay et al. 2009). In our study, the DEGs related to ET, JA and SA showed the close relationship between the defence response and HL. Especially in SA signal transduction, the upregulated DEGs and bursting SA content suggested that SA signalling may increase after HL. The same result was reported in Arabidopsis epistatic interactions, in which more SA accumulated in those plants than in the Arabidopsis recombinant inbred line (Rubén et al. 2009). Thus, we conclude that SA pathway activation may be needed to achieve complete HL. Moreover, endogenous and exogenous SA and JA measurements further confirmed the close relationship between plant hormones and HL. Alcazar et al. (2009) indicated that SA accumulation is necessary for growth retardation and the enhanced response in plant. Thus SA accumulation may be triggered by the defence response network of HL in our study, which is a similar result of SA accumulation in Arabidopsis hybrid necrosis (Alcazar et al. 2009). For the exogenous SA measurements result, some studies showed that SA treatment can enhance the activity of antioxidant enzymes and maintain the integrity of cell membrane, delaying cell death and senescence (Ton et al. 2002 Imran et al. 2007). Phytohormone treatment of hybrid weakness in Rice showed that SA could restore the height of plants expressing hybrid weakness, while other phytohormones (BRs, zeatin and GA3) appear to have little effect (Chen et al. 2013). Thus, we hypothesise that the enhanced defence by SA is temporarily resistant to immune system disorders and activity of antioxidant enzymes in HL. There was no change in JA content, but JA treatment accelerated HL. Antoniw et al. (1980) reported that exogenous JA treatment also can enhance plant defence resulting from the upregulation of PR genes. Then we infer that some genes or pathways regulating the HL may be triggered by exogenous JA. Their antagonistic roles of SA/JA in HL are similar to plant disease resistance reaction, which can modulate defence genes expression to regulate HL (Hideki et al. 2004). Moreover, a complex network of cross-talk between the SA and JA pathways further fine-tunes plant defence responses (Kunkel 2002). For example, the application of JA depressed the activation of the genes for the acidic PR proteins, which are SA dependent (Tomoya et al. 1998). For the complex results about the SA/JA content and the exogenous treatments, additional work is needed to reveal this genetic mechanism.
EDS1 is a key SA-related gene and also contributes to effectors-triggered immunity (ETI) which is mainly mediated by R proteins and PTI (Falk et al. 1999; Wagner et al. 2013). EDS1 was upregulated during HL stages (Fig. 4c), which implied that HL had a close relationship with defence responses. EDS1 also is a key node of the plant immune signalling network (Beckers et al. 2006). In the ‘plant–pathogen interaction’ pathway, complex with EDS1 and PAD4 led to PCD and defence amplification (Fig. 6a). In the ‘plant hormone signal transduction’ pathway, EDS1 can regulate the intracellular oxygen burst and SA accumulation as well as inhibits the JA/ET pathway (Beckers and Spoel 2006; Straus et al 2010). Thus, higher H2O2 content in HL plants may be regulated by EDS1. The results showed that SA/EDS1 may play an important role in defence responses network induced by HL.
ROS and HL
ROS has been proposed as important inducers of different types of developmental and/or environmental PCD. PCD controlled by ROS occurs during developmental processes as the aleurone cell death and leaf senescence, various forms of abiotic stress, the HR and allelopathic plant–plant interactions (Apel and Hirt 2004). Plant cells have well-developed defence systems against ROS and are able to remove them through non-enzymatic and enzymatic antioxidant processes (Cho et al. 2009). The major ROS-scavenging enzymes in plants consist of superoxide dismutase (SOD) and catalase (CAT) (Mittler et al. 2004). In Fig. 6a, significantly upregulated CDPK and Rboh which can participate in ROS accumulation indicated that ROS may increase during HL. In ‘MAPK signalling pathway-plant’ pathway, eight upregulated DEGs triggered H2O2 production (Fig. S5). Moreover, H2O2 content of lethal individuals gradually became higher, which further confirmed accumulation of ROS during the HL stages. Then key ROS-scavenging genes SOD and CAT in cabbage also were analysed. Among which, SOD (Bo4g165150/Bo9g014230) was downregulated and CAT (Bo1g006740, Bo3g167210 and Bo5g030530) performed no differential expression at HLW1 and HLW2 stages and downregulated at HLW3 and HLW4 stages. These results indicated that SOD and CAT enzymes may be repressed or inactivated during HL. Thus, the balance between ROS-producing and -scavenging system was broken, leading to PCD in HL.
Author contribution statement
ZX wrote and revised the manuscript. ZX and XL isolated the samples and performed the trait measurements, molecular experiments and marker assays. MZ, HL and ZF conceived the idea and critically reviewed the manuscript. LY, YZ and YW coordinated and designed the study. All the authors have read and approved the final manuscript.
Abbreviations
- DEG:
-
Differentially expressed gene
- HL:
-
Hybrid lethality
- HR:
-
Hypersensitive response
- JA:
-
Jasmonic acid
- PCD:
-
Programmed cell death
- SA:
-
Salicylic acid
- TF:
-
Transcription factor
References
Aleo M (2002) Endoplasmic reticulum Ca2+ signaling and calpains mediate renal cell death. Cell Death Differ 9:734–741 https://doi.org/10.1038/sj.cdd.44010291
Alcazar R, Garcia AV, Parker JE, Reymond M (2009) Incremental steps toward incompatibility revealed by Arabidopsis epistatic interactions modulating salicylic acid pathway activation. P Natl Acad Sci 106:334–339. https://doi.org/10.1073/pnas.0811734106
Antoniw J, Ritter C, Pierpoint W, Van Loon L (1980) Comparison of three pathogenesis-related proteins from plants of two cultivars of tobacco infected with TMV. J Gen Virol 47:79–87. https://doi.org/10.1099/0022-1317-47-1-79
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant 55:373–399. https://doi.org/10.1146/annurev.arplant.55.031903.141701
Bari R, Jones J (2009) Role of plant hormones in plant defence responses. Plant Mol Biol 69:473–488. https://doi.org/10.1007/s11103-008-9435-0
Beckers G, Spoel S (2006) Fine-tuning plant defence signalling: salicylate versus jasmonate. Plant Biol 8:1–10. https://doi.org/10.1055/s-2005-872705
Bomblies KJ, Lempe P, Epple N, Warthmann C, Lanz J, Dangl L, Weigel D (2007) Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol 5:1962–1972. https://doi.org/10.1371/journal.pbio.0050236
Chae E, Bomblie K, Kim S, Karelin D, Zaidem M, Ossowski S, Martín-Pizarro C, Laitinen R, Rowan B, Tenenboim H, Lechner S, Demar M, Habring-Müller A, Lanz C, Rätsch G, Weigel D (2014) Species-wide genetic incompatibility analysis identifies immune genes as hot spots of deleterious epistasis. Cell 159:1341–1351. https://doi.org/10.1016/j.cell.2014.10.049
Chen C, Chen H, Shan J, Zhu M, Shi M, Gao J, Lin H (2013) Genetic and physiological analysis of a novel type of interspecific hybrid weakness in rice. Mol Plant 6:716–728. https://doi.org/10.1093/mp/sss146
Chini A, Fonseca S, Fernández G, Adie B, Chico J, Lorenzo O, García-Casado G, López-Vidriero I, Lozano F, Ponce M, Micol J, Solano R (2007) The JAZ family of repressors is the missing link in jasmonate signalling. Nature 448:666–671. https://doi.org/10.1038/nature06006
Cho D, Shin D, Jeon B, Kwak J (2009) ROS-mediated ABA signaling. J Plant Biol 52:102–113. https://doi.org/10.1007/s12374-009-9019-9
Clay N, Adio A, Carine D, Georg J, Ausubel F (2009) Glucosinolate metabolites required for an Arabidopsis innate immune response. Science 323:95–101. https://doi.org/10.1126/science.1164627
Deng J, Fang L, Zhu X, Zhou B, Zhang T (2019) CC-NBS-LRR gene induces hybrid lethality in cotton. J Exp Bot 70:5145–5156. https://doi.org/10.1093/jxb/erz312
Dhirendra K (2014) Salicylic acid signaling in disease resistance. Plant Sci 228:127–134. https://doi.org/10.1016/j.plantsci.2014.04.014
Dobzhansky T (1959) Genetics and the origin of species. Nature 184:587–588. https://doi.org/10.1093/aibsbulletin/2.2.14-b
Falk A, Feys BJ, Frost LN, Jones JD, Daniels MJ, Parker JE (1999) EDS1, an essential component of R gene-mediated disease resistance in Arabidopsis has homology to eukaryotic lipases. Proc Natl Acad Sci USA 96:3292–3297. https://doi.org/10.1073/pnas.96.6.3292
Flor H (2003) Current status of the gene-for-gene concept. Annu Rev Phytopathol 9:275–296. https://doi.org/10.1146/annurev.py.09.090171.001423
Häffner E, Karlovsky P, Splivallo R, Traczewska A, Diederichsen E (2014) ERECTA, salicylic acid, abscisic acid, and jasmonic acid modulate quantitative disease resistance of Arabidopsis thaliana to Verticillium longisporum. BMC Plant Biol 14:85. https://doi.org/10.1186/1471-2229-14-85
Hannah M, Krmer K, Geffroy V, Kopka BM, Erban A, Vallejos C, Heyer A, Sanders F, Millneret P (2007) Hybrid weakness controlled by the dosage-dependent lethal (DL) gene system in common bean (Phaseolus vulgaris) is caused by a shoot-derived inhibitory signal leading to salicylic acid-associated root death. New Phytol 176:537–549. https://doi.org/10.1111/j.1469-8137.2007.02215.x
Hollingshead L (1929) Lethal factor in Crepis effective only in an interspecific hybrid. Genetics 15:114–140. https://doi.org/10.1007/BF02983371
Hu Y, Xue Y, Liu J, Fang Z, Yang L, Zhang Y, Lv H, Liu Y, Li Z, Zhuang M (2016) Hybrid lethality caused by two complementary dominant genes in cabbage (Brassica oleracea L). Mol Breed 36:73. https://doi.org/10.1007/s11032-016-0498-3
Imran H, Zhang Y, Du G (2007) Effect of salicylic acid (SA) on delaying fruit senescence of Huang Kum pear. Front Agric China 1:456–459. https://doi.org/10.1007/s11703-007-0075-y
Jeuken MJ, Zhang NW, McHale LK, Pelgrom KE, Lindhout B, Michelmore R, Vissera R, Niksa R (2009) Rin4 causes hybrid necrosis and race-specific resistance in an interspecific lettuce hybrid. Plant Cell 21:3368–3378. https://doi.org/10.1105/tpc.109.070334
Kjaersgaard T, Jensen M, Christiansen M, Gregersen P, Skriver K (2011) Senescence-associated barley NAC (NAM, ATAF1, 2, CUC) transcription factor interacts with radical-induced cell death 1 through a disordered regulatory domain. J Biol Chem 286:35418–35429. https://doi.org/10.1074/jbc.M111.247221
Koo A, Gao X, Jones A, Howe G (2009) A rapid wound signal activates the systemic synthesis of bioactive jasmonates in Arabidopsis. Plant J 59:974–986. https://doi.org/10.1111/j.1365-313X.2009.03924.x
Kunkel B (2002) Cross talk between signaling pathways in pathogen defense. Curr Opin Plant. https://doi.org/10.1016/S1369-5266(02)00275-3
Livak K, Schmittgen T (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402. https://doi.org/10.1006/meth.2001.1262
Maheshwari S, Barbash D (2010) The genetics of hybrid incompatibilities. Annu Rev Genet 45:331–355. https://doi.org/10.1146/annurev-genet-110410-132514
Mitsuhara I, Takayoshi I, Shigemi S, Yuki Y, Hiroyuki K, Sakino H (2008) Characteristic expression of twelve rice PR1 family genes in response to pathogen infection, wounding, and defense-related signal compounds (121/180). Mol Genet Genomics 279:415–427. https://doi.org/10.1007/s00438-008-0322-9
Mittler R, Vanderauwera S, Gollery M, Breusegem F (2004) Reactive oxygen gene network of plants. Trends Plant Sci 9:490–498. https://doi.org/10.1016/j.tplants.2004.08.009
Mou Z, Fan W, Dong X (2003) Inducers of plant systemic acquired resistance regulate NPR1 function through redox changes. Cell 113:935–944. https://doi.org/10.1016/S0092-8674(03)00429-X
Muller HJ (1942) Isolating mechanisms, evolution and temperature. Biol Symp 6:71–125
Orr HA, Presgraves DC (2000) Speciation by postzygotic isolation: forces, genes and molecules. BioEssays 22:1085–1094. https://doi.org/10.1002/1521-1878(200012)22:12%3c1085::AID-BIES6%3e3.0.CO;2-G
Pauwels P, Josée E, Janssen P (1991) Ca++ and Na+ channels involved in neuronal cell death protection by flunarizine. Life Sci 48:1881–1893. https://doi.org/10.1016/0024-3205(91)90220-6
Pickersgill B (1971) Relationships between weedy and cultivated forms in some species of Chili peppers (genus Capsicum). Evolution 25:683–691. https://doi.org/10.2307/2406949
Pre M, Atallah M, Champion A, De Vos M, Pieterse C, Memelink J (2008) The AP2/ERF domain transcription factor ORA59 integrates jasmonic acid and ethylene signals in plant defense. Plant Physiol 147:1347–1357. https://doi.org/10.1104/pp.108.117523
Rubén A, Ana V, Jane E, Matthieu R (2009) Incremental steps toward incompatibility revealed by Arabidopsis epistatic interactions modulating salicylic acid pathway activation. Proc Natl Acad Sci USA 106:334–339. https://doi.org/10.1073/pnas.0811734106
Shii C, Mok T, Temple S, Mok D (1980) Expression of developmental abnormalities in hybrids of Phaseolus vulgaris L. Interaction between temperature and allelic dosage. J Hered 71:219–222. https://doi.org/10.1093/oxfordjournals.jhered.a109353
Straus MR, Rietz S, Ver L, Themaat E, Bartsch M, Parker J (2010) Salicylic acid antagonism of EDS1-driven cell death is important for immune and oxidative stress responses in Arabidopsis. Plant J 62:628–640. https://doi.org/10.1111/j.1365-313X.2010.04178.x
Tang D (2005) Regulation of plant disease resistance, stress responses, cell death, and ethylene signaling in Arabidopsis by the EDR1 protein kinase. Plant Physiol 138:1018–1026. https://doi.org/10.1104/pp.105.060400
Tezuka T, Marubashi W (2006) Hybrid lethality in interspecific hybrids between Nicotiana tabacum and N. suaveolens: evidence that the Q chromosome causes hybrid lethality based on Q-chromosome-specific DNA markers. Theor Appl Genet 112:1172–1178. https://doi.org/10.1007/s00122-006-0219-0
Ting Z, Xu P, Wang W, Wang S, Caruana J, Yang H (2018) Arabidopsis g-protein β subunit AGB1 interacts with BES1 to regulate brassinosteroid signaling and cell elongation. Front Plant Sci 8:2225. https://doi.org/10.3389/fpls.2017.02225
Tomoya N, Ichiro Shigemi S, Norihiro O, Yuko O (1998) Antagonistic effect of salicylic acid and jasmonic acid on the expression of pathogenesis-related (PR) protein genes in wounded mature tobacco leaves. Plant Cell Physiol 5:1662–1670. https://doi.org/10.1086/498022
Verbert L, Devogelaere B, Parys J, Missiaen L, de Smedt H (2007) Proteolytic mechanisms leading to disturbed Ca2+ signaling in apoptotic cell death. Calcium Bind Proteins 2(1):21–29
Wagner S, Stuttmann J, Rietz S, Guerois R, Brunstein E, Bautor J, Niefind K, Parker J (2013) Structural basis for signaling by exclusive EDS1 heteromeric complexes with SAG101 or PAD4 in plant innate immunity. Cell Host Microbe 14:619–630. https://doi.org/10.1016/j.chom.2013.11.006
Wang Y, Hao X, Lu Q, Wang L, Qian W, Li N, Ding C, Wang X, Yang Y (2018) Transcriptional analysis and histochemistry reveal that hypersensitive cell death and H2O2 have crucial roles in the resistance of tea plant (Camellia sinensis (L.) O. Kuntze) to anthracnose. Hortic Res 1:18. https://doi.org/10.1038/s41438-018-0025-2
Wiebe G (1934) Complementary factors in barley giving a lethal progeny. J Hered 25:273–274. https://doi.org/10.1093/oxfordjournals.jhered.a103943
Xiao Z, Hu Y, Zhang X, Xue Y, Fang Z, Yang L, Zhang Y, Liu Y, Li Z, Liu X, Liu Z, Zhuang M (2017) Fine mapping and transcriptome analysis reveal candidate genes associated with hybrid lethality in cabbage (Brassica oleracea). Genes 5:8. https://doi.org/10.3390/genes8060147
Xing M, Su H, Liu X, Yang L, Zhang Y, Wang Y, Fang Z, Lv H (2019) Morphological, transcriptomics and phytohormone analysis shed light on the development of a novel dwarf mutant of cabbage (Brassica oleracea). Plant Sci 290:110283. https://doi.org/10.1016/j.plantsci.2019.110283
Yamada T, Marubashi W (2003) Overproduced ethylene causes programmed cell death leading to temperature-sensitive lethality in hybrid seedlings from the cross Nicotiana suaveolens × N. tabacum. Planta 217:690–698. https://doi.org/10.1007/s00425-003-1035-2
Yamauchi T, Yoshioka M, Fukazawa A, Mori H, Nishizawa N, Tsutsumi N, Yoshioka H, Nakazono M (2017) An NADPH oxidase RBOH functions in rice roots during lysigenous aerenchyma formation under oxygen-deficient conditions. Plant Cell 29:775–790. https://doi.org/10.1105/tpc.16.00976
Yang C, Li W, Cao J, Meng F, Liu J (2016) Activation of ethylene signaling pathways enhances disease resistance by regulating ROS and phytoalexin production in rice. Plant J 89:338. https://doi.org/10.1111/tpj.13388
Zhou W, Yoshida K, Shintaku Y, Takeda G (1991) The use of IAA to overcome interspecific hybrid inviability in reciprocal crosses between Nicotiana tabacum L. and N. repanda Willd. Theor Appl Genet 82:657–661. https://doi.org/10.1007/BF00226805
Zuellig M, Sweigart A, Malik H (2018) Gene duplicates cause hybrid lethality between sympatric species of Mimulus. PLoS Genet 14:e1007130. https://doi.org/10.1371/journal.pgen.1007130
Acknowledgements
The work reported here was performed at the Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, Beijing 100081, China.
Funding
This work was financially supported by grants from the National Natural Science Foundation of China (31572139), the Key Projects of the National Key Research and Development Program of China (2016YFD0100307), the Central Public-interest Scientific Institution Basal Research Fund (Y2020PT01), and the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-IVFCAAS).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Dorothea Bartels.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiao, Z., Liu, X., Fang, Z. et al. Transcriptome and plant hormone analyses provide new insight into the molecular regulatory networks underlying hybrid lethality in cabbage (Brassica oleracea). Planta 253, 96 (2021). https://doi.org/10.1007/s00425-021-03608-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00425-021-03608-1